

Abstract
Although harboring the apolipoprotein E4 (APOE4) allele is a well known risk factor in Alzheimer’s disease (AD), the mechanism by which it contributes to disease risk remains elusive. To investigate the role of proteolysis of apoE4 as a potential mechanism, we designed and characterized a site-directed cleavage antibody directed at position D151 of the mature form of apoE4 and E3. Characterization of this antibody indicated a high specificity for detecting synthesized recombinant proteins corresponding to the amino acid sequences 1-151 of apoE3 and E4 that would generate the 17 kDa (p17) fragment. In addition, this antibody also detected a ~17 kDa amino-terminal fragment of apoE4 following incubation with collagenase and matrix metalloproteinase-9 (MMP-9), but did not react with full-length apoE4. Application of this amino-terminal apoE cleavage-fragment (nApoECFp17) antibody, revealed nuclear labeling within glial cells and labeling of a subset of neurofibrillary tangles in the human AD brain. A quantitative analysis indicated that roughly 80% of labeled nuclei were microglia. To confirm these findings, cultured BV2 microglia cells were incubated with the amino-terminal fragment of apoE4 corresponding to the cleavage site at D151. The results indicated efficient uptake of this fragment and trafficking to the nucleus that also resulted in significant cell death. In contrast, a similarly designed apoE3 fragment showed no toxicity and primarily localized within the cytoplasm. These data suggest a novel cleavage event by which apoE4 is cleaved by the extracellular proteases, collagenase and MMP-9, generating an amino-terminal fragment that is then taken up by microglia, traffics to the nucleus and promotes cell death. Collectively, these findings provide important mechanistic insights into the mechanism by which harboring the APOE4 allele may elevate dementia risk observed in AD.
Keywords: Apolipoprotein E, protease, Alzheimer’s disease, neurofibrillary tangles, BV2 cells, collagenase, microglia, oligodendrocytes, PHF-1, nuclear localization
Introduction
Human apolipoprotein E (apoE) is a polymorphic gene that includes apoE2, apoE3, and apoE4, which differ by single amino acid substitutions involving cysteine-arginine replacements at positions 112 and 158 [1]. Variations in the APOE gene sequence results in three common alleles, E2, E3, and E4, represented with frequencies of 8%, 77%, and 15%, respectively, in the population [2]. Inheritance of one copy of the APOE4 allele increases Alzheimer’s disease (AD) risk fourfold, while two copies raises the risk tenfold [3]. CNS apoE is synthesized locally in the brain by neurons [4], astrocytes [5], and microglia [6]. Although apoE4 enhances the risk for AD, the mechanism by which apoE4 contributes to AD pathogenesis is not known. ApoE4 is a 34 kDa protein composed of 299 amino acids that plays a major role in cholesterol transport in the CNS, and may also help clear beta-amyloid from the brain [3]. Impairment in these two processes following inactivation of apoE4 may help drive the underlying pathology associated with AD. In this regard, apoE4 is more susceptible to proteolysis compared to apoE3 and E2 [7] and apoE4 fragments (14-20 kDa) have been identified in the AD brain [8]. Thus, proteolysis of apoE4 may promote AD pathogenesis through a loss of function [9]. Importantly, to date, the exact nature of the protease involved in cleaving apoE4 is unknown although several candidates have been reported including cathepsin D [10], a chymotrypsin-like protease [7], and aspartic proteases [11].
To assess whether cleavage of apoE occurs, we took the approach of synthesizing a site-directed cleavage antibody to an amino-terminal fragment of apoE41-151 and apoE31-151. Application of this antibody indicated it recognized the predicted 17 kDa amino-terminal cleavage fragment of apoE4 following incubation of full-length apoE4 with collagenase. In situ, application of the antibody revealed localization predominantly within neurons, neurofibrillary tangles (NFTs), and the nucleus of microglia in the human AD brain. In vitro studies using the BV2 microglia cell line indicated efficient uptake of the amino-terminal fragment corresponding to the cleavage site of apoE4 at position D151. In addition, trafficking of this fragment to the nucleus was evident and resulted in significant cell death. Based on these findings, it is suggested that proteolytic cleavage of apoE4 is a critical event that links harboring the APOE4 allele to disease risk associated with AD.
Materials and methods
Materials
Human recombinant apoE4 and E3 proteins purified from E. coli were purchased from ProSci Inc. (Poway, CA). The anti-apoE4 N-terminal rabbit polyclonal antibody was purchased from Aviva Systems Biology Corp. (San Diego, CA). PHF-1 was a generous gift from Dr. Peter Davies (Albert Einstein College of Medicine, Bronx, NY). Type I collagenase was from Sigma-Aldrich (St. Louis, MO). The rabbit polyclonal 6× His tag-ChIP grade antibody was purchased from Abcam Inc. (Cambridge, MA). The monoclonal antibodies, Olig-1 and Iba1/AFI1 were purchased from EMD Millipore (Billerica, MA).
Generation of the polyclonal site-directed cleavage antibody to ApoE4 and ApoE3
Polyclonal antibodies were synthesized using the 7-mer peptide C-RKRLLRD, which represents the N-terminal upstream neoepitope fragment of apoE4 and E3 that would be generated following cleavage after the terminal aspartic acid residue at position D151. The cysteine residue was included to facilitate coupling to sulfolink columns. Following synthesis, this peptide was coupled to KLH and injected into rabbits. The resulting sera (verified by ELISAs) were used to affinity purify antibodies using a sulfolink column (Thermo Scientific) coupled with the peptide used as an immunogen. For this antibody, synthesis of peptides, injections of immunogens, and collection of antisera were contracted out to Bethyl laboratories (Montgomery, TX).
Cell-free digestion of apoE4 with various proteases
To determine whether apoE4 and E3 is subject to proteolytic cleavage, 40 g of purified human recombinant apoE4 was incubated with collagenase (10 µg/ml) utilizing a reaction buffer containing 100 mM Tris-HCL, 10 mM CaCl2, pH 7.8 at 37°C for 2 hours. Full-length ApoE3 or E4 were incubated with active MMP-9 (.01 µg/ml) in the same reaction buffer overnight at 37°C. Similar experiments were performed using cHisApoE31-151. Reactions were terminated by the addition of 5× sample buffer and stored at -20°C until analyzed.
Generation of apoE4 and E3 fragments
The Target DNA sequence for nApoE41-151 or nApoE31-151 was designed with a 6× His tag at carboxy-terminal end of the protein to facilitate purification. An additional construct in which the 6× His tag was coupled to the amino-terminal end of nApoE41-151 was also synthesized. The complete sequences for all proteins were subcloned into a target vector for E. coli expression. E. coli BL21 (DE3) were transformed with recombinant plasmids and a single colony was inoculated into LB medium containing related antibiotic. Cultures were incubated at 37°C at 200 rpm and harvested cells were lysed by sonication. Fractions were pooled and dialyzed followed by 0.22 µm filter sterilization. Construction and purification of these amino-terminal fragments for apoE4 and E3 were contracted out to GenScript (Piscataway, NJ).
Cell culture and treatment studies using BV2 cells
BV2, murine microglial cells, were maintained at 37°C and 6% CO2 in a humidified incubator. Cells were maintained in RPMI 1640 Media (Hyclone) supplemented with 10% standard fetal bovine serum (Hyclone), 10% Cellgro MEM Nonessential Amino Acid (Corning) and 10% Penicillin streptomycin (Hyclone). Cells were cultured in 50 mL T25 Flasks. For experimentation cells were grown on BioCoat poly-D-lysine glass multiwell culture slides (Corning). All supplies were purchased from ThermoFisher Scientific Inc. (Waltham, MA). Treatment of BV2 cells was undertaken by incubation with ApoE41-151 or ApoE31-151 at a final concentration of 50 µg/ml for 24 hours prior to immunocytochemical studies.
Immunocytochemistry
Following treatment studies, BV2 cells were fixed by incubating cells in 4% paraformaldehyde for 23 minutes. For antibody labeling, cells were washed with 0.1 M Tris-buffered saline (TBS), pH 7.4, and pretreated with 3% hydrogen peroxide in 10% methanol to block endogenous peroxidase activity. Slides were subsequently washed in TBS with 0.1% Triton X-100 (TBS-A) and then blocked for thirty minutes in TBS-A with 3% bovine serum albumin (TBS-B). Slides were further incubated overnight at room temperature with anti-His rabbit antibody (1:2,000). Following two washes with TBS-A and a wash in TBS-B, slides were incubated in anti-rabbit HRP-Conjugated secondary antibody. Visualization was accomplished by using a tyramide signal amplification kit (Molecular Probes, Eugene, OR) consisting of Alexa Fluor 488-labeled tyramide (green, Ex/Em=495/519). Slides were mounted using Prolong Gold Antifade Mountant with DAPI (Molecular Probes).
