

Abstract
Background/Aims: Nicotinic acid (NA), a lipid-lowering drug, serves as a source of NAD+, the cofactor for Sirt1. Leucine (Leu) stimulates the AMPK/Sirt1 axis and amplifies the effects of other AMPK/Sirt1 activating compounds. Therefore, we tested the interactive effects of leucine and low dose NA on AMPK/Sirt1 signaling and downstream effects of lipid metabolism in cell culture, C. elegans and mice. Methods: LDL-receptor knockout mice were fed an atherogenic Western diet supplemented with leucine (24 g/kg diet) and sub-therapeutic NA combinations (50 mg/kg diet and 250 mg/kg diet) or low therapeutic NA (1000 mg/kg diet) for 8 weeks to evaluate markers of hyperlipidemia and atherosclerosis. Results: NA-Leu increased P-AMPK and Sirt1 in adipocytes and myotubes. In C. elegans, NA-Leu increased P-AMPK and DAF-16 (FOXO), reduced lipid accumulation and increased median survival under mild oxidative stress conditions. In the mice, NA-Leu reduced total cholesterol, cholesterol esters, plasma triglycerides, atherosclerotic lesion size, lipid area, and aortic macrophage infiltration, similar to the therapeutic NA dose. Conclusion: Leu amplifies the effects of NA on lipid metabolism, hyperlipidemia and atherosclerosis in mice, at least in part by activation of the AMPK/Sirt1 axis. This combination may be a potential therapeutic alternative for hyperlipidemia and atherosclerosis.
Keywords: AMPK, atherosclerosis, C. elegans, leucine, lipid metabolism, nicotinic acid, Sirt1
Introduction
Atherosclerosis is a slowly progressive disease which is the main cause of morbidity and mortality in developed countries [1,2]. It is caused by the accumulation of cholesterol and triglycerides in the arterial wall which initiates an inflammatory and cell death signaling cascade, resulting over time in reduced wall elasticity and decreased blood flow through the arteries. Therefore, dyslipidemia and hypercholesterolemia are considered major risk factors for disease development. Consequently, lipid lowering treatments are one of the most successful strategies to reduce cardiovascular disease risk [2]. Statins are the most widely used drugs and have significantly reduced the number of major cardiovascular events; however sub-optimal dosing or drug discontinuation occurs in 10 to 20% of patients due to the development of side effects. In addition, accumulating data shows a relationship between statin use, elevation of blood glucose levels and incidence of new-onset diabetes [3,4].
Nicotinic acid (NA) is a form of vitamin B3 (niacin) and has been used for decades as an effective treatment of dyslipidemias, as it lowers total and LDL-cholesterol, triglycerides and lipoprotein, raises HDL-cholesterol and reduces atherosclerotic plaque progression [5]. In addition, its anti-inflammatory, anti-thrombotic and anti-oxidant activities contribute to its overall beneficial effects [4]. However, therapeutic doses (1-3 g/day) are generally poorly tolerated because of severe vasodilation and flushing response [5]. In addition, the use of niacin is constrained in diabetic patients due to its potential hyperglycemic adverse effects [4,6]. While side effects are milder with the sustained and extended release preparations, they are still sufficient to limit patient acceptance.
The effects of niacin have been widely thought to be mediated by activation of the G-protein-coupled hydroxycarboxylic acid receptor 2 (HCAR2 or GPR109A), thereby suppressing adipocyte lipolysis and reducing substrate availability for hepatic lipid synthesis [7]. However, recent evidence suggests that niacin’s lipid lowering effects are mediated by GPR109A independent mechanisms since treatment with GPR109A agonists in mice lowered plasma free fatty acids comparable to niacin without any effects on triglycerides and other lipid parameters [7]. Moreover, GPR109A receptors are not expressed in liver cells and therefore can not exert niacin’s effect on lipoproteins.
NA is also a substrate for NAD+ biosynthesis, the cofactor for Sirt1, and treatment with NA increases cellular NAD+ levels and Sirt 1 activity [8,9]. Sirt1 is a well-known regulator of glucose and lipid metabolism. In addition, it deacetylates and thereby activates the liver X receptor (LXR), a regulator of cholesterol and lipid homeostasis [10]. Moreover, Sirt1 has direct beneficial effects on vascular endothelium-dependent function such as vasodilation via eNOS derived nitric oxide (NO), reversal of cholesterol transport in macrophages, prevention of foam cell production, as well as antithrombotic and anti-inflammatory effects by downregulation of the NFκB pathway [10]. Therefore, it is likely that some effects of NA are mediated by Sirt 1 activation.
We have previously demonstrated that leucine activates Sirt1, at least in part, by lowering the activation energy for NAD+, thereby co-activating and amplifying the effects of other sirtuin activators while reducing their required effective concentration [11,12]. Therefore, we assessed in this study whether leucine also amplifies the effects of NA on lipid metabolism and atherosclerosis and enables a meaningful dose reduction.
Materials and methods
Cell culture
Murine 3T3-L1 pre-adipocytes (Zenbio, Research Triangle Park, NC, USA) were grown in the absence of insulin in Dulbecco’s modified Eagle’s medium (DMEM, 25 mM glucose) containing 10% fetal bovine serum (FBS) and antibiotics (1% penicillin-streptomycin) (= adipocyte medium) at 37°C in 5% CO2 in air. Confluent pre-adipocytes were induced to differentiate with a standard differentiation medium (DM2-L1, Zen-Bio Inc., NC). Pre-adipocytes were maintained in this differentiation medium for 3 days and subsequently cultured in adipocyte medium for further 8 to 10 days to allow at least 90% of cells to reach full differentiation before treatment. Medium was changed every 2-3 days; differentiation was determined microscopically via inclusion of fat droplets.
Human hepatoma HepG2 cells (ATCC, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle’s medium (DMEM, 5.5 mM glucose) containing 10% fetal bovine serum (FBS) and antibiotics (1% penicillin-streptomycin) at 37°C in 5% CO2 in air. Medium was replaced with fresh medium every 2 to 3 days. Cells were split at a 1:4 ratio at 70 to 80% confluency.
C2C12 muscle cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and antibiotics (1% penicillin-streptomycin) at 37°C in 5% CO2 in air. Cells were grown to 100% confluence, changed into differentiation medium (DMEM with 2% horse serum and 1% penicillin-streptomycin), and fed with fresh differentiation medium every other day until myotubes were fully formed (6 days).
Caenorhabditis elegans (C. elegans) maintenance
Worms (N2 Bristol wild-type) were obtained from the Caenorhabditis Genetics Center (CGC) at the University of Minnesota and grown on standard NGM plates with E. coli (OP50) as food source at 21°C. For treatments, adult worms were bleached to obtain eggs, which were allowed to hatch overnight in M9 buffer. Then synchronized L1 larvae were transferred to E. coli fed NGM plates containing indicated treatments for about 35 hours to reach L4/young adult stage. All treatments were added to the agar in indicated concentrations.
