

Abstract
NADH shuttles, including malate-aspartate shuttle (MAS) and glycerol-3-phosphate shuttle, mediate the transfer of the reducing equivalents of cytosolic NADH into mitochondria. In our current study, we used BV2 microglia as a cellular model to determine the roles of NADH shuttles in lipopolysaccharides (LPS)-induced microglial activation. We found that aminooxyacetic acid (AOAA), a widely used MAS inhibitor, significantly attenuated LPS-induced increases in the levels of nitric oxide-a hallmarker of microglial activation. Our Western Blot assays also showed that AOAA blocked the LPS-induced increases in the protein levels of iNOS, TNF-α and COX-2. Furthermore, we found that AOAA decreased LPS-induced nuclear translocation of NF-κB. Collectively, our study has suggested that AOAA may be a new agent for inhibiting microglial activation. Our study has also suggested that MAS may be a novel target for modulating microglial activation under pathological conditions.
Keywords: AOAA, NADH shuttles, microglia, neuroinflammation
Introduction
NADH, the reduced form of nicotinamide adenine dinucleotide (NAD), plays important roles in multiple biological processes including energy metabolism, calcium homeostasis and antioxidation [1]. Since the inner mitochondrial membrane is impermeable to cytosolic NADH, NADH shuttles, including malate-aspartate shuttle (MAS) and glycerol 3-phosphate shuttle, transfer the reducing equivalents of cytosolic NADH into mitochondria thus increasing mitochondrial ATP production [1,2]. MAS is considered the major NADH shuttles in the brain [3]. Previous studies have suggested that MAS plays important roles in multiple biological processes, including glial synthesis of glutamate and glutamine [4], glucose-stimulated insulin secretion from β-islet cells [5], and calcium-dependent regulation of mitochondrial respiration in intact cortical neurons [6]. Aspartate aminotransferase (AST), a pyridoxal phosphate-dependent transaminase, is the rate-limiting enzyme of MAS [7]. AOAA is the inhibitor of AST, which is a widely used inhibitor of MAS [8].
Microglial activation is one of the key events in neuroinflammation in multiple major neurological diseases, including cerebral ischemia, Alzheimer’s disease and Parkinson’s disease [9,10]. A number of studies have suggested that neuroinflammation can produce both detrimental effects and beneficial effects in such diseases as ischemic brain injury [11,12]. Therefore, it is of significant theoretical and clinical significance to search for the new strategies for modulating microglial activation.
So far there has been no direct evidence indicating the roles of NADH shuttles in microglial activation. Because NADH plays significant roles in various biological processes including calcium homeostasis and antioxidation [1], which may significantly affect microglial activation [2], we proposed our hypothesis that NADH shuttles may affect microglial activation. In our current study, we used a BV2 microglia as a cellular model to test this hypothesis. Our study has suggested that AOAA may be a new agent for inhibiting microglial activation under pathological conditions. Our study has also provided direct evidence suggesting that MAS may play a significant role in LPS-induced microglial activation.
Materials and methods
Cell cultures
Microglia BV2 cells were plated into 6-well, 12-well or 24-well cell cultures plates at the initial density of 1×106 cells/mL in Dulbecco’s Modified Eagle’s Medium containing 4500 mg/L D-glucose, 584 mg/L L-glutamine (Thermo Scientific, Waltham, MA, USA), 1% penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA), supplemented with heat-inactivated (56°C for 30 min) 10% fetal bovine serum (Gemini, USA). The cells were maintained at 37°C in a 5% CO2 incubator.
Nitric oxide assay
Nitric oxide (NO) content was assessed by determining the levels of the nitrite accumulated in the culture media using Griess Reagent (Beyotime, Jiangsu, China), according to the manufacturer’s instructions. Briefly, 50 µl of culture media of the cells or culture media containing various concentrations of sodium nitrite (as standards) was mixed with 50 µl of Griess reagent I, and then with 50 µl of Griess reagent II. After 5 min at room temperature, A540nm of the samples and standards was assessed using a plate reader (Biotek Instruments Inc., Vermont, USA). The nitrite concentrations of the samples were calculated using the standards and normalized to the protein concentrations of the samples, which were assessed by the Bicinchoninic Acid (BCA) assay.
