

Graphical abstract
Keywords: Genomic profile, Essential thrombocythemia, JAK2, Genetic variants
Abstract
Clonal analysis of patients with triple negative myeloproliferative neoplasm (MPN) has provided evidence of additional aberrations, including epigenetic alterations. To discover such novel genetic aberrations, patients were screened through next-generation sequencing using a myeloid sequencing panel of 54 genes using a genetic analyser. Genetic variants in 28 genes, including TET2, BCOR, BCR, and ABL1 were identified in a triple negative essential thrombocythemia (ET) patient. The individual role of some of these variants in disease pathogenesis has yet to be studied. Somatic mutations in the same genes have been reported with variable frequencies in myeloid malignancies. However, no pathogenic impact of these variants could be found; therefore, long-term follow up of patients with genetic analysis of a large cohort and the use of whole genome sequencing is required to assess the effects of these variants.
Introduction
Essential thrombocythemia (ET) is a clonal myeloproliferative disorder characterized by the abnormal or dysregulated proliferation of megakaryocytes in a normocellular marrow. It is a rare disorder, with an estimated annual incidence of 0.6–2.5 per 100,000 people [1].
The phenotypic behaviour of this disease varies widely from asymptomatic or incidental discovery of thrombocytosis to presentation with major vascular occlusive events. The primary concerns of ET that require prompt diagnosis and management are the risk of thrombosis and progression into myelofibrosis or acute leukaemia [2]. The clonal nature of the disease is explained to a certain extent by the presence of the JAK2 V617F mutation in up to 50% of patients [3]. In a small subset of patients (1–2%), somatic mutations in the thrombopoietin receptor cMPL are found [4]. In the recent past, CALR mutations were found in 73% of ET cases, irrespective of JAK2 or MPL mutations [5]. Cytogenetic abnormalities, including del (20q) and trisomy 8, are detected in fewer than 10% of cases at the time of diagnosis [6]. Improvement in next-generation sequencing technology has led to the study of the genomic profile of MPN patients in more detail, and new genetic alterations, including epigenetic events in pathogenesis of MPN, have been identified. The effects of these genetic alterations on the clinical phenotype of the disease are uncertain and require further evaluation.
Case scenario
A 34-year-old gentleman with isolated thrombocytosis (platelet count 1800 × 109/L), was discovered incidentally during a workup for a viral illness. There was no history of blood disorders in the patient’s family. The peripheral smear was essentially normal except for marked platelet anisocytosis. There was no splenomegaly on examination. Bone marrow biopsy was consistent with essential thrombocythemia (Fig. 1A). Reticulin fibrosis was grade 0 (Fig. 1B). During initial molecular screening, screened MPL exon 10 (W515X), Jak2 exon 12 by Sanger sequencing and JAK2 (V617F) by PCR. Molecular testing for JAK2, CALR exon 9 and cMPL mutations were negative. Cytogenetic analysis showed a normal male karyotype. The patient was initially managed with two sessions of plateletpheresis. Hydroxyurea was started at 1 g/day, and the dose was then gradually escalated to 2 g/day. He achieved only partial remission with maximum doses of hydroxyurea. He was switched to anagrelide at 2 mg/day. During the course of the disease, he did not experience any haemorrhagic or thrombotic complications, but constitutional symptoms such as pruritus, fatigue, numbness, and tingling in fingers affected his quality of life. Interferon alpha 2A was started at 3 million units three times a week, but due to adverse events (severe myalgias and flu-like symptoms), it could not be continued. Considering the lack of response to first- and second-line therapies, Ruxolitinib (a JAK2 inhibitor) was started. The platelet count did not respond to Ruxolitinib, but there was a clinically significant improvement noticed in the constitutional symptoms.
Fig. 1A.

Bone marrow biopsy.
Fig. 1B.

Reticulin stain.