C. elegans Oil Red O staining
Treated L4/young adult worms were washed off from plates three times with PBS and collected in a 15 ml conical tube, then centrifuged at 1000 g for 30 sec. The supernatant was discarded and the pellet was washed with 10 ml PBS. After centrifugation, the supernatant was discarded except 400 µl, which was transferred to a new 1.5 ml eppendorf tube. Then 500 µl of 2x MRWB (160 mM KCl, 40 mM NaCL, 14 mM Na2EGTA, 1 mM Spermidine HCl, 0.4 mM Spermine, 30 mM NaPIPES pH 7.4, 0.2% b-Mercaptoethanol) and 100 µl of 20% Paraformaldehyde was added and gently rocked for 60 min at room temperature. Then tubes were centrifuged at 1500 g for 30 sec, then aspirated and washed with PBS once, centrifuged again and aspirated to 300 µl. Then 700 µl of isopropanol was added, mixed by inverting the tube and incubated with gentle shaking for 15 min at room temperature. Tubes were centrifuged, and isopropanol was removed. Then 1 ml of 60% filtered Oil Red O dye solution (0.5 g Oil Red O in 100 ml anhydrous isopropanol, equilibrated for 2 days by stirring at RT, then 4 vol ddH2O was mixed with 6 vol dye solution and equilibrated for 15 min at RT, then filtered with 0.2 M pore size) was added to worms and rotated on shaker overnight. Worms were pelleted by centrifugation (1200 g for 30 sec), dye was removed and washed 4 times with ddH2O to remove any unbound stain. For quantification, the Oil Red O was eluted from the cells by addition of 100% isopropanol and the optical density of 200 µl aliquots (triplicates/sample) was determined at a wavelength of 540 nm using a Biotek Synergy HT Microplate Reader (BioTek, Winooski, VT, USA). Data were normalized to protein content using the Pierce BCA protein assay kit.
Lifespan study protocol
50 to 60 synchronized adult worms per group were placed on NGM agar plates seeded with E. coli strain OP-50 (= counted as day 4 of study). To study lifespan under mild oxidative stress, 2 mM paraquat was added to the NGM agar in addition to the treatments (10 nM NA, 0.5 mM Leu, 10 μM NA or NA (10 nM)-Leu (0.5 mM) combination). For the lifespan study under normal condition, the worms were maintained on regular NGM plates (control) or NGM plates containing NA (10 nM)-Leu (0.5 mM). The worms were maintained at 21°C throughout the duration of the study. Live worms were placed on new plates every day to eliminate progeny. Worms were scored as dead if they did not respond to repeated touches with the platinum pick and scored as censored if they crawled off the plate.
Protein preparation for western blot
To obtain worm lysate, treated L4/young adult worms were washed off from plates with M9 buffer and collected into microcentrifuge tubes. After centrifugation (500 g for 5 min) supernatant was removed to about 100 µl. Then 250 µl RIPA buffer plus Protease and Phosphatase inhibitor mix was added.
To obtain cell lysate, cells were grown on 6-well plate and treated as described above. Cells were washed with PBS and then lysed with RIPA buffer plus Protease and Phosphatase inhibitor mix, incubated on ice for 10 minutes. Cell or worm lysates were homogenized, then centrifuged at 16,000 g for 10 min at 4°C. The clear supernatant was used for further experiments. Protein content was determined using the Pierce BCA protein assay kit.
Western blot
AMPK, Phospho-AMPKα (Thr172)- and Sirt1 antibodies were obtained from Cell Signaling (Danvers, MA, cat # 2532, 2535 and 9475, respectively), Sir2 and DAF-16 antibody were obtained from MyBiosource Inc. (San Diego, CA, cat # MBS621556) and Santa Cruz Biotechnology Inc. (Dallas, TX, cat # sc-9229), respectively. Protein levels of cell extracts were measured by BCA kit (Thermo Scientific, Pittsburgh, PA). Equal amounts of protein (10 to 30 ug) were resolved on 10% Tris/HCL polyacrylaminde gels (Criterion precast gel, Bio-Rad Laboratories, Hercules, CA), transferred to PVDF membranes, incubated in blocking buffer (3% BSA in TBS) and then incubated with primary antibody (1:1000 dilution for AMPK, Phospho-AMPK and Sirt1, 1:3000 dilution for Sir2, and 1:200 dilution for DAF-16), washed and incubated with secondary (anti-rabbit for AMPK, Phospho-AMPK and Sirt1, and anti-goat for DAF-16) horseradish peroxidase-conjugated antibody. Visualization and chemiluminescent detection was conducted using BioRad ChemiDoc instrumentation and software (Bio-Rad Laboratories, Hercules, CA) and band intensity was assessed using Image Lab 4.0 (Bio-Rad Laboratories, Hercules, CA), with correction for background. Band intensities were normalized to stain-free blots to control for loading, as described [13,14].
Seahorse fatty acid oxidation
The palmitate-stimulated oxygen consumption rate (OCR) was measured with the Seahorse XF 24 analyzer (Agilent Technologies Inc., Santa Clara, CA, USA) as previously described [15]. Cells were seeded at 40,000 cells per well, differentiated as described above, treated for 24 hours with the indicated treatments, washed twice with non-buffered carbonate-free pH 7.4 low glucose (2.5 mM) DMEM containing carnitine (0.5 mM), equilibrated with 550 mL of the same media in a non-CO2 incubator for 30 minutes, and then inserted into the instrument for 15 minutes of further equilibration. O2 consumption was measured in three successive baseline measures at eight-minute intervals prior to injection of palmitate (200 μM final concentration). Post-palmitate-injection measurements were taken over a 3-hour period with cycles consisting of 10 min break and three successive measurements of O2 consumption.
Sirt1 activity
The Sirt1 FRET-based screening assay kit (Cayman Chemical Company, Ann Arbor, MI, USA) was used for the measurement of Sirt1 activity in 3T3L1 adipocytes. The protocol was modified for the measurements in cells as follows; cell lysate (5 uL) of differentiated 3T3L1 treated with Leu (0.5 mM), NA (10 nM) or combination for 24 hours was incubated with peptide substrate under low NAD+ (250 uM) concentrations. The fluorescence was measured with excitation and emission wavelengths of 360 nm and 450 nm, respectively. The increase in fluorescence is proportional to the amount of deacetylated substrate and thus Sirt1 activity.
Animals
This study and all animal procedures were performed under the auspices of an Institutional Animal Care and Use Committee-approved protocol of the Georgia State University and in accordance with PHS policy and recommendations of the Guide (Protocol number A12024).
LDL receptor knockout (LDLRKO) mice were obtained from Jackson Laboratories (Bar Harbor, ME), and housed in groups under room temperature in a humidity-controlled environment with a regular light and dark cycle. All mice were provided free access to an atherogenic western diet (WD) containing 0.21% cholesterol (by weight) and 40% calories from fat and water for 4 weeks prior to treatment. After this induction period, the mice were randomized to one of the following treatment groups (n=12/group): (a) the WD diet alone, (b) WD and 24 g of leucine/kg diet, (c) WD and 24 g of leucine/kg diet and 50 mg nicotinic acid/kg diet, (d) WD and 24 g of leucine/kg diet and 250 mg nicotinic acid/kg diet, or (e) WD and 1000 mg nicotinic acid/kg diet. The treatments were administered for an additional eight weeks (total study duration 12 weeks); then the mice were humanely euthanized with CO2 inhalation. Blood and tissues were collected for further experiments as described below.