Cell toxicity studies
To test for cell-mediated cytotoxicity, an LDH Cytotoxicity Assay Kit was purchased from Pierce, ThermoFisher Scientific Inc. (Waltham, MA). BV2 cells were cultured on sterile 12-well plates until confluent. Treatment of BV2 cells was undertaken by incubation with apoE41-151, full-length human recombinant apoE4, or apoE31-151 at a final concentration of 50 µg/ml for 24 hours prior to cytotoxicity studies. Control wells consisted of cells incubated in the absence of apoE1-151 and media only wells. Following treatment, 50 µl of medium was collected and transferred to a 96-well flat bottom plate and assayed for LDH activity according to the Manufacturer’s instructions. The absorbance at 490 nm and 680 nm was collected with the 680 nm reading subtracted from the 490 nm reading to account for background per manufacturer’s instructions.
Western blot analysis
Western blot analysis was performed as previously described using the reducing agent DTT in the sample buffer to eliminate any potential covalent bonding [12]. Proteins were separated by 15% SDS-PAGE and transferred to nitrocellulose. Transferred slabs were stained in Coomassie blue to verify equal loading between samples. Membranes were incubated in nApoECFp17 (1:500) or a full-length N-terminal apoE4 antibody (1:500) overnight at 4°C and primary antibodies were visualized using goat anti-rabbit HRP-linked secondary, incubated for 1 hour at room temperature (1:5,000); Jackson’s Laboratory, West Grove, PA), followed by ECL detection. Frozen tissue from frontal cortex was homogenized in TPER buffer (ThermoFisher) and centrifuged (18,000×g, 10 min). The soluble fraction was removed and protein concentration was determined by the BCA method (Pierce). For each sample, 3 μg of protein were separated by SDS-PAGE (15% gels), transferred to nitrocellulose, and probed with the nApoECFp17 (1:500). To verify equal loading between samples, transferred slabs were stained in Coomassie blue and in addition Western blot analyses was performed using an anti-beta actin rabbit antibody (1:400) purchased from Abcam Inc. (Cambridge, MA). Densitometry analysis was performed using Image Studio software, version 5.2.5 (LI-COR Biosciences).
Nuclear fractionation experiments
Cells were fractionated using a cell fractionation kit (Abcam Inc., Cambridge, MA) according to the manufacturer’s protocol. Cytoplasmic, nuclear, and mitochondrial cell fractionations were loaded on an SDS-PAGE gel at 15 µg per sample and Western blotting performed using antibodies against 6× His-tag (1:1000), MEK1/2 (Cell Signaling; 1:1000), Histone H3 (Cell Signaling; 1:2000), Rab5 (Cell Signaling; 1:1,000), and apoptosis inducing factor (AIF) (Cell Signaling; 1:1000).
Immunoprecipitation and mass spectrometry
Immunoprecipitation experiments were conducted by incubating the nApoECF antibody with apoE4 samples digested with Type I collagenase. To terminate the actions of Type I collagenase, 100 mM EDTA was added at the conclusion of the experiment. Samples were incubated overnight at 4°C with nApoECFp17 antibody and then 50 µl of Protein A agarose beads were added for 2 hours at 4°C. The agarose beads were then spun down and washed 3× in PBS. Samples were prepared for Mass spectrometry experiments as described below.
For Mass spectrometry experiments samples were separated on 15% SDS Gels and gel slices were excised from SDS-PAGE gels and digested with trypsin as previously described [13]. Briefly, excised bands were destained in 50 mM ammonium bicarbonate/50% acetonitrile. Gel slices were dehydrated in 100% acetonitrile for 10 minutes at room temperature, followed by reduction in 10 mM DTT for 60 minutes at 56°C. Next, alkylation was performed in 55 mM iodoacetamide for 60 minutes at room temperature in the dark. Protein samples were digested with trypsin in 50 mM ammonium bicarbonate overnight at 30°C. Digested peptides were separated using reversed phase chromatography (Thermo Scientific Easy-nLC II) and eluted into a Velos Pro Dual-Pressure Linear Ion Trap Mass Spectrometer for MS/MS analysis CID fragmentation. Results were analyzed with Thermo Scientific’s Proteome Discoverer software (v1.4) and the Mascot search engine (v2.4) probing the human SwissProt database. Search parameters included carbamidomethyl (C) fixed modification, oxidation (M) variable modification, maximum of 2 missed cleavages, and 1.5 Da peptide mass tolerance.
Human subjects
Autopsy brain tissue from the front cortex of nine neuropathologically confirmed AD cases and three age-matched control cases were studied. Human brain tissues used in this study were provided by the Institute for Brain Aging and Dementia Tissue Repositories at the University of California, Irvine. Approval from Boise State University Institutional Review Board was not obtained due to the exemption granted that all tissue sections were fixed and received from University of California, Irvine. Brain tissue obtained from University of California, Irvine were anonymized and never identified except by case number. Tissue donors or their next of kin provided informed signed consents to the Institute for Memory Impairments and Neurological Disorders for the use of their tissues in research (IRB 2014-1526). Case demographics are presented in Table 1 for immunohistochemical studies and Table 2 for Western blot analyses.
Table 1.
Case Demographics for immunohistochemistry
| Group | Age (yrs) | Sex | Cause of death | APOE Genotype |
|---|---|---|---|---|
| AD | 71 | M | AD | 3/3 |
| AD | 61 | F | AD | 3/4 |
| AD | 80 | F | Dehydration | 4/4 |
| AD | 83 | F | Cardiac Arrest | 3/3 |
| AD | 75 | F | AD | 3/4 |
| AD | 65 | F | AD | N/A |
| AD | 76 | M | AD | 3/3 |
| AD | 77 | F | Cardiac Arrest | N/A |
| AD | 77 | F | Sepsis | 4/4 |
| NPN | 74 | F | Cancer | N/A |
| NPN | 65 | F | Cardiac Arrest | 3/4 |
| NPN | 67 | F | Heart Failure | 3/3 |
NPN, neuropathologically normal.
Table 2.
Case Demographics for Western blot analysis
| Group | Age (yrs) | Sex | APOE Genotype |
|---|---|---|---|
| AD | 87 | M | 3/4 |
| AD | 87 | M | 3/4 |
| AD/VD | 78 | M | 4/4 |
| AD/VD | 76 | M | 4/4 |
| AD/VD | 79 | M | 4/4 |
| AD/VD | 79 | M | 4/4 |
| NPN | 87 | F | 3/4 |
| NPN | 91 | F | 3/4 |
| NPN | 90 | M | 3/4 |
NPN, neuropathologically normal.
Immunohistochemistry and confocal microscopy
Free-floating 40 µm-thick serial sections were used for immunohistochemical and immunofluorescence studies as previously described [14]. Confocal microscopy was as previously described [14]. Briefly, for confocal immunofluorescence imaging, the primary antibodies were visualized with secondary antibodies tagged with either Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen, Carlsbad, CA). Images were taken with a Zeiss LSM 510 Meta system combined with the Zeiss Axiovert Observer Z1 inverted microscope and ZEN 2009 imaging software (Carl Zeiss, Inc., Thornwood, NY). Confocal Z-stack and single plane images were acquired with an Argon (488 nm) and a HeNe (543 nm) laser source. Z-stack images were acquired utilizing the Plan-Apochromat 63×/NA 1.4 and alpha Plan-Fluar 100×/NA1.45 Oil objectives and with the diode (405 nm) and Argon (488 nm) laser sources., emission band passes of 505-550 nm for the detection of the nApoECFp17 (green channel, Alexa Fluor 488) and 550-600 nm for detection of Iba1, GFAP, Olig-1, MMP-9, AT8, and PHF-1 (red channel, Alexa Fluor 555).