Western blotting
BV2 microglia were harvested and lysed in Radio-Immunoprecipitation Assay buffer (Millipore, Temecula, California, USA) containing complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) and 1 mM phenylmethanesulfonyl fluoride. After quantifications of the protein samples using BCA Protein Assay Kit (Pierce Biotechnology, Rockford, Illinois, USA), 30 µg of total protein was electrophoresed through a 10% SDS-polyacrylamide gel and then transferred onto a 0.45-µm pore-size nitrocellulose membrane. The blots were incubated overnight at 4°C with primary antibodies (rabbit polyclonal iNOS antibody, 1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, California, USA; rabbit polyclonal COX-2 antibody, 1:800 dilution, WANLEI Biotechnology, Shenyang, China; rabbit polyclonal NF-κB p65 antibody, 1:800 dilution, Abcam; goat polyclonal Lamin A/C antibody, 1:500 dilution, WANLEI Biotechnology, Shenyang, China; and rabbit polyclonal TNF-α antibody, 1:800 dilution, WANLEI Biotechnology, Shenyang, China) in TBST containing 1% bovine serum albumin (BSA) and then incubated with horse radish peroxidase-conjugated secondary antibody (1:2000 dilution, Hua An Biotechnology, Zhejiang Province, China) in TBST containing 1% BSA at room temperature for 1 h. Protein signals were detected using the ECL detection system (Pierce Biotechnology, Rockford, Illinois, USA). An anti-β-Actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to normalize sample loading and transfer. The intensities of the bands were quantified by densitometry using the Gel-Pro Analyzer (Media Cybernetics, Rockville, Maryland, USA).
Intracellular lactate dehydrogenase (LDH) assay
Cell survival was quantified by measuring the intracellular LDH activity of the cells. In brief, cells were lysing buffer containing 0.04% Triton X-100, 2 mM HEPES, 0.2 mM dithiothreitol, and 0.01% BSA (pH 7.5). Subsequently 50 µl cell lysates were mixed with 150 µl 500 mM potassium phosphate buffer (pH 7.5) containing 1.5 mM NADH and 7.5 mM sodium pyruvate. The changes of A340nm were monitored over 90 s, using a plate reader (Synergy 2, BioTek). Percentage of cell survival was calculated by normalizing the LDH values of sample to LDH activity measured in the lysates of control (wash only) culture wells.
Statistical analyses
All data are presented as mean ± SEM. Statistical analyses were assessed by one-way ANOVA, followed by Student-Newman-Keuls post-hoc test. P values less than 0.05 were considered statistically significant.
Results
AOAA treatment dose-dependently decreased the LPS-induced NO production by BV2 microglia
We determined the effects of treatment of 0.1, 0.25 and 0.5 mM AOAA on LPS-induced NO production in the BV2 cell cultures. AOAA was shown to dose-dependently attenuate the LPS-induced NO production by the BV2 cells (Figure 1). While AOAA is a most widely used MAS inhibitor, its major side effect is acting as an inhibitor of GABA-T [7]. Because GABA-T is present in both astroglia and microglia [13], we conducted experiments to exclude the possibility that modulation of GABA-T by AOAA is involved in the effect of AOAA on the LPS-induced NO production. BV2 cells were co-treated with 1 mg/ml LPS and 150 µM Vigabatrin, a specific GABA-T inhibitor at its widely used concentration in reported studies [13-15]. Our study did not show that Vigabatrin was capable of decreasing either the levels of LPS-induced NO production by the BV2 cells (Supplementary Figure 1A) or the cell survival (Supplementary Figure 1B). Therefore, our study has suggested that MAS may be a new mediator of LPS-induced NO production by BV2 microglia.
Figure 1.
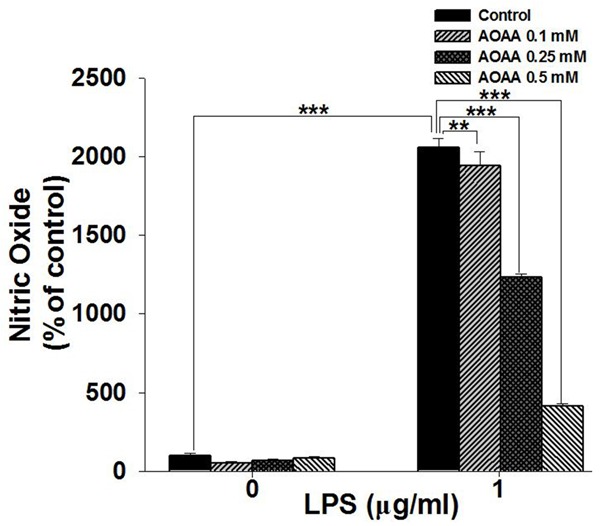
AOAA treatment significantly attenuated LPS-induced NO production by BV2 microglia. BV2 microglia were pre-treat with 0.1, 0.25 or 0.5 mM AOAA for 0.5 h, followed by co-treatment with 1 μg/mL LPS for 23.5 h. The NO levels in the medium were determined by using Griess agent. Data were collected from three independent experiments. N = 12. **P < 0.01; ***P < 0.001.