Plasma cholesterol and triglyceride levels
After four and eight weeks of treatment, mice were fasted for 4 hours and blood samples in all groups were obtained from the tails and collected into EDTA-coated tubes to analyze plasma lipid and cholesterol profiles. Plasma total cholesterol (TC, Pointe Scientific, Canton, MI), free cholesterol (FC, Wako, Richmond, VA) and triglyceride (TG, Wako, Richmond, VA) concentrations were measured using enzymatic assays according to manufacturer’s instructions. Cholesterol ester (CE) was calculated by multiplying the difference between TC and FC by 1.67.
Atherosclerosis assessment
Following euthanasia, the circulatory system was perfused with phosphate-buffered saline (PBS), then the heart and aorta were removed. The upper one-third of the heart was dissected and embedded in Optimal Cutting Temperature Compound (Sakura Tissue-Tek, Torrance, CA), frozen, and stored at -80°C. Blocks were serially cut at 8 µm intervals and stained with hematoxylin and 0.5% Oil Red O (Sigma-Aldrich) to evaluate aortic sinus atherosclerotic intimal area. Atherosclerotic lesion area and Oil Red O positive area were quantified using Image-Pro Plus software (Media Cybernetics, Bethesda, MD). Whole aorta (from sinotubular junction to iliac bifurcate) was dissected and fixed in 10% formalin, and the adventitia was cleaned. Aortas were opened along the longitudinal axis and pinned onto black silicon elastomer (Rubber-Cal, Santa Ana, CA) for the quantification of atherosclerotic lesion area. The percentage of total aortic surface covered with atherosclerotic lesions was quantified by Image-Pro Plus software (Media Cybernetics, Bethesda, MD).
Macrophage infiltration
Sections of the aortic sinus were immuno-stained with rat monoclonal antibody against macrophage-specific CD68 (Clone FA11, 1:75, AbD Serotec, Raleigh, NC) followed by staining with alkaline phosphatase-conjugated mouse anti-rat (for CD68, 1:50) secondary antibodies (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Control slides contained no primary antibody. The CD68-positive areas were analyzed using Image-Pro Plus software (Media Cybernetics, Bethesda, MD).
Statistical analysis
Data were analyzed via one-way analysis of variance and least significant difference test was used to separate significantly different group means using GraphPad Prism version 6 (GraphPad Software, La Jolla California USA, www.graphpad.com). All data are expressed as mean ± SEM.
For the statistical analysis of the lifespan study with C. elegans, data were analyzed via the standard approach for survival analysis using Kaplan-Meier estimate, as described [16,17]. Statistical significance between curves was determined with the Gehan-Breslow Wilcoxon test using Prism 6 (GraphPad Software, La Jolla California, USA, www.graphpad.com); this test gives more weight to deaths at early time points [18]. The hazard ratio describes the rate of deaths in the treatment group compared to the control group, assuming that the rate is consistent over time.
Results
We previously demonstrated that leucine synergizes with other Sirt1/AMPK activators, such as resveratrol or metformin, to increase muscle, adipocyte and liver fat oxidation in vitro and in vivo [12,19]. To test whether these effects could be also achieved with a combination of a low concentration of NA, an indirect Sirt1 activator, we explored Sirt1 and AMPK signaling in C2C12 muscle cells, 3T3L1 adipocytes and HepG2 liver cells. As demonstrated in Figure 1, leucine combined with low dose NA increased AMPK phosphorylation and Sirt1 activity in 3T3L1 adipocytes, and C2C12 muscle cells more than the individual components, although leucine and NA (10 nM) also had modest effects on AMPK and Sirt1. Since fat oxidation is a downstream effect of this signaling pathway, we measured the oxygen consumption rate (OCR) after palmitate stimulation in muscle and liver cells (Figure 2), and found them to be significantly increased by the NA-Leu combination by 41% and 16% in C2C12 and HepG2 cells, respectively, compared to control untreated cells.
Figure 1.
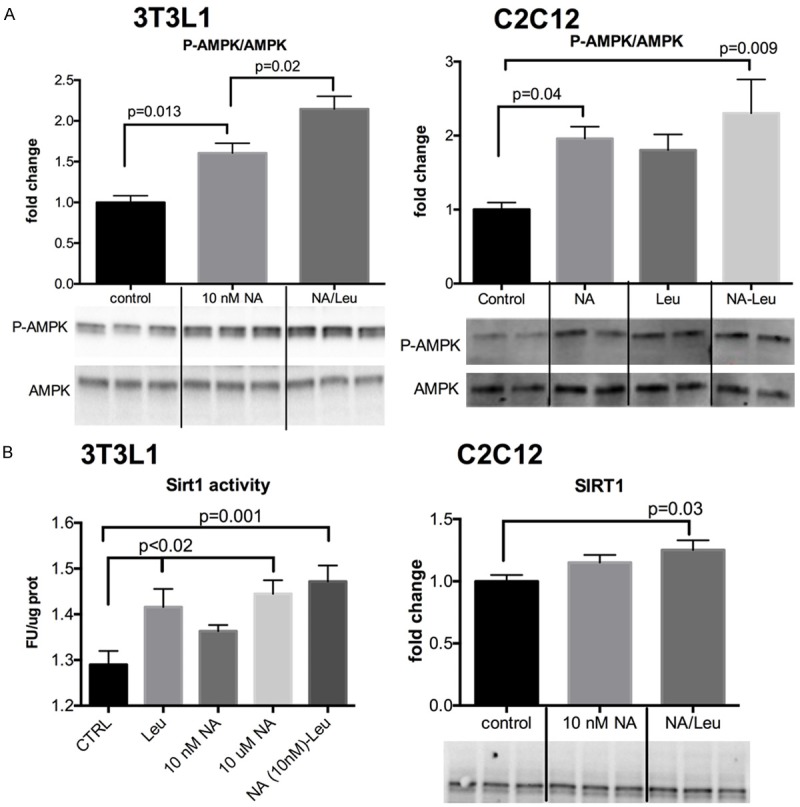
NA-Leu combination increases AMPK and Sirt1 activity in muscle cells and fat cells. Differentiated 3T3L1 adipocytes and C2C12 muscle cells were treated with indicated treatments for 4 or 24 h. Protein expression of Phospho-AMPK and total AMPK (A) and Sirt1 activity in 3T3L1 and Sirt1 protein expression in C2C12 (B) were measured, as described under Methods. Quantitative data and representative blots are shown. Data are represented as mean ± SEM (n=3 to 6).
Figure 2.