Quantification and statistical analysis
To quantify potential differences between AD (N=9) and neuropathologically normal cases (N=3) (NPN), the number of nApoECFp17-labeled nuclei was counted in a 40× field (N=3 fields) for each case. Data were averaged and expressed ± S.E.M. To determine the percent co-localization, a quantitative analysis was performed as described previously [15] by taking 40× immunofluorescence, overlapping images from three different fields in frontal cortex sections in representative AD cases. Capturing was accomplished by using a 2.5× photo eyepiece, and a Sony high resolution CCD video camera (XC-77). As an example, to determine the percent co-localization between Iba1 and nApoECFp17, photographs were analyzed by counting the total number of double-labeled cells per 40× field for each case, and the number of cells labeled with each antibody alone. An example of a representative field used to quantify percent co-localization is shown in Supplementary Figure 4. Statistical differences in this study were determined using Student’s two-tailed T-test employing Microsoft Office Excel.
Results
We designed a site-directed cleavage antibody based on a putative caspase-cleavage recognition site at position D151 (LLRD) of the mature, full-length form of apoE4 and E3 (299 amino acids). Following affinity purification of this polyclonal rabbit antibody from sera, Western blot analysis was performed to determine if this antibody (herein termed the amino-terminal apoE cleavage fragment (nApoECFp17 antibody) could detect a predicted 17 kDa fragment following incubation of apoE4 with collagenase. There was no immunoreactivity of nApoECFp17 with full-length apoE4, nor were we able to detect any fragments following similar cell-free experiments utilizing recombinant apoE3 (Figure 1A). It is important to note that the lack of immunoreactivity of nApoECFp17 following incubation of full-length apoE3 with collagenase does not imply the antibody is incapable of reacting to apoE31-151 but more likely reflects the inability of collagenase to produce this fragment under these experimental conditions. Indeed, we would predict the nApoeCFp17 antibody would react with all allelic forms of apoE due to the fact the sequence upstream of residue 151 is the same for apoE4, apoE3, and apoE2. To confirm that the 17 kDa band identified in Figure 1A indeed represents the amino-terminal fragment of apoE4 following cleavage at position 151, immunoprecipitation experiments were performed using nApoECFp17-pull down experiments followed by mass spectrometry. Mass spectrometry of the 17 kDa band immunoprecipitated by nApoECFp17 indicated 77% coverage of the amino acid sequence between amino acids 1-151.
Figure 1.
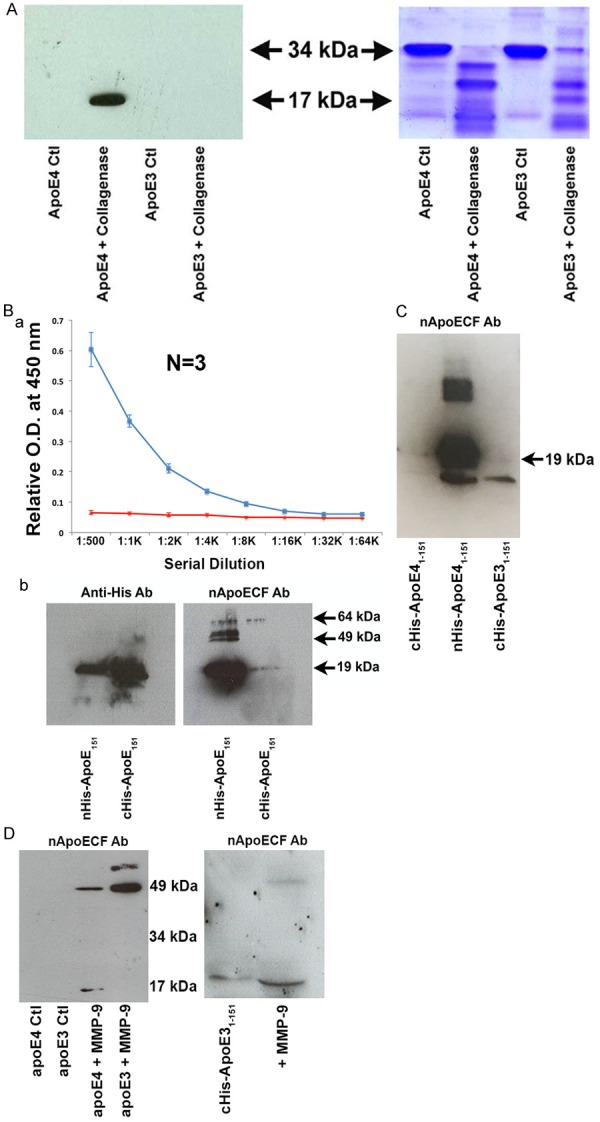
Characterization of a novel site-directed cleavage antibody to fragmented apoE4. (A) Preincubation of human recombinant apoE4 but not apoE3 with Type I collagenase for 1.5 hours at 37°C results in the generation of a 17 kDa fragment recognized by affinity-purified nApoECFp17 antibody (lane 2) following Western blot analysis. The right panel depicts the transferred gel slab that was stained with Coomassie blue revealing full-length apoE4 and E3 (34 kDa) as well as the associated fragmentation pattern following incubation with Type I collagenase. (B) Specificity of nApoECFp17 to the C-terminal end of apoE41-151. Top panel: ELISA assays in which 96-well plates were coated overnight at 4°C with either apoE41-151 with the His-Tag attached to the C-terminal end (red line), or with the His-Tag attached to the amino-terminal end (blue line). The results indicated that the nApoECFp17 antibody only recognized apoE41-151 in which the His-tag was attached to the amino-terminal end. Similar experiments as shown in the top panel for ELISA results were performed using Western blot analysis. In this case, the nApoECFp17 antibody (1:500 dilution) preferentially immunolabeled the apoE4 fragment in which the His-tag was localized to the amino-terminal end (ride side of bottom panel). The left panel shows the identical experiment in utilizing an anti-His antibody that easily labeled both protein fragments regardless of the location of the His-tag. (C) Specificity of nApoECFp17 antibody to the C-terminal end of apoE41-151. Identical Western blot experiments as presented in Panel Bb with the exception that an amino-terminal fragment of apoE31-151 with the His-tag located on the C-terminal end was included as a comparison. The results show that the nApoECFp17 antibody preferentially immunolabeled only the apoE fragment in which the His-tag was localized to the amino-terminal end (middle panel). For both (B and C) the following amounts of protein were loaded on SDS gels: cHis-ApoE41-151, 72 µg, cHis-ApoE31-151, 72 µg, and nHis-ApoE41-151, 29 µg. (D) Left Panel, Full-length human recombinant apoE3 or apoE4 was incubated with activated MMP-9 for 24 hours at 37°C and reactions were terminated by the addition of 5X sample buffer. Proteins were separated on 15% SDS-PAGE gels, transferred to nitrocellulose, and then probed with the nApoECFp17 overnight at 1:500. The “Ctl” lanes represent full-length apoE4 or E3 proteins incubated in the absence of MMP-9. Generation of the p17 band and a high-molecular weight band at ~49 kDa was observed following incubation of full-length apoE4 with MMP-9, while only a high molecular weight band ~49 kDa was observed following cleavage of full-length apoE3. The right panel depicts a similar experiment using the cHis-ApoE31-151 fragment in which the 6× His tag was coupled to the C-terminal end (lane 1). Incubation of cHis-ApoE31-151 with active MMP-9 (lane 2) resulted in a stronger and slightly smaller p17 band. These results suggest that cleavage of the 6× His tag by MMP-9 produced a slightly smaller fragment that now lacking the 6×-His tag allowed for better access of the epitope for the nApoECF antibody and stronger immunolabeling (see right panel D lane marked “+MMP-9”).
