

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a major health problem worldwide, and is often associated with lipotoxic injury, defective mitochondrial function, and insulin resistance. Thyroid hormones (THs) are important regulators of hepatic lipid metabolism. Among the THs, diiodothyronine (T2) and triiodothyronine (T3) have shown promising results in lowering hepatic fat content in various models of NAFLD. In this study, we used a targeted metabolomics approach to investigate the differential effects of T2 and T3 on the early metabolic adaptation in the livers of rats fed high fat diet (HFD), a period when hepatosteatosis is reversible. Our results showed that both T2 and T3 strongly induced autophagy and intra-hepatic acylcarnitine flux but prevented the generation of sphingolipid/ceramides in animals fed HFD. Interestingly, although both T2 and T3 decreased hepatic fat content, only T2 was able to rescue the impairment in AKT and MAPK/ERK pathways caused by HFD. In summary, we have identified and characterized the effects of T2 and T3 on hepatic metabolism during short-term exposure to HFD. These findings illuminate the common and divergent metabolic pathways by T2 and T3 that also may be important in the prevention and treatment of NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a major health problem associated with obesity and diabetes. It affects more than 40% of the U.S. population and currently is the single largest cause of liver transplants worldwide1. NAFLD is a spectrum of diseases that starts with hepatosteatosis and can progress to non-alcoholic steatohepatitis (NASH) that eventuates in cirrhosis. The molecular mechanism(s) for NAFLD and its progression still are not fully understood and currently there are no approved drug therapies for NAFLD2. Recent studies suggest that NAFLD is not only due to excess triglyceride storage but also to the accumulation of other lipid species that potentially are cytotoxic and induce inflammation in hepatocytes3. Cytotoxic lipid species such as ceramides can induce mitochondrial dysfunction resulting in enhanced Reactive oxygen species (ROS) production, which is thought to represent the central abnormality responsible for the progression from simple fatty liver to NASH and impaired hepatic insulin action4, 5. These changes are associated with other metabolic features such as defective amino acid metabolism, impaired TCA cycle flux, and unregulated hepatic glucose production6.
Thyroid hormones (THs) and their analogues have long been recognized as important regulators of hepatic lipid metabolism, and studies in rodents suggest that they can reduce hepatosteatosis7. TH actions were thought to be classically mediated by 3,3′5-triiodothyronine (T3) and levothyroxine (T4); however, in recent years, another TH-related compound, 3,5-diiodothyronine (T2), has been shown to be bioactive. Both T3 and T2 can influence several aspects of energy expenditure8. In particular, one of the prominent characteristics of TH signaling is a rapid induction in increasing oxygen consumption and ATP production. Recent studies also have linked this signaling pathway to fatty acid oxidation (FAO) in a variety of tissues9, 10. Although T3 has long been thought to exert its effects primarily by binding to nuclear TH receptors (TRs)11, 12, the mechanism of action for T2 still remains not entirely clarified even if some mechanisms have been proposed such as the activation of sirtuin1 (SIRT1)13 and a direct interaction with the subunit Va of the cytochrome oxidase in bovine heart14, 15. Initial reports described T2 showing a difference in the non-genomic effects on oxygen consumption when compared to those induced by T3 16. Subsequent studies showed that T2 also is able to act at the genomic level and modulate gene expression, suggesting that it could be an alternative ligand for TRβ116.
Metabolomics is a novel technology that has emerged as a powerful tool for identifying metabolite biomarkers associated with NAFLD pathogenesis. Metabolomics provides a comprehensive view of the changes in several metabolic and signaling pathways and their interactions17–23. In this connection, THs such as T3 and T2 have been shown to reduce hepatic fat accumulation in both animal and cell culture models7, 24–28. However, a comprehensive and comparative metabolomic analysis of T2 and T3 is lacking. Most of our current knowledge of the actions of T2 and T3 were demonstrated independently without a side-by-side comparison of their effects on hepatic metabolism. Additionally, our understanding of the events associated with the early stages of NAFLD is very limited since the focus of the field has been on the diet-induced or genetic models of chronic obesity and NAFLD. The early adaptive stages of NAFLD are important since they may not only help understand the metabolic changes that lead to progression of NAFLD, but also identify potential drug targets for the treatment of hepatosteatosis when it still is reversible. In this paper, we focused on the relative effects of T2 and T3 in hepatic adaptation to acute feeding of high fat diet in rats. For this purpose, we sought to gain a comprehensive metabolic view, analyzing different hepatic metabolites related to lipid, ceramide, and amino acid metabolism in response to short-term HFD and its modulation by T2 or T3 treatment. Our results provide novel insights into the differential effects of T2 and T3 on hepatic lipid metabolism, mitochondrial function, and insulin signaling pathways after a short-term HFD regimen.