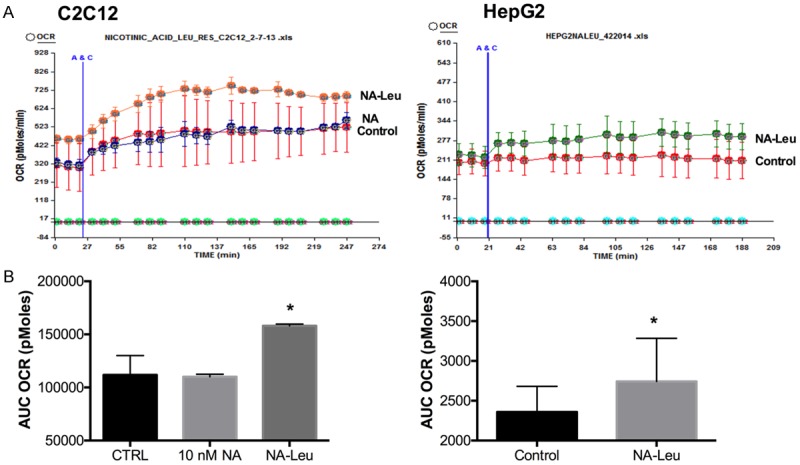
NA-Leu combination increases palmitate-induced fat oxidation in muscle cells and liver cells. Differentiated C2C12 muscle cells (n=3) (A) and HepG2 liver cells (n=10) (B) were treated with indicated treatments for 24 h. Oxygen consumption rate (OCR) after 200 μM palmitate injection (A&C) was measured and the area under the curve (AUC) was calculated. Data are represented as mean ± SEM. *significantly different from control (P<0.05).
Next, we explored the overall effects of the NA-Leu combination in the simple organism C. elegans, which is considered a well-defined model for obesity and lipid metabolism [20]. Figure 3 demonstrates that Sir2, the ratio of Phospho-AMPK/AMPK as well as DAF-16, the ortholog of the transcription factor FOXO, were increased in C. elegans by 85%, 43% and 100%, respectively. This was associated with a 50% decrease in lipid accumulation in the worms (Figure 3D). Although the individual compounds exhibited a small effect on these parameters, only the NA-Leu combination resulted in a significant reduction of lipid accumulation. To test whether these effects can influence overall life expectancy, we conducted lifespan studies in C. elegans. Since metabolic diseases such as obesity and diabetes are associated with an increase in oxidative stress, we conducted the lifespan studies under normal and mild oxidative stress conditions (2 mM paraquat) (Figure 4). There was no difference in the survival curves under normal conditions (Figure 4D). However, under mild oxidative conditions, the NA-Leu combination significantly increased median survival by 25% and reduced the hazard ratio by 20% (Figure 4A and 4C).
Figure 3.
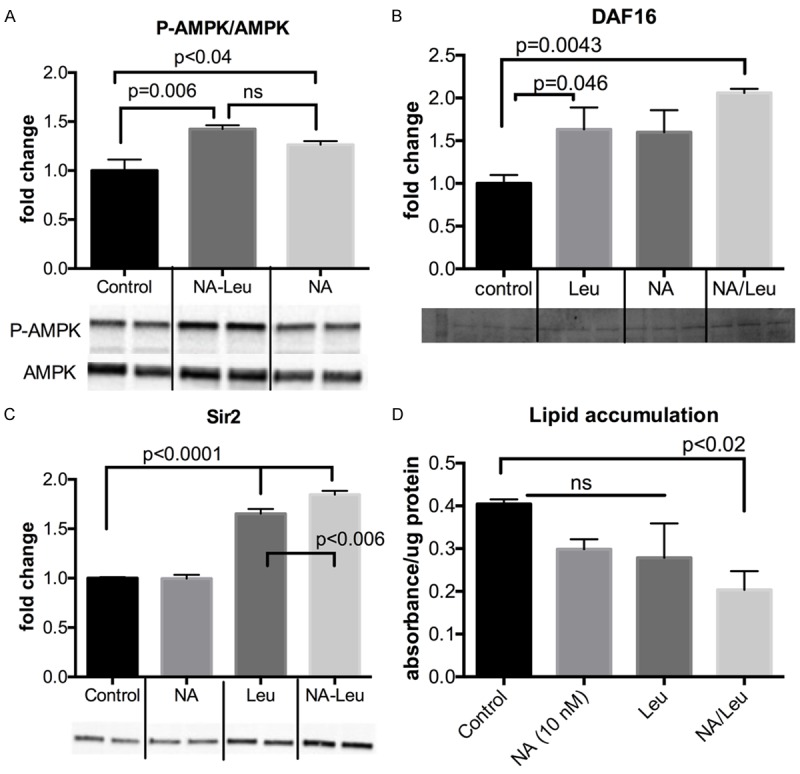
NA-Leu combination increases P-AMPK and DAF-16 protein expression and reduces lipid accumulation in C. elegans. C. elegans L1 larvae were maintained on agar plates containing indicated treatments until adulthood. Protein expression of P-AMPK/AMPK (A), DAF-16 (B) and Sir2 (C) are shown as quantitative data and representative blots (n=2-4). (D) Quantitative data of lipid accumulation after Oil Red O Staining (n=4). Data are represented as mean ± SEM.
Figure 4.
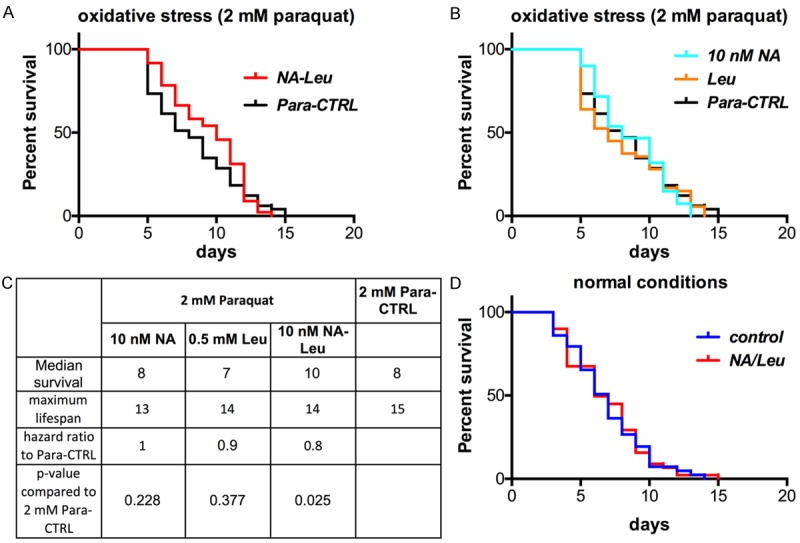
NA-Leu increases the median lifespan in C. elegans under mild oxidative conditions. Survival curves of synchronized L4 worms maintained on NGM agar plates until death with or without NA-Leu under mild oxidative conditions (2 mM paraquat (A and B), n=60 per group) or without paraquat (= normal conditions (D), n=50 per group). (B) Survival curves of individual treatment groups under mild oxidative conditions (n=60 per group). (C) Lifespan table summarizing median survival, maximum lifespan and hazard ratio using the Kaplan-Meier survival analysis and Gehan-Breslow-Wilcoxon test to determine statistical significance between curves (P<0.05), as described under Material & Methods.