Further validation of the specificity of the nApoECFp17 antibody was determined following ELISA and Western blot experiments utilizing the recombinant amino-terminal fragment corresponding to the predicted cleavage site of apoE4 at D151. To accomplish this, the amino-terminal fragment of apoE4 was generated with the addition of a 6× His tag added either to the amino- or carboxyl-terminal end (see methods for details). We hypothesized that if the epitope for nApoECFp17 antibody was indeed in the nearby upstream region of apoE4 following cleavage at D151, then the addition of a 6× His tag at the carboxyl end may alter the ability of the antibody to bind and be recognized following ELISA or Western blot analysis. Results in Figure 1B confirmed this hypothesis: the nApoECFp17 antibody only recognized the fragment either by ELISA or Western blot analysis when the 6× His tag was on the amino-terminal end of the fragment. Thus, the presence of the 6× His tag on the carboxyl end completely abolished the ELISA signal (Figure 1Ba, red line) and greatly attenuated the detection of the fragment by Western blot in contrast to what was observed for an anti-His antibody that strongly immunolabeled both His-tagged fragments (Figure 1Bb, bottom panel). Identical experiments to Panel B were repeated with the exception that an apoE31-151 amino-terminal fragment was included in which the His-tag was attached to the C-terminal of the protein. It is important to note that the only difference between apoE31-151 and apoE41-151 in this case was a single amino acid substitution at position 112 (cysteine for arginine), which lies far upstream to the putative epitope of nApoECFp17 located at the C-terminal end (RKRLLRD). Because the His-tag was attached to the C-terminal end of apoE31-151 we predicted the nApoECF antibody would not be able to bind to its’ epitope much like for cHis-ApoE41-151. This was indeed the case, as Figure 1C depicts the nApoECF antibody strongly recognized only nHis-ApoE41-151 even though we loaded ~3 fold less protein as compared to the cHis-ApoE fragments. It is noteworthy that for nHis-ApoE41-151 we were also able to detect ~49 kDa band in addition to the predicted 17 kDa band (Figure 1Bb and 1C). These results would seem to support the ability of cleaved apoE4 to aggregate into higher molecular weight species.
To determine whether the nApoECFp17 antibody could immunoreact with apoE31-151, experiments were undertaken using active MMP-9 due its high degree of homology with collagenase. Incubation of full-length apoE4 with MMP-9 resulted in the formation of both the p17 band as well as a higher molecular band running at 49 kDa (Figure 1D, left panel). Interesting, similar experiments with full-length apoE3 resulted primarily in the immunodetection of the 49 kDa band (Figure 1D, left panel). In a final set of experiments we sought to test whether MMP-9 could cleave the 6× His tag from the c-terminal end of cHisApoE31-151 and thereby allow access of the binding epitope for the nApoECF antibody. As shown in Figure 1D (right panel, lane marked “+ MMP-9”), incubation with active MMP-9 led to a slightly smaller fragment that was more strongly labeled with nApoECF as compared to the control. The smaller fragment was presumably as a result of cleaving off the 6 additional histidine residues thereby causing a shift in the mobility of this slightly smaller fragment. Collectively, these data strongly support the ability of the nApoECF antibody to recognize both the apoE3 and E4 (1-151) amino-terminal fragment.
Our preliminary findings validated the specificity of the nApoECFp17 antibody and suggested a novel cleavage event of apoE4 in vitro. To test whether a similar cleavage event occurs in the AD brain, immunohistochemistry experiments were undertaken using postmortem frontal cortex brain sections from AD subjects. As an initial approach, we first characterized the staining profile in AD sections using the nApoECFp17 antibody in single label, bright-field experiments. Application of nApoECFp17 revealed labeling in all AD cases examined (N=9), with the strongest labeling observed in neurons, neuropil threads in plaque-rich regions, blood vessels, and small circular structures present in both white and gray matter (Figure 2). It is noteworthy that we detected labeling by the nApoECFp17 antibody in AD cases that had the APOE genotype of 3/3 (Figure 2B). This was not surprising given the fact that these two proteins differ by on a single amino acid at position 112 [1]. Thus, the putative carboxy-terminal epitope recognized by nApoECFp17 would be identical for both protein 1-151 fragments. Additional experiments supported the specificity of the nApoECFp17 antibody including a lack of staining observed following application of preimmune serum, and preadsorption of the affinity-purified antibody with the immunogenic peptide, which significantly reduced labeling in serial AD sections (Figure 3). To determine if there was a difference in nApoECFp17 labeling between NPN and AD subjects, irrespective of the APOE genotype, a quantitative analysis was performed. We observed a significant increase in the number of labeled-nuclei in AD versus neuropathologically normal cases (P=.0004) (Figure 3Fa). Moreover, there was a trend for a higher number of labeled nuclei in APOE 4/4 AD cases as compared to APOE 3/3 or 3/4 AD cases, although statistical significance was only seen between the 4/4 versus the 3/4 APOE groups (Figure 3Fb).
Figure 2.
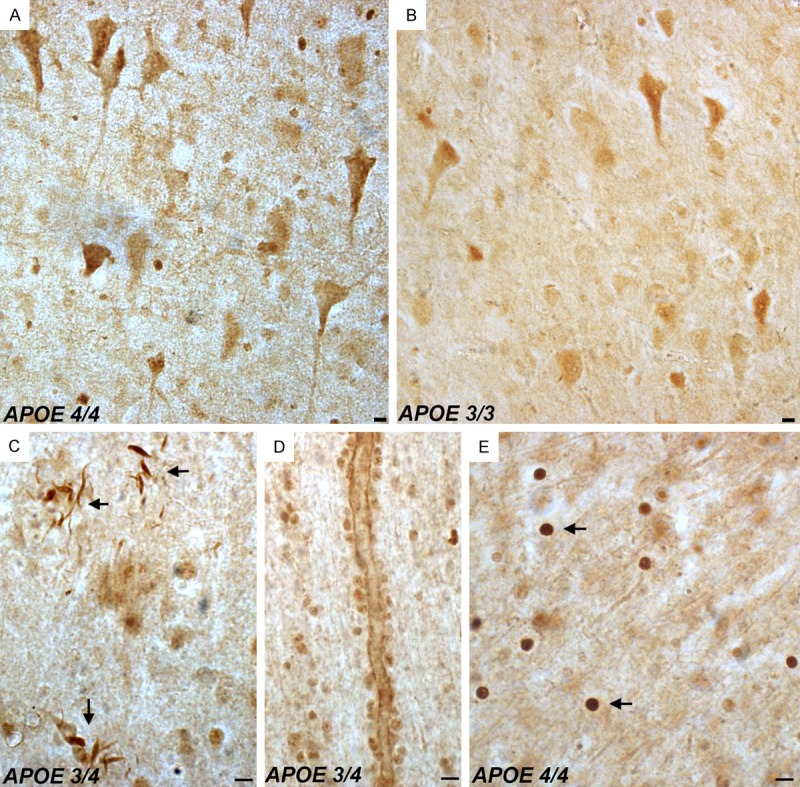
Detection of fragmented apoE in the frontal cortex of the Alzheimer’s disease brain. Representation bright-field staining in AD frontal cortex tissue sections following application of the nApoeCFp17 antibody (1:100). (A and B) Representative immunostaining of neurons by nApoECFp17 in two separate AD cases that differ in the APOE allele genotype as depicted in (A and B). Staining was also observed in neuropil threads within plaque-rich regions (arrows, C), along blood vessels (D), and the strongest labeling was within small circular structures throughout both gray and white matter (arrows, E). All scale bars represent 10 µm.
Figure 3.