We also determined if 0.1-0.5 mM AOAA may affect the survival of LPS-treated BV2 microglia. Our experiments did not find that 0.1 and 0.25 mM AOAA can produce significant decreases in the intracellular LDH levels of the LPS-treated BV2 microglia, while 0.5 mM AOAA decreased the intracellular LDH of the cells by less than 20% (Supplementary Figure 2).
AOAA blocked the LPS-induced increases in the protein levels of iNOS, TNF-α and COX-2
We determined the effects of LPS and AOAA on the protein levels of iNOS and TNF-α in BV2 microglia, which are also major hallmarkers of microglial activation [16,17]. We found that LPS induced significant increases in the proteins levels of iNOS (Figure 2A and 2B) and TNF-α (Figure 2C and 2D), which were prevented by co-treatment of LPS with 0.5 mM AOAA (Figure 2A-D). We also determined the effects of LPS and AOAA on the protein levels of COX-2 in BV2 microglia, showing that AOAA significantly attenuated the LPS-induced increase in the COX-2 level of the cells (Figure 3).
Figure 2.
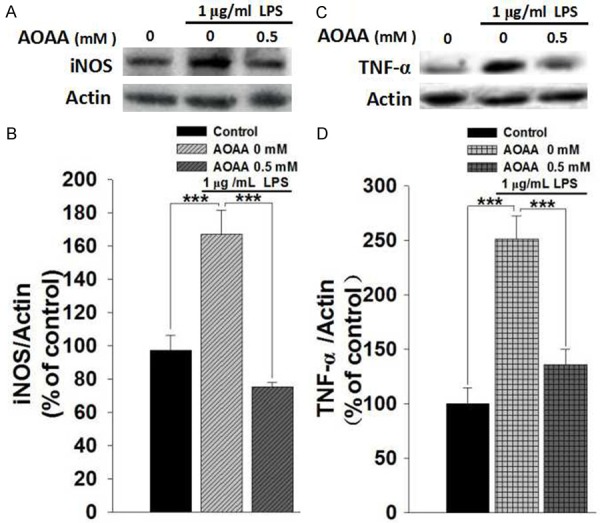
AOAA treatment prevented LPS-induced increases in the protein levels of iNOS and TNF-α of BV2 microglia. A. Representative Western blots showing the effect of AOAA on the iNOS level of the BV2 cells. B. Quantifications of the Western blots indicated that 0.5 mM AOAA treatment prevented 1 μg/mL LPS-induced increase in the iNOS level of the cells. C. Representative Western blots showing the effect of AOAA on the TNF-α level of the BV2 cells. D. Quantifications of the Western blots indicated that 0.5 mM AOAA treatment prevented 1 μg/mL LPS-induced increase in the TNF-α level of the cells. The BV2 microglia were pre-treat with 0.5 mM AOAA for 0.5 h, followed by co-treatment with 1 μg/mL LPS for 23.5 h. The protein levels of iNOS and TNF-α of the cells were determined by Western blots. Data were collected from three independent experiments. N = 6. ***P < 0.001.
Figure 3.