Since NA has been used for decades as a therapeutic drug for dyslipidemia and atherosclerosis, we next tested the effects of the NA-Leu combination on lipid metabolism and atherosclerosis in LDLRKO mice, a mouse model of atherosclerosis. Concentrations of about 100-300 mg/kg body weight are used in the literature to achieve niacin effects on lipid metabolism and atherosclerosis [21-24]. Therefore, we selected a concentration of 1000 mg/kg diet as the therapeutic dose; this is equivalent to about 100 mg NA/kg body weight (based on a 30 g mouse and a food intake of 3 g/day) and approximately equivalent to a low therapeutic dose of nicotinic acid in hypercholesterolemic humans (~1,500 mg/day). A four-times and twenty-times lower dose of NA was used for the combination with leucine which we assumed to have no effect by itself. As expected, the atherogenic western diet resulted in profound elevations in plasma cholesterol, cholesterol esters, and trigylcyerides in the LDLRKO mice. Addition of a therapeutic dose of NA (1,000 mg/kg diet) for four weeks resulted in 20% and 28% decreases in total cholesterol and cholesterol esters, respectively (P<0.01) (Figure 5A). Although leucine exerted no independent effect on these parameters, addition of leucine to sub-therapeutic doses of NA (50 or 250 mg/kg diet) resulted in comparable decreases in total cholesterol and esterified cholesterol to that found with the therapeutic dose (P<0.01). These differences were sustained at eight weeks time point (Figure 5B). Similarly, addition of a therapeutic dose of NA (1,000 mg/kg diet) resulted in a 32% decrease in plasma triglycerides (P<0.01) (Figure 5A) at four weeks, and a statistically comparable decrease in triglycerides was found when leucine was added to sub-therapeutic doses of NA (50 or 250 mg/kg diet) (P<0.01). However, these reductions were not sustained at 8 weeks, although there was still a trend for the NA-Leu 250 mg combination (P=0.058) (Figure 5B).
Figure 5.
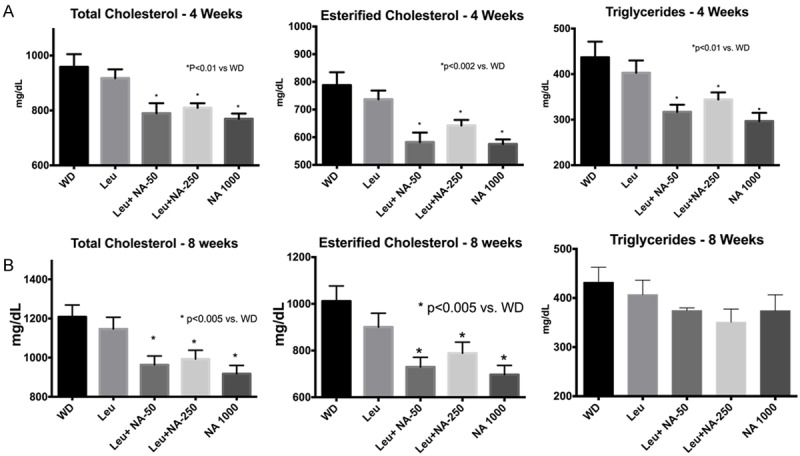
Plasma cholesterol and triglycerides in LDLRKO mice. LDLRKO mice were fed an atherogenic western diet (WD) for eight weeks supplemented with different sub-therapeutic doses NA (50 or 250 mg NA) combined with leucine, or a low full-therapeutic dose of NA (1000 mg NA). Plasma lipids (total cholesterol, esterified cholesterol and triglycerides) were measured at 4 weeks (A) and 8 weeks (B) of treatment (n=12).
To assess the degree of atherosclerosis, sections of the aorta were histologically examined and stained for lipid deposition (Oil Red O staining) and macrophage infiltration (CD68 immunostaining). Addition of a therapeutic dose of NA (1,000 mg/kg diet) to the Western diet resulted in a ~50% decrease in atherosclerotic lesion size, as illustrated in the representative Oil Red O stained aortic histology slides and quantified as total lesion and lipid area (Figure 6). Also addition of leucine to a sub-therapeutic dose of NA (50 mg/kg diet) resulted in a comparable decrease in lesion and lipid area to that found with the therapeutic dose (P<0.0001). Although leucine exerted an independent effect on plaque area, this effect was significantly less than that of the sub-therapeutic NA-Leu combination. Similarly, addition of a therapeutic dose of NA (1,000 mg/kg diet) to the Western diet also resulted in a ~50% decrease in aortic macrophage infiltration, represented as CD68-positive area in the histology slides and quantified as % CD68 positive area in the lesion (Figure 7). Leucine combined to the sub-therapeutic dose of NA (50 mg/kg diet) resulted in a comparable decrease in macrophage infiltration to that found with the therapeutic dose (P<0.0001). However, leucine also exerted a significant independent effect on reducing macrophage infiltration; this effect was not as great as the therapeutic dose of NA, but was also not significantly different from the NA-Leu combination.
Figure 6.
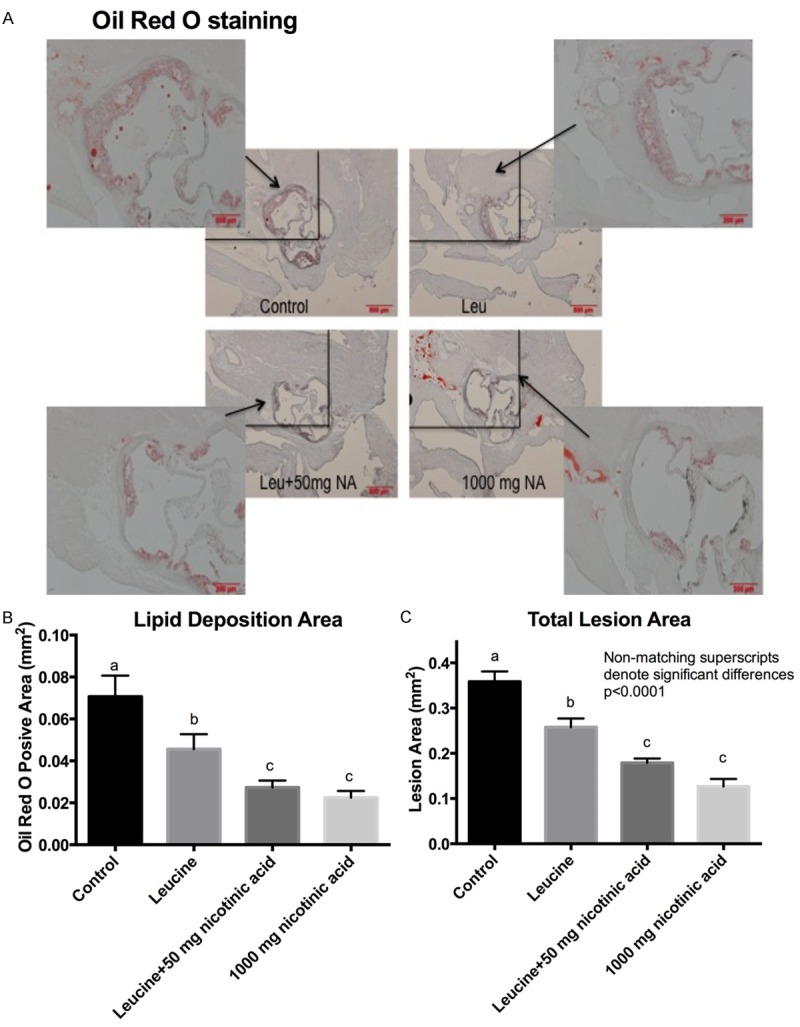
Lipid deposition in aortic plaques of LDLRKO mice. LDLRKO mice were fed an atherogenic western diet (WD) for eight weeks supplemented with different sub-therapeutic doses NA (50 or 250 mg NA) combined with leucine, or a low full-therapeutic dose of NA (1000 mg NA). After eight weeks of treatment, a subset of six mice per group was euthanized and three sections of the aortic sinus from each animal were evaluated for atherosclerotic plaque development after Oil Red O staining. A. Representative images of aortic plaque lipid deposition with 10x magnification of the left upper corner. B. Quantification of aortic lipid deposition (Oil Red O positive area). C. Quantification of atherosclerotic lesion area of the whole aorta (from sinotubular junction to iliac bifurcate). Differing letters above the bars denote significant differences between groups, P≤0.05 (n=6).