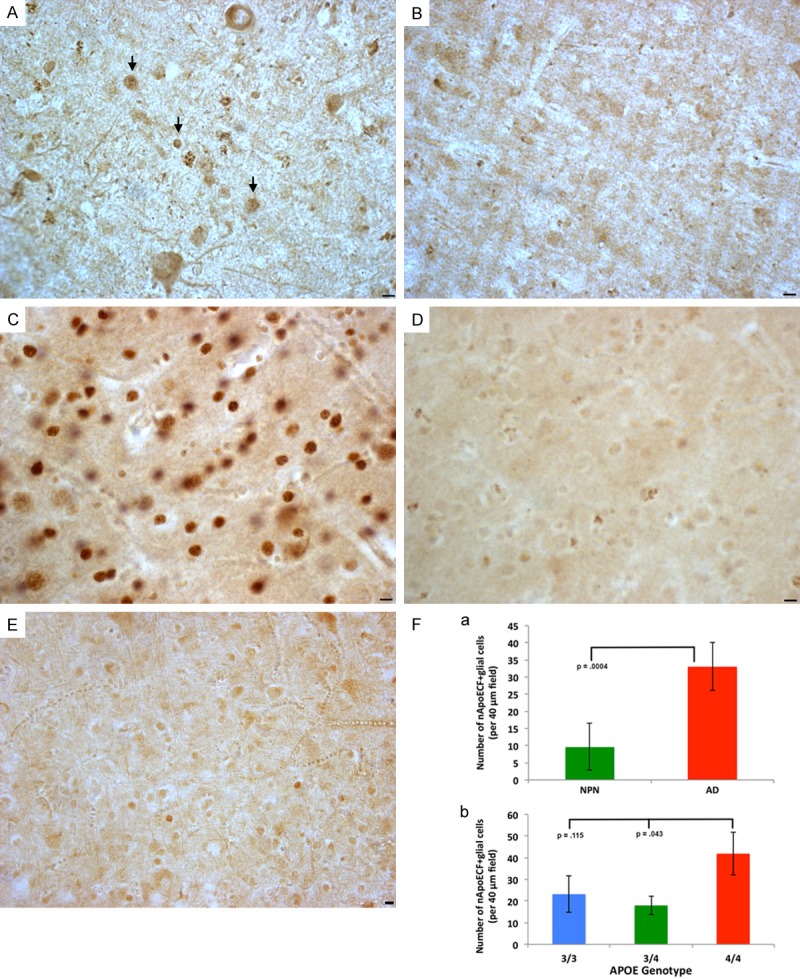
Specificity of the nApoECFp17 antibody labeling in the Alzheimer’s disease brain. (A and B) Serial AD frontal cortex sections were immunolabeled with immunized serum (A) or preimmune serum (B) with specific staining only observed following application of immunized serum. Arrows in (A) point to stained nuclei following application of immunized serum. (C and D) Serial AD frontal cortex sections were incubated with nApoECFp17 antibody alone (C), or following preadsorption with the peptide used as an immunogen (D). Staining was largely prevented following preadsorption with peptide (D). (E) We did observe labeling by nApoECFp17 in age-matched control cases, however, in general the number of cells labeled were fewer and the intensity of staining less. Except for the NPN case shown in (E) where the APOE status was not available, all other panels depict AD sections in which the APOE genotype was 4/4. All scale bars represent 10 µm. (F) a: Depicts quantitative results of the number of nApoECFp17-labeled nuclei in neuropathologically normal cases (NPN, green bars, N=3) and in AD cases (red bars, N=9) following staining of frontal cortex sections with nApoECFp17. For each case, the number of labeled nuclei was counted in a 40× field (N=3 fields, ± SEM). The data indicated a significant increase in the number of nuclei labeled in AD cases (P=.0004). b: Illustrates quantitative analysis of the number of nApoECFp17-positive nuclei (per 40 µm field, 3 different fields, ± SEM) in specific APOE allele AD cases. Although higher numbers of nApoECFp17-positive nuclei were observed for the APOE 4/4 group as compared to the other genotypes, a significant difference (P<0.5) was only observed following comparison of the APOE 4/4 group to 3/4. All scale bars represent 10 µm.
To confirm the immunohistochemical findings, Western blot analysis was undertaken utilizing postmortem brain extracts and the nAPoECFp17 antibody (see Table 2 for case demographics). We were able to detect the predicted 17 kDa fragment in all cases examined including neuropathological normals who were identified as having an APOE genotype of 3/4 (Figure 4A). Densitometry analysis indicated no significant difference between any of the groups when normalized to beta-actin loading (Figure 4B). The results depicted in Figure 4A underscore the extreme specificity of the nApoECFp17 antibody in recognizing only the 17 kDa fragment. To confirm the identification of the p17 fragment, Western blot experiments were replicated using a commercial antibody to full-length polyclonal antibody to apoE4 whose epitope is at the extreme amino terminus of the protein. In this case identification of the p17 fragment, full-length apoE running at 34 kDa, as well as an assortment of higher and lower molecular weight bands were identified (Supplementary Figure 1).
Figure 4.
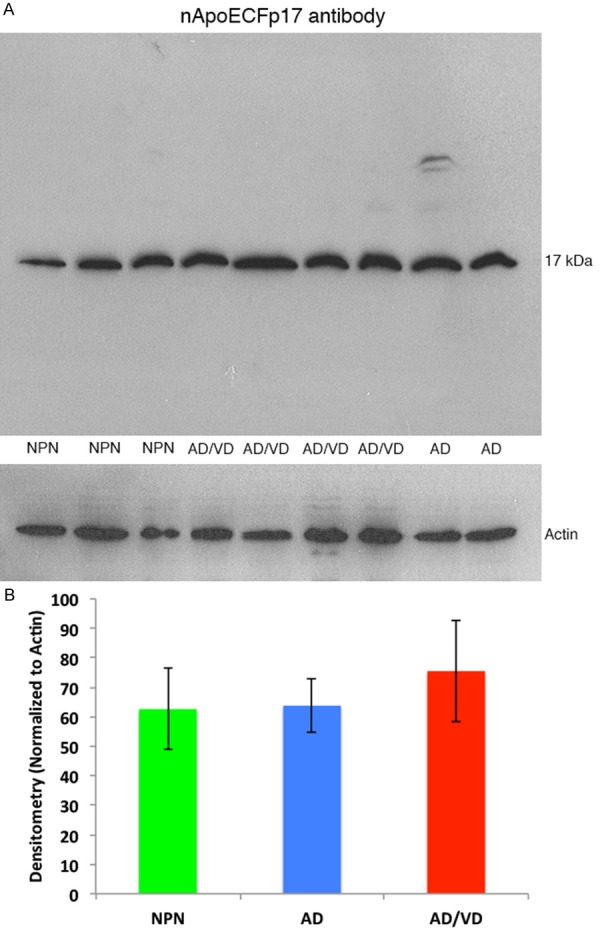
Western blot analysis of human brain extracts with the nApoECFp17 antibody. (A) Western blot analysis was performed utilizing protein extracts prepared from frozen, frontal cortex brain tissue from neuropathological controls, (NPN, APOE genotype 3/4), AD/VD subjects (APOE genotype 4/4), or AD subjects (APOE genotype 3/4). Soluble, human brain lysates were separated on 15% SDS-PAGE gels, transferred to nitrocellulose, and then probed with the nApoECFp17 antibody (1:500). (A) A prominent band at the predicted molecular weight for the nApoE1-151 fragment was observed in all cases examined. The lower panel in (A) represents a loading control blot using a rabbit antibody to beta-actin (1:400). (B) Densitometry analysis of the p17 band normalized to actin in NPN subjects, AD, and AD/VD subjects indicated no significant differences between any of the cohorts tested. Data represent the combined densitometry average for two independent experiments, ± S.D.
Based on the small size of the circular structures stained with the nApoECFp17 antibody, we hypothesized labeling was occurring in the nuclei of glia cells. Therefore, immunofluorescence confocal studies were carried out utilizing the nApoECFp17 antibody and various glial markers including Iba1 to detect microglia, Olig-1 to detect oligodendrocytes, and GFAP to specifically label astrocytes. In addition, the nuclear stain, DAPI, was employed to verify whether staining was nuclear. We were able to demonstrate triple labeling of DAPI, nApoECFp17 and Iba1 and Olig-1 in representative frontal cortex AD sections (Figure 5A-H). A quantitative analysis indicated that approximately 80% and 27% of microglia and oligodendrocytes, respectively, were labeled with the nApoECFp17 antibody (Figure 5I and 5J). For representative staining in a low-field image used determine the percent localization see Supplementary Figure 4. No colocalization of nApoECFp17 with GFAP was observed under similar experimental conditions (Figure 5K-M). Similar experiments were performed with nApoECFp17 together with a full-length apoE4 polyclonal antibody to determine whether co-localization was evident within the nucleus of glia cells. Co-localization of the two antibodies within the nucleus was evident under these conditions (Supplementary Figure 2A-D).
Figure 5.

Nuclear localization of an amino-terminal fragment of apoE within microglia of the AD brain. (A-D) Representative images from confocal immunofluorescence in AD utilizing the nuclear stain, DAPI, (A), nApoECFp17 (B), the microglial specific marker, Iba1 (C), with the overlap image shown in (D). Note the strong nuclear localization of the nApoECFp17 antibody (arrow) within labeled microglia. The arrowhead in (A-D) depicts a single microglial cell labeled with Iba1 that was not strongly labeled with the nApoECFp17 antibody. (E-H) Identical to (A-D) with the exception that the oligodendrocyte marker, Olig-1, was employed to specifically label oligodendrocytes. (I, J) Quantification of microglia (I) or oligodendrocytes (J) double-labeled with nApoECFp17 indicated co-localization in 80% and 27.5% of microglia and oligodendrocytes, respectively. Data show the number of Iba1- or olig-1-labeled cells alone (blue bars), number of nApoECF-labeled nuclei (green bars), and the number of cells with both antibodies (red bars) identified in a 40× field within frontal cortex AD sections by immunofluorescence microscopy (n=3 fields for 3 different AD cases, ± S.E.M.). (K-M) In contrast to the nuclear localization of nApoECFp17 within microglia and oligodendrocytes, confocal immunofluorescence double-label experiments with nApoECFp17 and the astrocytic marker, GFAP, failed to produce any co-localization between these two markers.