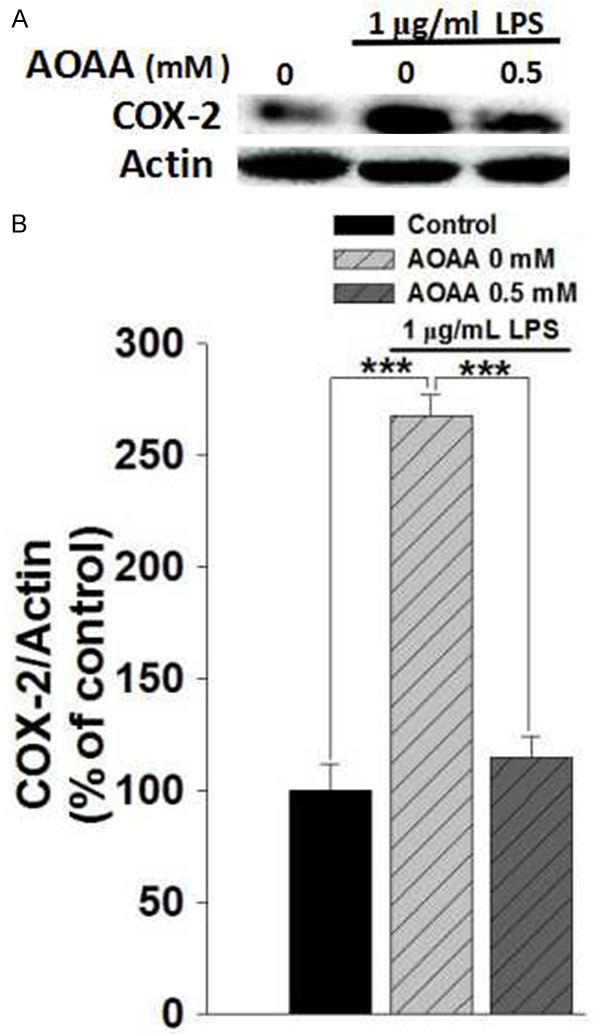
AOAA treatment prevented LPS-induced increases in the protein levels of COX-2 of the BV2 microglia. A. Representative Western blots showing the effect of AOAA on the COX-2 level of the BV2 cells. B. Quantifications of the Western blots indicated that 0.5 mM AOAA treatment prevented 1 μg/mL LPS-induced increase in the COX-2 level of the cells. The BV2 microglia were pre-treat with 0.5 mM AOAA for 0.5 h, followed by co-treatment with 1 μg/mL LPS for 23.5 h. The protein level of COX-2 of the cells was determined by Western blots. Data were collected from three independent experiments. N = 6. ***P < 0.001.
AOAA treatment decreased the LPS-induced nuclear translocation of NF-κB protein levels
We further determined the effects of LPS and AOAA on the nuclear translocation of NF-κB-a key factor in neuroinflammation-in microglia BV2 cells [18,19]. LPS induced a significant increase in the nuclear level of NFκB, which was significantly attenuated by co-treatment of LPS with 0.5 mM AOAA (Figure 4).
Figure 4.
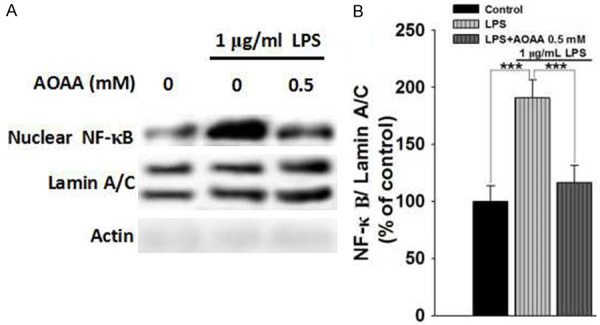
AOAA attenuated LPS-induced nuclear translocation of NF-κB in BV2 microglia. A. Representative Western blots showing the effect of AOAA on the nuclear level of NF-κB of the BV2 cells. B. Quantifications of the Western blots indicated that 0.5 mM AOAA treatment significantly attenuated 1 μg/mL LPS-induced increase in the nuclear level of NF-κB of the BV2 cells. The BV2 microglia were pre-treated with 0.5 mM AOAA for 0.5 h, followed by co-treatment with 1 μg/mL LPS for 23.5 h. The nuclear protein level of NF-κB of the cells was determined by Western blots. Data were collected from three independent experiments. N = 6. ***P < 0.001.
Since previous studies have suggested that CD38/cADP-ribose signaling system plays a significant role in microglial activation [20] and survival [21,22], we also determined the potential roles of CD38/cADP-ribose signaling system in the AOAA-mediated changes of LPS-induced microglial activation. Our study did not observe significant effects of 30 µM Br-cADPR, an antagonist of cADPR-activated ryanodine receptors, on the AOAA-produced changes of LPS-induced NO production (Supplementary Figure 3A and 3B).