Figure 7.
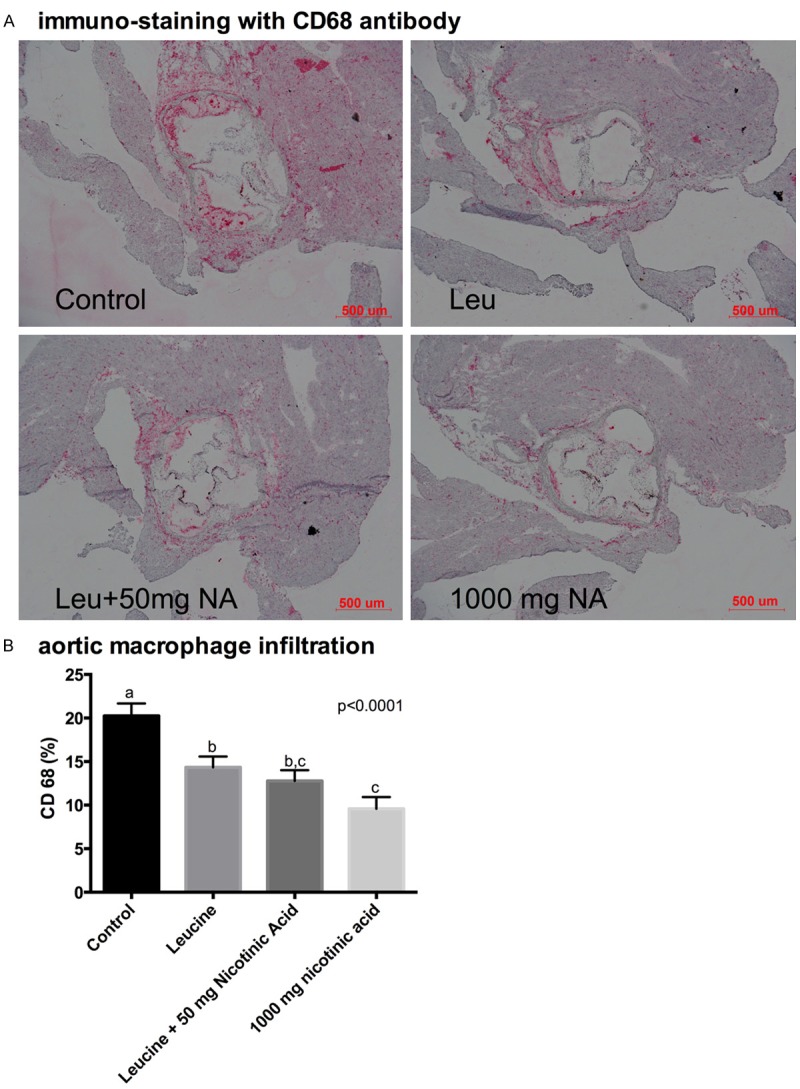
Aortic macrophage infiltration in LDLRKO mice. LDLRKO mice were fed an atherogenic western diet (WD) for eight weeks supplemented with different sub-therapeutic doses NA (50 or 250 mg NA) combined with leucine, or a low full-therapeutic dose of NA (1000 mg NA). After eight weeks of treatment, a subset of six mice per group was euthanized and three sections of the aortic sinus from each animal were immuno-stained with rat monoclonal antibody against macrophage-specific CD68. (A) Representative images of CD68 immune-stained aortic slides and (B) quantification of the CD68-positive areas. Differing letters above the bars denote significant differences between groups, P≤0.05 (n=6).
Discussion
In this study we demonstrate that the combination of a low dose of NA with leucine results in effects on lipid metabolism and atherosclerosis in vitro and in vivo comparable to a full therapeutic dose of NA alone.
The pathophysiology of atherosclerosis is a multi-step process. Since the accumulation of lipoprotein-derived lipids in the arterial wall is considered the first step, normalization of hypercholesterolemia and dyslipidemia are important therapeutic goals to stop progression of atherosclerotic plaque development. Clinical evidence over the last decades have clearly demonstrated a robust relationship between LDL-cholesterol reduction and the decrease in cardiovascular disease (CVD) with every 1 mmol/L (~40 mg/dl) reduction in LDL-cholesterol resulting in a ~22% decrease in CVD events [25]. Therefore, lipid-lowering drugs are considered first-line treatment options when other lifestyle changes are not sustainable. Most lipid lowering drugs achieve a reduction of 15% (fibrates) to 50% (potent statins) in LDL-cholesterol [3]. However, despite their potent effects on LDL-cholesterol reductions, statins often need to be combined with other drugs to achieve improvement on other lipid parameters and an overall reduction in arteriosclerotic lesion size [26]. For example, although 100 mg/kg/day atorvastatin feeding in mice for 2 months resulted in a ~45% reduction in total cholesterol, LDL-cholesterol and inflammation, the reduction of atherosclerotic lesion size was only mild and non-significant [26]. The NA-Leu combination in the present study achieved a ~20% reduction in total cholesterol; however, this was also associated with a significant reduction (~50%) in total lipid deposition and total lesion area.
Sirt1 and AMPK are well-known regulators of glucose and lipid metabolism and, via a bidirectional interaction, they stimulate fatty acid oxidation and inhibit hepatic lipid accumulation [27-29]. We have demonstrated that leucine activates Sirt1 by reducing the required concentration for the cofactor NAD+, thus enabling Sirt1 activation particularly under replete metabolic conditions such as obesity which are associated with decreased Sirt1 activation [30,31]. Also leucine enables co-activation with other AMPK/Sirt1 activators thereby reducing their necessary concentration [12,30]. Since NA acts as substrate for NAD+ biosynthesis and activates Sirt1 by increasing cellular NAD+ levels, we hypothesized that leucine would also amplify the effects of NA on Sirt1 activation. In accordance with this, Leu combined with a low concentration of NA activated Phospho-AMPK and Sirt1 significantly in adipocytes, muscle cells and C. elegans, while the individual components had little or no effect. This resulted in increased fatty acid oxidation in muscle cells and liver HepG2 cells as well as in a reduction in lipid accumulation in C. elegans.
Inflammation plays also a major role in the development and progression of atherosclerosis. Oxidized lipids and lipoproteins induce an immune response in the endothelial cells with the production of adhesion molecules, consecutively recruiting and activating monocytes and finally transforming them into cholesterol-laden foam cells [32,33]. Sirt1 exerts an anti-inflammatory effect by deacetylating RelA/p65, thus reducing NF-κB signaling and the expression of endothelial adhesion molecules such as ICAM-1 and VCAM-1 [34]. Si et al. reported that niacin attenuated aortic vascular inflammation induced by high fat diet in guinea pigs and lowered the number of macrophages in the arterial wall by ~66%, measured as CD68 positive stained cells. This was also associated with a reduction in NF-κB signaling and other inflammatory markers such as IL-6, TNF-α and CRP [35]. Accordingly, we saw in our study about a 50% reduction in aortic macrophage infiltration (CD68 staining) with the NA-Leu combination, comparable to the full dose NA.