Application of the nApoECFp17 antibody to AD tissue sections indicated a lack of labeling of extracellular plaques. To confirm these findings, double-label immunofluorescence experiments were carried out using the anti-beta-amyloid antibody 6E10. As shown in Supplementary Figure 3, although the 6E10 antibody strongly labeled numerous extracellular plaques, there was no evidence of co-localization with the nApoECFp17 antibody. Previous studies have shown that amino-terminal fragments of apoE4 localize specifically within NFTs, while C-terminal fragments are confined to plaques within the AD brain [12,16]. To confirm whether nApoECFp17 localizes to NFTs, double-label confocal experiments were carried out using the standard tangle markers, AT8 and PHF-1. We confirmed the presence of nApoECFp17 in a subset of NFTs; however, we also observed staining of nApoECFp17 in neurons that were negative for AT8 or PHF-1 (Figure 6). In addition, we did observe nuclear labeling of nApoECFp17 within neurons, however, the staining appeared less intense than in glial cells and displayed a much broader distribution throughout the cell and apical dendrite (See Figure 6A and 6G).
Figure 6.
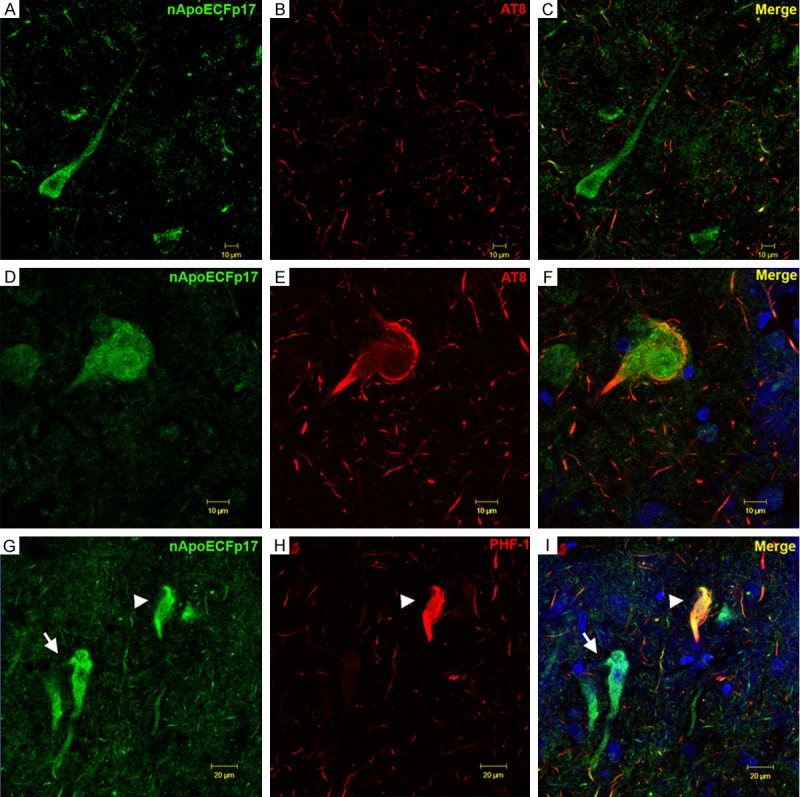
Co-localization of a fragment of apoE within a subset of neurofibrillary tangles in the AD brain. (A-F) Confocal immunofluorescence double-labeling in representative frontal cortex sections of the AD brain utilizing the nApoECFp17 antibody (green) and AT8 (red) revealed co-localization of the two antibodies in a subset of neurons (F), but not in the majority of neurons labeled with nApoECFp17 (C). Note that even though the two antibodies localized to the same NFT, there appeared to be a spatial separation with AT8 labeling staining the periphery and nApoECFp17 staining the interior of the cell (F). (G, H) Identical to (A-F) with the exception that the mature tangle marker, PHF-1 (red, H) was employed. Similar results with PHF-1 were observed with only a subset of fibrillar NFTs showing co-localization of the two antibodies (arrowhead, I). In contrast to AT8 labeling, strong co-localization of PHF-1 and nApoECFp17 was evident within the NFT (arrowhead, I). The blue fluorescence in (F and I) represents the nuclear stain, DAPI.
We next sought to provide functional data to support the in vivo findings by utilizing an in vitro model system consisting of BV2 microglial cells treated with the nHisApoE41-151 construct used for Western blot analysis in Figure 1C and 1D. Immunofluorescence examination of cells incubated with the apoE41-151 indicated uptake into BV2 cells and apparent nuclear localization (Figure 7B). To further corroborate these findings, confocal analysis was performed to assess the nuclear localization of apoE41-151-treated BV2 cells. As shown in Figure 7E-G, nuclear localization of apoE41-151 was evident under these conditions although much of the staining appeared to be localized within the nuclear rim.
Figure 7.
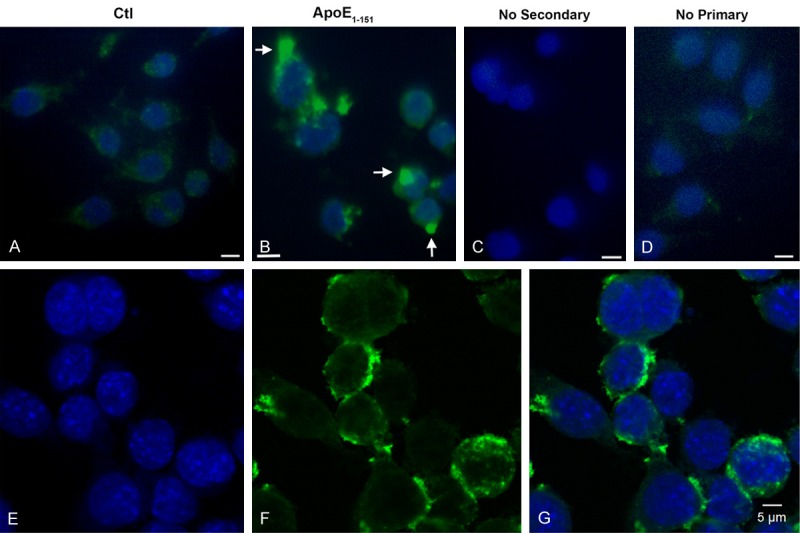
Treatment of BV2 cells with an amino-terminal fragment of apoE4 leads to cellular uptake and nuclear localization. BV2 cells were plated on glass chamber slides in normal growth media and treated for 24 hours with an amino-terminal fragment of apoE corresponding to the cleavage site at D151 of the full-length apoE4 protein. Following treatment, cells were fixed and fluorescence immunocytochemistry was carried out using an anti-His rabbit, polyclonal antibody at 1:2,000, followed by HRP-conjugated secondary antibody at 1:200 (see methods for details). (A) Control, no treatment. (B) BV2 cells treated for 24 hours with apoE41-151. (C and D) represent control experiments in the absence of secondary (C) or primary antibody (D). (E, F) Representative fluorescence, confocal images in BV2 cells following treatment with apoE41-151 confirmed the nuclear localization of the fragment. Panels are DAPI alone (E), anti-His antibody labeling alone (F), and the merged panel in (G). In this case, a similar trafficking of the fragment to the nucleus (as in B) was noted with the majority of apoE41-151 labeling appeared to localize around the nuclear rim.
Because it was difficult to ascertain by immunocytochemistry whether nuclear localization of apoE41-151 was indeed occurring following treatment of BV2 cells, fractionation experiments were performed followed by Western blot analysis. Following treatment of BV2 cells with apoE41-151, cytoplasmic, nuclear, and mitochondrial fractions were assayed by Western blot analysis using an anti-His-tag antibody. As shown in Figure 8, the presence of the apoE41-151 was evident in all three fractions albeit weakly in the cytoplasmic fraction (Figure 8, top panel). The kit used to separate the various fractions separates nuclear, cytosolic, and mitochondrial fractions. However, due to the same vesicular size it is possible the mitochondrial fraction also may contain endosomes as a result of co-fractionation. This would be predicted if apoE41-151 is taken up by receptor-mediated endocytosis. Therefore, we also probed this “mitochondrial” fraction with a specific endosomal marker, Rab5, and were able to identify identical bands under these conditions (Figure 8, bottom panel). Further studies will be required to elucidate the exact nature of this fraction. Taken together, the data presented in Figures 7 and 8 supported our immunohistochemical findings of nuclear localization of an amino-terminal fragment of apoE4 corresponding to the amino acid sequence 1-151 in microglial cells of the AD brain.