Results
T2 and T3 effects on hepatic TG accumulation and acylcarnitines in rats fed HFD
Previous studies showed that T2 and T3 influenced hepatic lipid metabolism by virtue of their abilities to stimulate fatty acid β-oxidation (FAO) both in vitro and in vivo 16, 29. Thus, we sought to characterize the primary effect of T2 and T3 on lipid catabolism through analysis of β-oxidative intermediates in hepatic tissues. Accordingly, we first measured hepatic triglyceride (TG) levels and found a significant decrease in both T2- and T3-treated rats fed HFD vs. rats fed HFD alone (Fig. 1), consistent with earlier reports25–28. The body weight, serum iodothyronine, and blood glucose levels of animals in each group are provided (Suppl Fig. 1).
Figure 1.
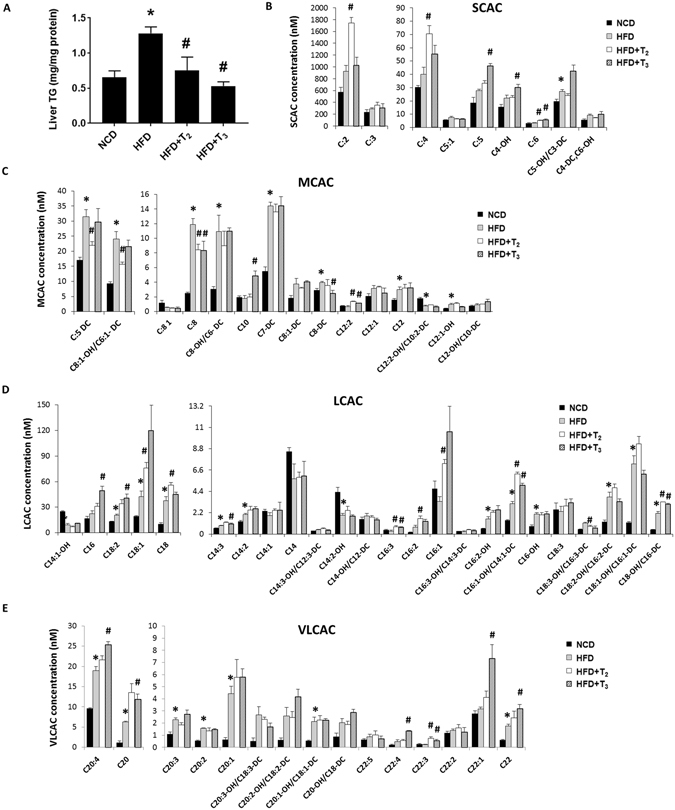
T2 and T3 reduce hepatic lipid accumulation in HFD fed rats associated with increased acylcarnitine flux. (A) Representative graph showing Triglycerides (TGs) content in rats liver treated respectively with NCD, HFD, HFD+T2, HFD+T3 for 1 week. Values are means ± SEM (n = 4). *P < 0.05 in NCD Vs HFD; #P < 0.05 in HFD Vs HFD+T2/HFD+T3. Metabolomics profiles of (B) short chain acylcarnitines (SCAC), (C) medium chain acylcarnitines (MCAC), (D) long chain acylcarnitines (LCAC) and (E) very long chain acylcarnitines (VLCAC) in NCD, HFD, HFD+T2 and HFD+T3 treated rats. Values are means ± SEM (n = 4). *P < 0.05 in NCD Vs HFD; #P < 0.05 in HFD Vs HFD+T2/HFD+T3.