Oxidative stress is a causative or exacerbating factor for many human metabolic diseases including atherosclerosis, and thus influences aging [36]. C. elegans is considered a good model to study metabolic disorders since it contains many key components related to metabolic and oxidative stress networks [37]. Paraquat is an oxidative stressor which interferes with the electron transfer in the mitochondrial respiratory chain and thus increases ROS production. The 2 mM paraquat concentration caused a mild oxidative stress in the worms without significantly changing the median survival and total survival curve, although the maximum survival was slightly (but not significantly) increased. Under these conditions, the NA-Leu combination increased significantly the median lifespan and reduced the hazard ratio by 20% in C. elegans while there was no difference under normal culture conditions. The NA-Leu combination also increased significantly the protein expression of DAF-16 in C. elegans, a transcription factor regulating insulin signaling and leading to lifespan extension under oxidative stress conditions [38,39].
In vitro experiments with nicotinic acid are generally conducted with concentrations in the μM to mM range [8,40]. Based on dose-response curves (data not shown), concentrations of NA ≤ 100 nM exerted no effect on the variables studied, and our experimental cell culture concentrations were therefore set below this level at 10 nM to be tested in combination with leucine at a concentration (0.5 mM) that we have previously demonstrated to be attainable in diet or supplement and which has no independent effect. Similarly, we reduced the nicotinic acid concentration in the leucine combination by up to 95% (50 mg/kg diet) in the animal diet with no loss of efficacy on the tested variables compared to the therapeutic dose (1000 mg/day). Moreover, the dose was sixty-fold lower compared to a similar study [23], which used a concentration of 0.3% NA (3 g NA/kg diet) to evaluate the effects of NA on atherosclerosis in LDLRKO mice fed a high fat-atherogenic diet. In this study, ~30% reduction in atherosclerotic lesion size was achieved, while the 50 mg NA-Leu combination in our study resulted in a ~50% reduction.
Limitations
Although the LDLRKO mouse is considered to be a good mouse model for atherosclerosis, it has some limitations. Development of marked hypercholesterolemia and atherosclerosis in this model requires the feeding of an atherogenic diet. However, the components of these diets are not standardized, a great variety with different concentrations of fat and cholesterol exists, which makes comparisons of results to other studies sometimes difficult [41]. In addition, we did not provide control groups for the NA 50 and 250 mg/kg diet in the absence of leucine; instead, we assumed that these concentrations would not have significant independent effects on the measured parameters since they are far below the therapeutic range of NA in mice, as described above [21-24].
Conclusion
These data demonstrate the amplifying effects of leucine on NA induced effects on atherosclerosis, as depicted in Figure 8. This allows a dose reduction of NA of ~95% without loss of efficacy which will lower the incidence of adverse effects. The effects of this combination are associated with activation of the AMPK/Sirt1 pathway.
Figure 8.
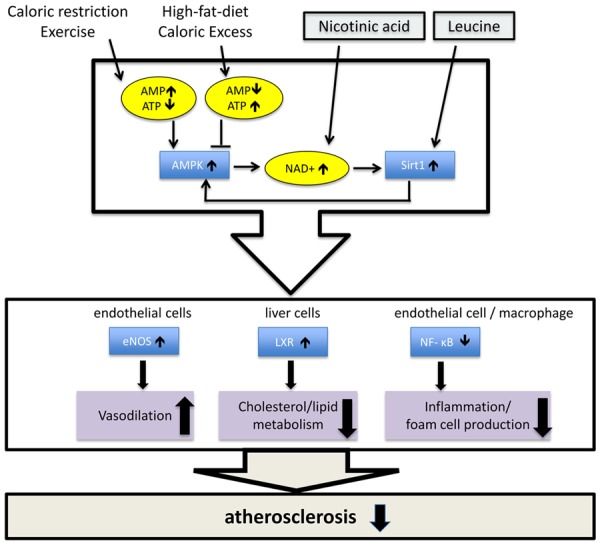
Proposed model of the synergistic effects of NA-Leu combination. AMPK and Sirt1 are nutrient sensors and bidirectionally interacting with each other. Caloric restriction or exercise increases AMP, thus activating AMPK, while high-fat diet or caloric excess increases ATP and consecutively inhibits AMPK. Low dose nicotinic acid in combination with leucine synergistically activates Sirt1, counteracting the inhibitory effects of overnutrition. AMPK/Sirt1 activation improves atherosclerosis by acting in multiple ways; activation of LXR in liver cells reduces hypercholesterolemia and hyperlipidemia, inhibition of NF-κB signaling in endothelial cells and macrophages prevents inflammation and foam cell production, and activation of eNOS improves endothelium-dependent vasodilation [10,42].
Acknowledgements
The C. elegans strain was provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This project was funded by NuSirt Biopharma Inc. (http://nusirt.com). BX and HS received funds from NuSirt Biopharma to conduct the animal study. The funders had no role in data collection and analysis, but were involved in study design, decision to publish and preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.American heart stroke association: 2015 Heart Disease and Stroke Statistics - At-aglance. https://www.heart.org.