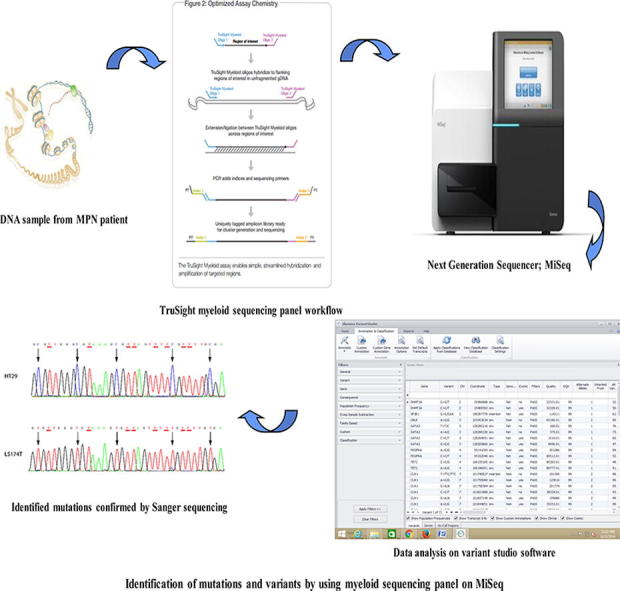
Considering the negativity for BCR-ABL1, JAK2, CALR and cMPL, prolonged thrombocythemia unresponsive to therapy in this case, performed next-generation sequencing to identify genetic markers in the 54 genes, including 15 full genes and 35 hotspots (ABL1, ASXL1, ATRX, BCOR, BCORL1, BRAF, CALR, CBL, CBLB, CBLC, CDKN2A, CEBPA, CSF3R, CUX1, DNMT3A, ETV6/TEL, EZH2, FBXW7, FLT3, GATA1, GATA2, GNAS, HRAS, IDH1, IDH2, IKZF1, JAK2, JAK3, KDM6A, KIT, KRAS, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PDGFRA, PHF6, PTEN, PTPN11, RAD21, RUNX1, SETBP1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, and ZRSR2) involved in the regulation of histone function and DNA methylation that may explain the phenotypic behaviour and provide potential therapeutic options to treat the disease.
Methodology
The DNA was extracted from the peripheral blood sample of the patient using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Research protocol was approved by the Institutional Review Board (ERC/IRB) and conformed to the tenets of the Declaration of Helsinki. Written informed consent was obtained from the patient.
The myeloid sequencing panel of 54 genes (complete coding exons of 15 genes and exonic hotspots of 39 genes) was sequenced. The panel focuses on ∼141 kb of genomic content consisting of ∼250 bp, and the medium coverage of the sample was >95% of amplicons at >500× coverage. Amplicon libraries were prepared using a TruSight myeloid sequencing panel (Illumina® Inc, San Diego, CA, USA) and paired-end sequencing runs were performed on a MiSeq (Illumina® Inc, San Diego, CA, USA) genome sequencer. Data analysis alignment was performed with on-instrument MiSeq reporter software. The mutations identified as pathogenic were confirmed using the Sanger method according to the standard protocol (BigDye® Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems®).
Results
Genomic analysis was performed using variant studio software v2.2 (Illumina, San Diego, CA, USA). The medium coverage of the sample was >95% of amplicons at >500× coverage. Several databases, such as dbSNP, COSMIC and Ensemble, were used to report mutations and search for variants.
A total of 71 variants were identified, including 6 deletions, 5 insertions and 60 variants identified as single nucleotide variants (SNVs). Of the SNVs discovered, 16 were synonymous variants, and 11 were missense variants in the TET2, BCOR, GATA2, CDKN2A, NOTCH1, TP53, CBLC, ASXL1 and BCORL1 genes (Table 1). The remaining 33 variants include 4 splice region variants, 1 3’ UTR variant, 1 downstream variants and 27 intronic variants with a MAF value of >1%, as shown in Supplementary Table 1. The variants c.86C > G, p.P29R (rs12498609) in TET2 and c.5102T > G, p.V1701G (rs200163930) in BCOR were found to be deleterious on SIFT and possibly damaging on polyphen. p.P29R is a missense variant in the TET2 gene, but this variant lies within a non-conserved region and is thus not regarded as a true missense mutation. p.V1701G is also a missense mutation but on the International Cancer Genome Consortium (ICGC) data portal p.V1701G is a low-functional impact missense mutation.
Table 1.
Missense variants identified by NGS in an ET case.