Figure 8.
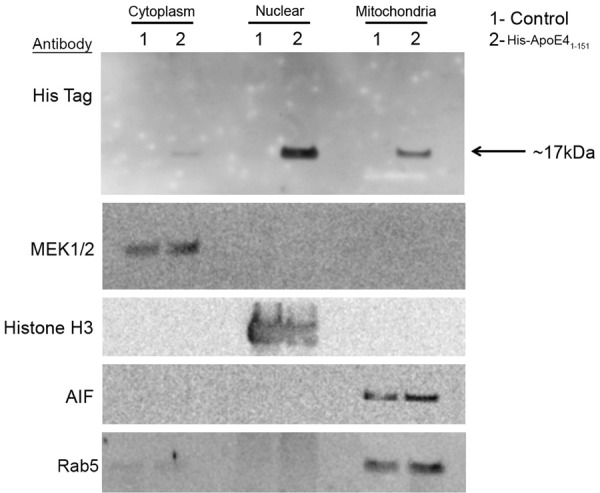
Trafficking of the amino-terminal fragment of apoE4 to the nucleus confirmed by nuclear fractionation. Separation of nuclear, cytoplasmic and mitochondrial proteins by differential centrifugation was undertaken following treatment of BV2 cells with apoE1-151 (50 µg/ml) for 24 hours. The upper panel in each section shows immunoblotting results following Western blot analysis utilizing an anti-His antibody showing the presence of the apoE41-151 faintly in the cytoplasm with much stronger labeling found in the nuclear and mitochondrial fractions (lanes marked ‘2’). MEK1/2 was employed as a cytoplasmic marker, Histone H3 as a nuclear marker, Rab5 as an endosomal marker, and AIF as a mitochondrial marker. Results are representative of two independent experiments.
In addition to the uptake and trafficking apoE41-151 in BV2 cells, apoE41-151 also led to significant cell toxicity following exogenous treatment, with an approximately 2 fold increase in cell death as compared to untreated, control cells (Figure 9, red bar). No toxicity was observed following treatment of BV2 cells with full-length apoE4 (Figure 9, blue bar). These data suggest the nuclear localization of apoE41-151 following exogenous treatment may be important step for stimulating cell death pathways in BV2 cells.
Figure 9.
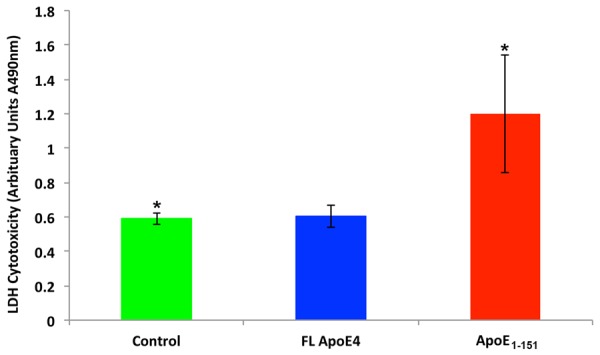
Treatment of BV2 cells with an amino-terminal fragment of apoE4 induces cytotoxicity. BV2 cells were cultured in sterile 12-well plates until confluent and then treated under various conditions for 24 hours. Following treatment, the supernatant from each condition was collected and analyzed for LDH activity as described in the Materials and Methods. Compared to untreated cells (green bar) or cells treated with 50 µg/ml full-length recombinant apoE4 (blue bar), treatment of BV2 cells with 50 µg/ml of apoE41-151 (red bar) resulted in a significantly greater level of cytotoxicity as assessed by the level of LDH activity found in the supernatant. *Denotes significant difference between the controls and apoE41-151-treated cells, P=.039. All results are representative of the average of three independent experiments, ± S.E.M.
In a final set of experiments we tested whether an amino-terminal fragment of apoE3 could traffic to the nucleus following treatment of BV2 cells. The only difference between the two amino-terminal fragments is the presence of a cysteine residue at position 112 for the apoE3 fragment in lieu of an arginine that is normally found in apoE4. As shown in Figure 10, treatment of BV2 cells with this apoE3 fragment did not lead to strong nuclear localization and the abundance of labeling appeared to be in distal cytoplasmic compartments (Figure 10A and 10C). In addition, cytotoxicity experiments actually demonstrated a small but significant protection following treatment of BV2 cells with the apoE31-151 fragment (Figure 10D).
Figure 10.
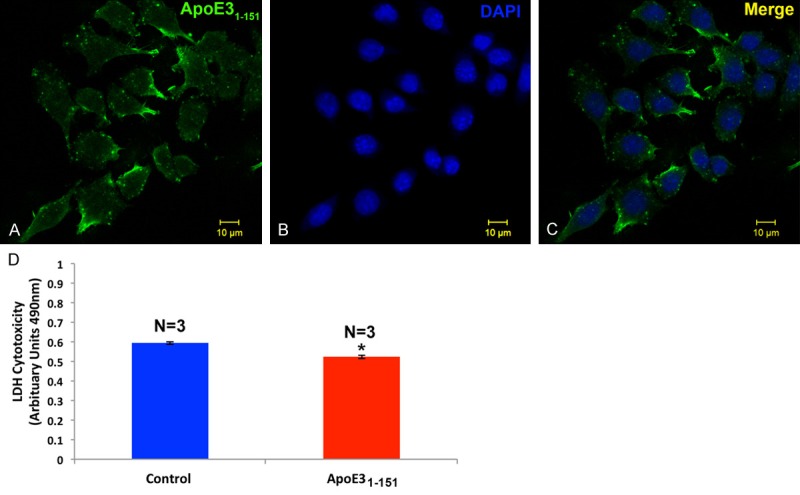
Treatment of BV2 cells with an amino-terminal fragment of apoE3 leads primarily to cytoplasmic localization and fails to induce cytotoxicty. (A) BV2 cells were plated on glass chamber slides in normal growth media and treated for 24 hours with apoE31-151. Following treatment, cells were fixed and immunocytochemistry was carried out using an anti-His rabbit, polyclonal antibody at 1:2,000, followed by HRP-conjugated secondary antibody at 1:200 (see methods for details). Localization of the apoE3 fragment appeared to be primarily within the cytoplasmic compartment (green, A and C). (B) Panel B depicts nuclear labeling only (blue, DAPI). (D) BV2 cells were cultured in sterile 12-well plates until confluent and then treated with or without 50 µg/ml apoE31-151 for 24 hours. Following treatment, the supernatant from each condition was collected and analyzed for LDH activity as described in the Materials and Methods. Compared to untreated, control cells (blue bar) cells treated with 50 µg/ml of apoE31-151 (red bar) resulted in a significantly lower level of cytotoxicity as assessed by the level of LDH activity found in the supernatant. Results are representative of the average of three independent experiments, ± S.E.M. *Denotes significant difference between the controls and apoE31-151-treated cells, P=.0001.
Discussion
A central question in AD is determining how harboring the APOE4 allele translates molecularly into an increase risk for dementia. One possibility is that apoE4 is more susceptible to proteolytic cleavage as compared to E2 and E3 and results in a loss of function such as beta-amyloid clearance [3,7,9]. To test whether apoE3 and E4 fragments can be identified in the AD brain, we designed a site-directed cleavage antibody at position D151 of the mature form of apoE4 and E3 (299 amino acids). Several lines of evidence supported the specificity of the nApoECFp17 antibody towards an amino-terminal fragment of apoE: 1) The antibody correctly identified the predicted ~17 kDa fragment that would occur following cleavage at position D151 and did not react with the full-length form of apoE4; 2) Mass spectrometry of the 17 kDa band immunoprecipitated by nApoECFp17 indicated 77% coverage of the amino acid sequence between amino acids 1-151; 3) Immunohistochemical experiments indicated the localization of the nApoECFp17 antibody within NFTs and not plaques. Previous studies have indicated a preferential localization of apoE4 antibodies such that N-terminal antibodies preferentially immunolabel tangles, while C-terminal antibodies immunolabel plaques only [12,16]; 4) Western blot analysis in human brain extracts indicated a high degree of specificity to the p17 fragment; 5) Immunolabeling by nApoECFp17 was largely prevented following preadsorption experiments with the immunogen corresponding to the upstream sequence (RKRLLRD) that would be generated following cleavage of apoE4 at position 151; 6) And finally, we show preferential immunoreactivity of nApoECFp17 to the C-terminus of nApoE41-151 corresponding to the RKRLLRD sequence, which was blocked if a 6× His-tag was present on this end of the fragment.