- 2.Alagona P, Ahmad TA. Cardiovascular disease risk assessment and prevention. Med Clin North Am. 2015;99:711–731. doi: 10.1016/j.mcna.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Preiss D, Packard CJ. Emerging therapeutic approaches to treat dyslipidemia. Curr Cardiol Rep. 2014;16:506. doi: 10.1007/s11886-014-0506-3. [DOI] [PubMed] [Google Scholar]
- 4.Zafrir B, Jain M. Lipid-lowering therapies, glucose control and incident diabetes: evidence, mechanisms and clinical implications. Cardiovasc Drugs Ther. 2014;28:361–377. doi: 10.1007/s10557-014-6534-9. [DOI] [PubMed] [Google Scholar]
- 5.Carlson L. Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. J Intern Med. 2005;258:94–114. doi: 10.1111/j.1365-2796.2005.01528.x. [DOI] [PubMed] [Google Scholar]
- 6.Creider JC, Hegele RA, Joy TR. Niacin: another look at an underutilized lipid-lowering medication. Nat Rev Endocrinol. 2012;8:517–28. doi: 10.1038/nrendo.2012.22. [DOI] [PubMed] [Google Scholar]
- 7.Lauring B, Taggart AK, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, Cote J, Khalilieh S, Liu J, Luo WL, Maclean AA, Peterson LB, Polis AB, Sirah W, Wu TJ, Liu X, Jin L, Wu K, Boatman PD, Semple G, Behan DP, Connolly DT, Lai E, Wagner JA, Wright SD, Cuffie C, Mitchel YB, Rader DJ, Paolini JF, Waters MG, Plump A. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4:148ra115. doi: 10.1126/scitranslmed.3003877. [DOI] [PubMed] [Google Scholar]
- 8.Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, Tsuchiya M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J Biol Chem. 2007;282:24574–24582. doi: 10.1074/jbc.M610357200. [DOI] [PubMed] [Google Scholar]
- 9.Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- 10.Stein S, Matter CM. Protective roles of SIRT1 in atherosclerosis. Cell Cycle. 2011;10:640–647. doi: 10.4161/cc.10.4.14863. [DOI] [PubMed] [Google Scholar]
- 11.Bruckbauer A, Zemel MB. Synergistic effects of polyphenols and methylxanthines with leucine on AMPK/Sirtuin-mediated metabolism in muscle cells and adipocytes. PLoS One. 2014;9:e89166. doi: 10.1371/journal.pone.0089166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruckbauer A, Zemel MB, Thorpe T, Akula MR, Stuckey AC, Osborne D, Martin EM, Kennel S, Wall JS. Synergistic effects of leucine and resveratrol on insulin sensitivity and fat metabolism in adipocytes and mice. Nutr Metab (Lond) 2012;9:77. doi: 10.1186/1743-7075-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colella AD, Chegenii N, Tea MN, Gibbins IL, Williams KA, Chataway TK. Comparison of Stain-Free gels with traditional immunoblot loading control methodology. Anal Biochem. 2012;430:108–110. doi: 10.1016/j.ab.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Gilda JE, Gomes AV. Stain-Free total protein staining is a superior loading control to b-actin for Western blots. Anal Biochem. 2013;440:186–188. doi: 10.1016/j.ab.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruckbauer A, Zemel MB. Synergistic effects of metformin, resveratrol, and hydroxymethylbutyrate on insulin sensitivity. Diabetes Metab Syndr Obes. 2013;6:93–102. doi: 10.2147/DMSO.S40840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goel MK, Khanna P, Kishore J. Understanding survival analysis: Kaplan-Meier estimate. Int J Ayurveda Res. 2010;1:274–278. doi: 10.4103/0974-7788.76794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rich JT, Neely JG, Paniello RC, Voelker CC, Nussenbaum B, Wang EW. A practical guide to understand Kaplan-Meier curves. Otolaryngol Head Neck Surg. 2010;143:331–336. doi: 10.1016/j.otohns.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez RLMC, Naranjo JD. A pretest for choosing between logrank and wilcoxon tests in the two-sample problem. Metron. 2010;68:111–125. [Google Scholar]
- 19.Fu L, Bruckbauer A, Li F, Cao Q, Cui X, Wu R, Shi H, Zemel MB, Xue B. Interaction between metformin and leucine in reducing hyperlipidemia and hepatic lipid accumulation in diet-induced obese mice. Metabolism. 2015;64:1426–1434. doi: 10.1016/j.metabol.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Zheng J, Greenway FL. Caenorhabditis elegans as a model for obesity research. Int J Obes (Lond) 2012;36:186–194. doi: 10.1038/ijo.2011.93. [DOI] [PubMed] [Google Scholar]
- 21.Su G, Sun G, Liu H, Shu L, Zhang J, Guo L, Huang C, Xu J. Niacin suppresses progression of atherosclerosis by inhibiting vascular inflammation and apoptosis of vascular smooth muscle cells. Med Sci Monit. 2015;21:4081–4089. doi: 10.12659/MSM.895547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lipszyc PS, Cremaschi GA, Zorrilla-Zubilete M, Bertolino ML, Capani F, Genaro AM, Wald MR. Niacin modulates pro-inflammatory cytokine secretion. A potential mechanism involved in its anti-atherosclerotic effect. Open Cardiovasc Med J. 2013;7:90–98. doi: 10.2174/1874192401307010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest. 2011;121:1163–1173. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kühnast S, Louwe MC, Heemskerk MM, Pieterman EJ, van Klinken JB, van den Berg SA, Smit JW, Havekes LM, Rensen PC, van der Hoorn JW, Princen HM, Jukema JW. Niacin reduces atherosclerosis development in APOE*3Leiden. CETP mice mainly by reducing NonHDL-Cholesterol. PLoS One. 2013;8:e66467. doi: 10.1371/journal.pone.0066467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stein EA, Raal FJ. Lipid-lowering drug therapy for CVD prevention: looking into the future. Curr Cardiol Rep. 2015;17:104. doi: 10.1007/s11886-015-0659-8. [DOI] [PubMed] [Google Scholar]
- 26.Nachtigal P, Pospisilova N, Jamborova G, Pospechova K, Solichova D, Andrys C, Zdansky P, Stanislav M, Semecky V. Atorvastatin has hypolipidemic and anti-inflammatory effects in apoE/LDL receptor-double-knockout mice. Life Sci. 2008;82:708–717. doi: 10.1016/j.lfs.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. AMPK and SIRT1: a longstanding partnership? Am J Physiol Endocrinol Metab. 2010;298:E751–E760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2009;19:291–302. doi: 10.1016/j.numecd.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 30.Bruckbauer A, Baggett B, Zemel MB. Synergistic effects of polyphenols with leucine and beta-hydroxy-beta-methylbutyrate (HMB) on energy metabolism. FASEB J. 2013;27 637.23. [Google Scholar]
- 31.Bruckbauer A, Zemel MB. Effects of dairy consumption on SIRT1 and mitochondrial biogenesis in adipocytes and muscle cells. Nutr Metab (Lond) 2011;8:91. doi: 10.1186/1743-7075-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore K, Sheedy F, Fisher E. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenfeld ME. Inflammation and atherosclerosis: direct versus indirect mechanisms. Curr Opin Pharmacol. 2013;13:154–160. doi: 10.1016/j.coph.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein S, Schäfer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U, Tanner FC, Lüscher TF, Matter CM. SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/- mice. Aging (Albany NY) 2010;2:353–360. doi: 10.18632/aging.100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Si Y, Zhang Y, Zhao J, Guo S, Zhai L, Yao S, Sang H, Yang N, Song G, Gu J, Qin S. Niacin inhibits vascular inflammation via downregulating nuclear transcription factor-κB signaling pathway. Mediators Inflamm. 2014;2014:263786. doi: 10.1155/2014/263786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tangvarasittichai S. Oxidative stress, insulin resistance, dyslipidemia and type 2 diabetes mellitus. World J Diabetes. 2015;6:456. doi: 10.4239/wjd.v6.i3.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreno-Arriola E, Cárdenas-Rodríguez N, Coballase-Urrutia E, Pedraza-Chaverri J, Carmona-Aparicio L, Ortega-Cuellar D. Caenorhabditis elegans: a useful model for studying metabolic disorders in which oxidative stress is a contributing factor. Oxid Med Cell Longev. 2014;2014:705253. doi: 10.1155/2014/705253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maiese K. FoxO transcription factors and regenerative pathways in diabetes mellitus. Curr Neurovasc Res. 2015;12:404–413. doi: 10.2174/1567202612666150807112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webb AE, Brunet A. FOXO flips the longevity SWItch. Nat Cell Biol. 2013;15:444–446. doi: 10.1038/ncb2749. [DOI] [PubMed] [Google Scholar]
- 40.Haas MJ, Alamir AR, Sultan S, Chehade JM, Wong NC, Mooradian AD. Nicotinic acid induces apolipoprotein A-I gene expression in HepG2 and Caco-2 cell lines. Metabolism. 2011;60:1790–1796. doi: 10.1016/j.metabol.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]