| Gene | Genotype | Variant allele freq | Protein position | Amino acids | Sift | PolyPhen | dbSNP ID |
|---|---|---|---|---|---|---|---|
| GATA2 | het | 0.6 | 164 | A/T | Tolerated (0.68) | Benign (0.008) | rs2335052 |
| TET2 | het | 0.5 | 29 | P/R | Deleterious (0.01) | Possibly damaging (0.628) | rs12498609 |
| TET2 | het | 0.5 | 1762 | I/V | Tolerated (0.32) | Benign (0.029) | rs2454206 |
| CDKN2A | het | 0.3 | 76 | A/V | Tolerated (0.34) | Benign (0.01) | – |
| NOTCH1 | het | 0.3 | 1731 | P/L | Tolerated (0.06) | Benign (0.143) | – |
| NOTCH1 | het | 0.3 | 1731 | P/S | Tolerated (0.3) | Benign (0.022) | – |
| TP53 | het | 0.2 | 379 | R/C | Tolerated (0.12) | Benign (0.003) | – |
| CBLC | het | 0.4 | 435 | P/S | Tolerated (0.09) | Benign (0.021) | rs116023028 |
| ASXL1 | hom | 1 | 815 | L/P | Tolerated (0.17) | Benign (0) | rs6058694 |
| BCOR | het | 0.2 | 1701 | V/G | Deleterious (0.02) | Benign (0.249) | rs200163930 |
| BCORL1 | hom | 1 | 111 | F/L | Tolerated (1) | Unknown (0) | rs4830173 |
Bold items mean that these variants were found to be deleterious on prediction models (SIFT and Polyphen).
Discussion
Essential thrombocythemia is a BCR-ABL1 negative myeloproliferative neoplasm that primarily involves megakaryocyte lineages, and the disease is manifested by sustained elevations of platelet counts in the peripheral blood. This proliferation is thought to occur as a consequence of somatic mutations in JAK2 V617F, MPL W515 K/L and CALR genes in up to 70–90% of ET cases [5], [7]. After excluding mutations in these three genes, a proportion of ET patients still do not harbour any identifiable mutation. New insights into the molecular pathogenesis of MPN have revealed the presence of additional acquired or inherited genetic modifiers outside of JAK2, MPL or CALR, which may be responsible for the phenotypic variation found in ET [8]. According to Tefferi, rare genetic polymorphisms/mutations other than JAK2/CALR/MPL may be detected in PV or ET, and these genetic alterations do not necessarily produce unfavourable impacts on the prognosis of the disease [9].
In the present case, the patient had very high platelet counts, absence of a clonal marker and a normal karyotype. The clinical course of the patient was insistent and he showed resistance to all therapies, including the novel agent offered to him. Symptoms such as tinnitus, numbness of fingers and paresthesias were developed later on. The patient’s clinical course and limited genetic information necessitated performing extensive genetic screening to identify new genetic markers. By using a myeloid sequencing panel, variants were identified in 28 genes, including TET2 and BCOR. Variants in these two genes were found to be deleterious and to exert a possibly damaging effect on prediction models such as SIFT and Polyphen.
Mutations in the gene TET2 (ten eleven translocation two) were first reported in MPN, myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML) by Bernard et al. in 2009 [10]. The identified variant c.86C > G, p.P29R (rs12498609) in the TET2 gene in this patient lies within a non-conserved region, and it may not be considered a true missense mutation, but according to Hanri, TET2 mutations situated outside of the conserved domains may also have the potential to alter protein function and may extra clinical impact [11]. Abdel Wahab et al. evaluated the largest set of MPN samples for somatic TET2 alterations by sequencing all coding exons of TET2 and found frame shift, nonsense, and non-synonymous alterations [12].
A study conducted by Patriarca et al. found 2(3.6%) ET patients with TET2 mutations in their cohort. The clinical course of these patients was indolent, with a mean platelet count <1000× 109/L and no significant history of thrombotic or haemorrhagic complications [13]. The effect of TET2 alterations on the clinical outcome of patients with different MPN is debatable. Thus far, TET2 mutations have not been shown to influence survival or prognosis in MPN patients [14].
Another variant identified in this particular ET patient was p.V1701G, a missense mutation in the BCOR gene, which is found to be a low-functional-impact variant on the ICGC portal. Its effect on the disease’s pathogenesis is unknown. Mutations in the BCOR gene have been reported in a small number of patients with MDS and AML. Sequencing of BCOR and related BCORL1 genes in a cohort of 354 MDS patients identified 4.2% and 0.8% of mutations, respectively [15]. Although low in frequency, BCOR might be considered as a key gene in risk stratification [16]. Deep sequencing technologies show that BCOR mutations commonly arise after other concomitant mutations in MDS. The study by Grossmann et al. [15] reported that BCOR mutations were associated with an inferior outcome in a cohort of 422 CN-AML patients (25.6% vs 56.7% overall survival at 2 years; P = 0.032).
Thus far, mutations or polymorphisms in BCOR have not been reported in MPN. The identified variants in this unique case are reported as neutral variants and no alteration in BCR, ABL1, JAK2, CALR, and cMPL are found. Thus, the genetic complexity of MPN will increase, and additional genetic causes need to be identified.