Discussion
The major observations of our current study include: First, AOAA, a widely used MAS inhibitor, significantly attenuated LPS-induced increases in the levels of nitric oxide-a hallmarker of microglial activation-by BV2 microglia. Second, AOAA prevented the LPS-induced increases in the protein levels of iNOS, TNF-α and COX-2. Third, AOAA decreased LPS-induced nuclear translocation of NF-κB. In addition, our study has shown that neither 0.1 mM AOAA nor 0.25 mM AOAA significantly affected the survival of BV2 microglia (Supplementary Figure 2), while both 0.1 mM AOAA nor 0.25 mM AOAA significantly decreased LPS-induced NO production (Figure 1), thus arguing against the possibility that AOAA decreased LPS-induced microglial activation by decreasing cell number. Collectively, our study has suggested that AOAA can significantly inhibit LPS-induced microglial activation. Our study has also suggested that MAS may play a significant role in LPS-induced microglial activation.
Because microglial activation is one of the key events in neuroinflammation in multiple major neurological diseases [10,21], it is of both theoretical and clinical importance to search for the new strategies for modulating microglial activation. Our current study has suggested that AOAA can markedly decrease LPS-induced microglial activation, suggesting that AOAA may become a new agent for inhibiting microglial activation.
AOAA is a most widely used MAS inhibitor, while its major side effect is acting as an inhibitor of GABA-T [7]. However, our current study did not observe that the inhibitor of GABA-T could affect the effects of AOAA on LPS-induced microglial activation (Supplementary Figure 1). Therefore, our study has suggested that MAS may be a new mediator of LPS-induced microglial activation. Previous studies have suggested that MAS plays important roles in various biological processes, such as glial synthesis of glutamate and glutamine [4] and calcium-dependent regulation of mitochondrial respiration in intact cortical neurons [6]. Our previous study has also found that AOAA can both decrease the intracellular ATP levels and induce apoptosis of resting and LPS-activated BV2 microglia [2]. In comparison with our previous study, our current study has provided the first evidence indicating that AOAA can block the increases in the multiple markers of LPS-induced microglial activation, including the levels of iNOS, TNF-α, COX-2, and nuclear translocation of NF-κB.
While our study has suggested a significant role of MAS in LPS-induced microglial activation, it remains unclear how MAS may be involved in mediating LPS-induced microglial activation. Because NADH plays significant roles in a variety of biological processes including calcium homeostasis and oxidative stress [23], which can affect microglial activation [2], MAS may affect microglial activation by modulating the cytosolic and mitochondrial levels of NADH. Future studies are warranted to investigate these mechanisms.
In summary, our study has suggested that AOAA may be a new agent for inhibiting microglial activation under pathological conditions. Our study has also suggested that MAS may be a novel target for modulating microglial activation.
Acknowledgements
This work is supported by National Natural Science Foundation of China #81271305 (to W. Y.), a Major Research Grant from the Science & Technology Committee of Shanghai Municipality (#16-JC1400500 and #16JC1400502) (to W. Y.), and a Shanghai Jiao Tong University Grant Interdisciplinary Research on Medicine and Engineering (to W. Y.). The authors declare that they have no competing interests.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ma Y, Nie H, Chen H, Li J, Hong Y, Wang B, Wang C, Zhang J, Cao W, Zhang M, Xu Y, Ding X, Yin SK, Qu X, Ying W. NAD+/NADH metabolism and NAD+-dependent enzymes in cell death and ischemic brain injury: current advances and therapeutic implications. Curr Med Chem. 2015;22:1239–1247. doi: 10.2174/0929867322666150209154420. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Wang C, Wei X, Ding X, Ying W. Malate-aspartate shuttle inhibitor aminooxyacetate acid induces apoptosis and impairs energy metabolism of both resting microglia and LPS-activated microglia. Neurochem Res. 2015;40:1311–1318. doi: 10.1007/s11064-015-1589-y. [DOI] [PubMed] [Google Scholar]