A somewhat surprising outcome of the current study was that in addition to the labeling of NFTs we found strong labeling of nApoECFp17 within the nuclei of glial cells, most predominantly, microglia. In a previous study, we designed a site-directed amino-terminal cleavage antibody at position D154 of the mature form of apoE4 and showed widespread labeling of this antibody (nApoE4CF) specifically within NFTs of the AD brain [12]. In the current study, the nApoECFp17 antibody only labeled a subset of NFTs and the most prominent labeling was within nuclei of glia cells. On the other hand, application of nApoE4CF was never observed within glia cells of the AD brain [12]. Thus, there were clear differences between these two antibodies even though the nApoECFp17 putative epitope is only three amino acids downstream to that recognized by nApoE4CF. One final difference was unlike the nApoECFp17 in the present study, incubation of full length apoE4 with collagenase did not produce a fragment that was recognized by nApoE4CF [12].
In the CNS, apoE is mainly synthesized and secreted by astrocytes and microglia [5,17], both of which are found to surround amyloid plaques. Based on the current findings, we hypothesize that apoE may be cleaved in the extracellular compartment and the remaining amino-terminal fragment is then taken up by neighboring microglia. The trafficking of this specific amino-terminal fragment of apoE4 to the nucleus was confirmed using an in vitro model system consisting of BV2 microglial cells incubated with the apoE41-151 fragment.
The apoE protein has several domains including the C-terminal domain that is known to bind to cholesterol and beta-amyloid, an amino-terminal domain that primarily includes the LDL receptor-binding region (residues 134-150), and a hinge region connecting the two domains that contains a number of putative proteolytic cleavage sites [18,19]. Previous studies have demonstrated that apoE4 is taken up into cells through a specific receptor-mediated pathway involving either the LDL receptor or LRP. We hypothesize that it is the LRP receptor expressed on the cell surface of both microglia and neurons that is responsible for mediating the uptake of apoE1-151 due to the fact that LRP is the major receptor for lipid-poor forms of apoE [20]. The lipid-binding region of apoE is located within the C-terminal domain (residues 244-272) [19], and thus apoE1-151 by lacking this domain would be lipid free. In addition, the LRP/LDL receptor-binding domain of apoE (residues 134-150) [18] corresponds exactly to the C-terminal end of apoE1-151 to which our nApoECF antibody recognizes and labels nuclei in the AD brain. Therefore, we hypothesize that this fragment may bind with high affinity to LRP and is readily taken up by cells expressing this receptor. Once internalized by the LRP receptor through receptor-mediated endocytosis, it would be expected that the fragment would then be delivered to endosomes and eventually lysosomes for degradation. Our data showing localization of apoE41-151 within Rab5-positive fractions (Figure 8) suggests that uptake did indeed occur through receptor-mediated endocytosis. How then the apoE1-151 fragment traffics to the nucleus is unknown to us at this time and will require further study.
We also tested whether a similarly designed amino-terminal fragment to apoE3 could be taken up and traffic to the nucleus following treatment in BV2 cells. In this case, our data supported the uptake of this fragment into cytoplasmic compartments but did not lead to strong nuclear localization. Moreover, extracellular treatment of BV2 cells with this apoE3 (1-151) fragment actually resulted in a small, but significant protection from cytotoxicity. These results contrast with the in vitro experiments using the apoE4 amino-terminal fragment and suggest that the nuclear localization and subsequent cytotoxicity may be related events.
In conclusion, harboring the APOE4 allele translates into a well-known risk factor for dementia, however, the mechanism by which this occurs is not known. Recent studies have supported a role for the proteolysis of apoE4 and a subsequent loss or gain of toxic function following the generation of N- and C-terminal fragments of the protein. We now present evidence that an amino-terminal fragment of apoE can localize to the nucleus of microglia cells in the AD brain. It is interesting to speculate on whether nuclear localization of this fragment alters gene expression, leading to the stimulation of cell death pathways. In support of this was a recent study demonstrating direct transcriptional activation in apoE4-transfected cells [21]. The authors were able to identify 76 genes activated by apoE4 and included among them genes involved in programmed cell death [21]. These data add the growing body of evidence that proteolytic cleavage of apoE4 may be the link associated with the heightened risk of dementia associated with the APOE4 allele in AD.
Acknowledgements
This work was funded by National Institutes of Health Grant 1R15AG042781-01A1 and AG0165773 to WWP. This study was also supported by the KO Alzheimer’s Dementia Foundation (Boise, ID). The project described was supported by Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grants #P20GM103408 and P20GM109095. We also acknowledge support from The Biomolecular Research Center at Boise State with funding from the National Science Foundation, Grants #0619793 and #0923535; the MJ Murdock Charitable Trust; and the Idaho State Board of Education. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Weisgraber KH, Rall SC Jr, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. 1981;256:9077–9083. [PubMed] [Google Scholar]
- 2.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 3.Eisenstein M. Genetics: finding risk factors. Nature. 2011;475:S20–22. doi: 10.1038/475S20a. [DOI] [PubMed] [Google Scholar]
- 4.Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. 1999;154:601–611. doi: 10.1016/S0002-9440(10)65305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76:1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu PT, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu HL, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD, Gilbert JR. Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis. 1996;3:229–245. doi: 10.1006/nbdi.1996.0023. [DOI] [PubMed] [Google Scholar]
- 7.Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho HS, Hyman BT, Greenberg SM, Rebeck GW. Quantitation of apoE domains in Alzheimer disease brain suggests a role for apoE in Abeta aggregation. J Neuropathol Exp Neurol. 2001;60:342–349. doi: 10.1093/jnen/60.4.342. [DOI] [PubMed] [Google Scholar]
- 9.Rohn TT. Proteolytic cleavage of apolipoprotein e4 as the keystone for the heightened risk associated with Alzheimer’s disease. Int J Mol Sci. 2013;14:14908–14922. doi: 10.3390/ijms140714908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Scott SA, Shelton SB, Crutcher KA. Cathepsin D-mediated proteolysis of apolipoprotein E: possible role in Alzheimer’s disease. Neuroscience. 2006;143:689–701. doi: 10.1016/j.neuroscience.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Marques MA, Owens PA, Crutcher KA. Progress toward identification of protease activity involved in proteolysis of apolipoprotein e in human brain. J Mol Neurosci. 2004;24:73–80. doi: 10.1385/JMN:24:1:073. [DOI] [PubMed] [Google Scholar]
- 12.Rohn TT, Catlin LW, Coonse KG, Habig JW. Identification of an amino-terminal fragment of apolipoprotein E4 that localizes to neurofibrillary tangles of the Alzheimer’s disease brain. Brain Res. 2012;1475:106–115. doi: 10.1016/j.brainres.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 14.Day RJ, Mason MJ, Thomas C, Poon WW, Rohn TT. Caspase-cleaved Tau co-localizes with early tangle markers in the human vascular dementia brain. PLoS One. 2015;10:e0132637. doi: 10.1371/journal.pone.0132637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohn TT, Day RJ, Sheffield CB, Rajic AJ, Poon WW. Apolipoprotein E pathology in vascular dementia. Int J Clin Exp Pathol. 2014;7:938–947. [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci U S A. 2001;98:8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahley RW, Huang Y. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein E4-associated neuropathology. J Med Chem. 2012;55:8997–9008. doi: 10.1021/jm3008618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narita M, Holtzman DM, Fagan AM, LaDu MJ, Yu L, Han X, Gross RW, Bu G, Schwartz AL. Cellular catabolism of lipid poor apolipoprotein E via cell surface LDL receptor-related protein. J Biochem. 2002;132:743–749. doi: 10.1093/oxfordjournals.jbchem.a003282. [DOI] [PubMed] [Google Scholar]
- 21.Theendakara V, Peters-Libeu CA, Spilman P, Poksay KS, Bredesen DE, Rao RV. Direct transcriptional effects of apolipoprotein E. J Neurosci. 2016;36:685–700. doi: 10.1523/JNEUROSCI.3562-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.