We next performed metabolomic profiling of hepatic acylcarnitines species ranging in size from 2 to 22 carbons. Acylcarnitine esters are synthesized from their respective acyl-CoA intermediates by enzymes known as carnitine acyltransferases residing in mitochondria to promote their import into mitochondria whereupon they undergo FAO. Long chain fatty acids represent substrates derived from lipolysis of triglycerides from adipose tissue, hepatic stores, and diet. Even chain acylcarnitine species from C6 to C22 represent fatty acid metabolites due to incomplete fatty acid oxidation. Odd short-chain species, such as C3 and C5 are mostly formed by amino acid catabolism whereas C4 is derived from both fatty acids and amino acids. Acylcarnitine C2, which is used as a proxy measure of acetyl-CoA, is generated by catabolism of fatty acids, amino acids, and/or glucose30. C4-OH (hydroxybutyrate) is the end product of fatty acid β-oxidation and also has been used as a marker for this process.
Hepatic acylcarnitine profiling showed that T2 and T3 had different effects on lipid metabolism in livers from rats fed HFD. We observed that T2- and T3-treated rats fed HFD displayed increased levels of hepatic short-chain acylcarnitines (SCAC), that are end products from β-oxidation of fatty acids and amino acids as well as from TCA cycle intermediates supplied by amino acids during anapleurosis (Fig. 1B). In this connection, the increase in hepatic C3, C4 and C5 SCACs in T2- and T3- treated rats occurred in parallel with a decrease in hepatic amino acids levels (Suppl Fig. 2). Our metabolomic analysis also revealed that HFD significantly increased intrahepatic levels of branched-chain amino acid valine (Suppl Fig. 2), which was abolished by T2 or T3 treatment (Suppl Fig. 2). Similar to the trend observed for valine, other amino acids such as alanine, aspartate, methionine, glutamine, and tryptophan also were increased in livers of rats fed HFD. Furthermore, this effect was reduced by T2 or T3 treatment, with significant decreases observed by T2 for alanine, methionine, glutamine and tryptophan (Suppl Fig. 2). In addition to SCACs, hepatic long-chain and very long-chain acylcarnitines (LCAC, VLCAC) tended to be increased in rats fed HFD, and increased even more in T2- and T3-treated rats, most likely due to hydrolysis of stored triglycerides as well as influx of FFAs supplied to the liver by lipolysis from adipose tissue (Fig. 1C,D). Medium-chain acylcarnitines (MCAC) also were increased by HFD; however, in contrast to SCACs and LCACs, they were mostly decreased by the two iodothyronines (Fig. 1B). This decrease in MCAC levels compared to the acylcarnitines of other lengths suggested that there might be a relative increase in MCAC flux in the β-oxidation pathway of the liver during treatment by T2 or T3.
T2 and T3 regulation of lipolysis, autophagy, FAO, mitochondrial biogenesis and anti-oxidant proteins in rats fed HFD
To further understand the increases in LCAC and VLAC levels after T2 or T3 treatment, we assessed the role of the THs on intrahepatic lipolysis. Hepatic TG breakdown is mediated by neutral extralysosomal lipases such as Adipose triglyceride lipase (ATGL)31 or via lipophagy, a form of autophagy in which lipids are sequestered into autophagosomes and degraded after autophagosomal fusion with lysosomes via lysosomal acid lipases32. Our results demonstrated that both T2 and T3 had no significant effects on ATGL phosphorylation, a marker for ATGL activity (Fig. 2A,B). In contrast, we found that both T2 and T3 induced autophagy proteins such as Microtubule-associated protein 1A/1B-light chain 3 B-II (LC3B-II), and Transcription factor E3 (TFE3) expression (Fig. 2A,B). T2 also increased Transcription factor EB (TFEB) expression significantly but T3 did not. These results suggested that the increases in lipolysis by both T2 and T3 were likely mediated by autophagy/lipophagy rather than by ATGL activity.
Figure 2.