- 3.McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol. 2006;71:399–407. doi: 10.1016/j.bcp.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Pardo B, Contreras L, Satrustegui J. De novo synthesis of glial glutamate and glutamine in young mice requires aspartate provided by the neuronal mitochondrial aspartateglutamate carrier Aralar/AGC1. Front Endocrinol (Lausanne) 2013;4:149. doi: 10.3389/fendo.2013.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubi B, del Arco A, Bartley C, Satrustegui J, Maechler P. The malate-aspartate NADH shuttle member Aralar1 determines glucose metabolic fate, mitochondrial activity, and insulin secretion in beta cells. J Biol Chem. 2004;279:55659–55666. doi: 10.1074/jbc.M409303200. [DOI] [PubMed] [Google Scholar]
- 6.Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, Pardo B, Satrustegui J. Calciumregulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci. 2013;33:13957–13971. 13971a. doi: 10.1523/JNEUROSCI.0929-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzpatrick SM, Cooper AJ, Duffy TE. Use of beta-methylene-D, L-aspartate to assess the role of aspartate aminotransferase in cerebral oxidative metabolism. J Neurochem. 1983;41:1370–1383. doi: 10.1111/j.1471-4159.1983.tb00835.x. [DOI] [PubMed] [Google Scholar]
- 8.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 9.Ying W. NAD+ and NADH in brain functions, brain diseases and brain aging. Front Biosci. 2007;12:1863–1888. doi: 10.2741/2194. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Wu D, Ding X, Ying W. SIRT2 is required for lipopolysaccharide-induced activation of BV2 microglia. Neuroreport. 2015;26:88–93. doi: 10.1097/WNR.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 11.Xia W, Han J, Huang G, Ying W. Inflammation in ischaemic brain injury: current advances and future perspectives. Clin Exp Pharmacol Physiol. 2010;37:253–258. doi: 10.1111/j.1440-1681.2009.05279.x. [DOI] [PubMed] [Google Scholar]
- 12.Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- 13.Lee M, Schwab C, McGeer PL. Astrocytes are GABAergic cells that modulate microglial activity. Glia. 2011;59:152–165. doi: 10.1002/glia.21087. [DOI] [PubMed] [Google Scholar]
- 14.Besse A, Wu P, Bruni F, Donti T, Graham BH, Craigen WJ, McFarland R, Moretti P, Lalani S, Scott KL, Taylor RW, Bonnen PE. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab. 2015;21:417–427. doi: 10.1016/j.cmet.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M, McGeer EG, McGeer PL. Mechanisms of GABA release from human astrocytes. Glia. 2011;59:1600–1611. doi: 10.1002/glia.21202. [DOI] [PubMed] [Google Scholar]
- 16.Kawahara K, Mori M, Nakayama H. [NO-induced apoptosis and ER stress in microglia] . Nihon Yakurigaku Zasshi. 2004;124:399–406. doi: 10.1254/fpj.124.399. [DOI] [PubMed] [Google Scholar]
- 17.Liu T, Zhang T, Yu H, Shen H, Xia W. Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood-brain barrier disruption. J Neuroinflammation. 2014;11:107. doi: 10.1186/1742-2094-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Chen Z, Yan M, He P, Dai H. The protective role of isorhamnetin on human brain microvascular endothelial cells from cytotoxicity induced by methylglyoxal and oxygen-glucose deprivation. J Neurochem. 2016;136:651–659. doi: 10.1111/jnc.13436. [DOI] [PubMed] [Google Scholar]
- 19.Michelucci A, Bithell A, Burney MJ, Johnston CE, Wong KY, Teng SW, Desai J, Gumbleton N, Anderson G, Stanton LW, Williams BP, Buckley NJ. The neurogenic potential of astrocytes is regulated by inflammatory signals. Mol Neurobiol. 2016;53:3724–3739. doi: 10.1007/s12035-015-9296-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayo L, Jacob-Hirsch J, Amariglio N, Rechavi G, Moutin MJ, Lund FE, Stein R. Dual role of CD38 in microglial activation and activationinduced cell death. J Immunol. 2008;181:92–103. doi: 10.4049/jimmunol.181.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma Y, Jiang J, Wang L, Nie H, Xia W, Liu J, Ying W. CD38 is a key enzyme for the survival of mouse microglial BV2 cells. Biochem Biophys Res Commun. 2012;418:714–719. doi: 10.1016/j.bbrc.2012.01.084. [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Cao W, Wang L, Jiang J, Nie H, Wang B, Wei X, Ying W. Basal CD38/cyclic ADP-ribose-dependent signaling mediates ATP release and survival of microglia by modulating connexin 43 hemichannels. Glia. 2014;62:943–955. doi: 10.1002/glia.22651. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Wei G, Wang D, Yung P, Ying W. Posttreatment with the Ca(2+)-Mg(2+)-dependent endonuclease inhibitor aurintricarboxylic acid abolishes genotoxic agent-induced nuclear condensation and DNA fragmentation and decreases death of astrocytes. J Neurosci Res. 2008;86:2925–2931. doi: 10.1002/jnr.21733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.