Conclusions
This is an unusual case report of ET resistance to both conventional and novel therapies. Next-generation sequencing identified genetic variants in 28 genes; however, no pathogenic impacts of these variants could be found in the literature. Thus, management of this patient requires longer follow up with a careful assessment of disease progression or transformation.
Conflict of Interest
There is no conflict of interest between authors.
Footnotes
Peer review under responsibility of Cairo University.
References
- 1.Beer P.A., Erber W.N., Campbell P.J., Green A.R. How I treat essential thrombocythemia. Blood. 2011;117(5):1472–1482. doi: 10.1182/blood-2010-08-270033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cervantes F. Management of essential thrombocythemia. Hematol Am Soc Hematol Educ Program. 2011;2011:215–221. doi: 10.1182/asheducation-2011.1.215. [DOI] [PubMed] [Google Scholar]
- 3.Levine R.L., Wadleigh M., Cools J., Wernig G., Huntly B.J., Boggon T.J. Activating mutation in the tyrosine kinase JAK2 in polycythemiavera, essential thrombocythemia and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 4.Vannucchi A.M., Antonioli E., Guglielmelli P., Pancrazzi A., Guerini V., Barosi G. Characteristics and clinical cor-relates of MPL 515W > L/K mutation in essential thrombocythemia. Blood. 2008;112:844–847. doi: 10.1182/blood-2008-01-135897. [DOI] [PubMed] [Google Scholar]
- 5.Klampfl T., Gisslinger H., Harutyunyan A.S., Nivarthi H., Rumi E., Milosevic J.D. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- 6.Gangat N., Tefferi A., Thanarajasingam G., Patnaik M., Schwager S., Ketterling R. Cytogenetic abnormalities in essential thrombocythemia: prevalence and prognostic significance. Eur J Haematol. 2009;83:17–21. doi: 10.1111/j.1600-0609.2009.01246.x. [DOI] [PubMed] [Google Scholar]
- 7.Cervantes F., Passamonti F., Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL-negative myelopro-liferative disorders. Leukemia. 2008;22(5):905–914. doi: 10.1038/leu.2008.72. [DOI] [PubMed] [Google Scholar]
- 8.Beer P.A., Jones A.V., Bench A.J., Goday-Fernandez A., Boyd E.M., Vaghela K.J. Clonal diversity in the myeloproliferative neoplasms: independent origins of genetically distinct clones. Br J Haematol. 2009;144(6):904–908. doi: 10.1111/j.1365-2141.2008.07560.x. [DOI] [PubMed] [Google Scholar]
- 9.Tefferi A., Lasho T.L., Guglielmelli P., Finke C.M., Rotunno G., Elala Y. Targeted deep sequencing in polycythemiavera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30. doi: 10.1182/bloodadvances.2016000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernard O.A., Delhommeau F., Fontenay M., Vainchenker W. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;25(10):785–788. doi: 10.1051/medsci/20092510785. [DOI] [PubMed] [Google Scholar]
- 11.Hanri DP. Mutational analysis of the TET2 gene in Philadelphia negative myeloproliferative neoplasms. Master’s thesis. Bloemfontein: University of the Free State; 2014. Available from <http://hdl. handle.Net/11660/1442>.
- 12.Abdel-Wahab O., Mullally A., Hedvat C., Garcia-Manero G., Patel J., Wadleigh M. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patriarca A., Colaizzo D., Tiscia G., Spadano R., Di Zacomo S., Spadano A. TET2 mutations in Ph-negative myeloproliferative neoplasms: identification of three novel mutations and relationship with clinical and laboratory findings. Biomed Res Int. 2013;2013:929840. doi: 10.1155/2013/929840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tefferi A., Lim K.H., Abdel-Wahab O., Patel J., Patnaik M.M., Hanson C.A. Detection of mutant TET2 in myeloid malignancies other than myelopro-liferative neoplasms: CMML, MDS. MDS/MPN AML Leukemia. 2009;23(7):1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grossmann V., Tiacci E., Holmes A.B., Kohlmann A., Martelli M.P., Kern W. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153–6163. doi: 10.1182/blood-2011-07-365320. [DOI] [PubMed] [Google Scholar]
- 16.Damm F., Chesnais V., Nagata Y., Yoshida K., Scourzic L., Okuno Y. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169–3177. doi: 10.1182/blood-2012-11-469619. [DOI] [PubMed] [Google Scholar]