Administration of T2 and T3 increase lipophagy and FAO regulatory proteins in HFD fed rats. Representative cropped Immunoblots and densitometry showing proteins content of lipolytic and autophagic markers (A,B), and β-oxidative markers (C,D). Values are means ± SEM (n = 4). *P < 0.05 in NCD Vs HFD; #P < 0.05 in HFD Vs HFD+T2/HFD+T3.
We next examined the presence of mitochondrial β-oxidation markers in liver samples from rats fed HFD and treated with T2 or T3 (Fig. 2C,D). T3 treatment significantly increased Carnitine palmitoyltransferase I alpha (CPT1α) protein expression and T2 tended to increase CPT1α protein expression in mice fed HFD although not significant (Fig. 2C,D). Moreover, HFD decreased levels of Uncoupling protein 2 (UCP2) whereas both T2 and T3 treatments reversed these effects (Fig. 2C,D). Acetyl-CoA carboxylase (ACC) phosphorylation increases FAO in hepatic cells.Interestingly, only T3 was able to significantly rescue the decrease in p-ACC/ACC levels induced by HFD, with T2 having only a minor effect (Fig. 2C,D). These data suggested that in general both T2 and T3 increased the levels of pro-FAO proteins to assist entry of fatty Acyl-CoAs into the mitochondria leading to increased FAO and oxidative phosphorylation. Taken together these data suggested that both iodothyronines increased hydrolysis of hepatic triglycerides and FAO to prevent the development of fatty liver. We also observed that that only T3 significantly increased expression of mitochondrial biogenesis markers such as Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), Nuclear respiratory factor 1(NRF-1), and Transcription factor A, mitochondrial (TFAM) (p = 0.06) in livers from rats fed HFD (Suppl Fig. 3A,B). Surprisingly, T2 had no effect on these markers. Furthermore, HFD induced the expression of the anti-oxidant enzyme, Superoxide dismutase 1(SOD1) (Suppl Fig. 4). T3 decreased SOD1 protein expression in livers from rats fed HFD whereas T2 maintained the same level as HFD alone (Suppl Fig. 4). Taken together, these data suggest that T3 and T2 may utilize different mechanisms to handle the increased ROS and oxidized mitochondrial proteins due to increased oxidative phosphorylation.
T2 and T3 effects on the induction of hepatic sphingolipid synthesis in rats fed HFD
A diet rich in saturated lipids leads to increased accumulation of cytotoxic lipid species such as the sphingolipid, ceramide and its metabolites. It now is well established that ceramides not only are key structural components of cellular membranes but also important sphingolipid second messengers involved in cellular stress responses5. Our metabolomic profiling showed a consistent increase in almost all the examined hepatic ceramide species from the livers of rats fed HFD compared to rats fed NCD (Fig. 3A). Interestingly, we found that rats treated with either of the two iodothyronines rescued the HFD effect by decreasing the levels of most of the ceramide species (Fig. 3A). Other sphingolipids such as sphingomyelin were stimulated by HFD but were reduced after T2 or T3 treatment (Fig. 3B).
Figure 3.

HFD-induced Sphingolipids accumulation is prevented by T2 and T3 treatment. Quantitative analysis of Ceramide (A), Sphingomylines and (B) Sphinganine after HFD and HFD+T2/HFD+T3 treatment. Values are means ± SEM (n = 4). *P < 0.05 in NCD Vs HFD; #P < 0.05 in HFD Vs HFD+T2/HFD+T3.
Selective T2 and T3 effects on MAPK and PI3K signaling in livers of rats fed HFD
HFD is associated with impaired insulin signaling in both animals and humans33. This effect generally is attributed to increased TG accumulation, ceramide signaling, and accumulation of branched-chain amino acids4, 34. Since we observed a protective effect of T2 and T3 on these parameters, we next investigated their effects on insulin/growth factor signaling effectors such as MAPK and PI3K. Taking phosphorylation of ERK and AKT as a marker for active MAPK and PI3K signaling we found that only T2 significantly increased ERK and AKT activity whereas T3 did not affect these proteins (Fig. 4A,B). These results, therefore, suggested that T2 had a very distinct effect on ERK and AKT signaling by T2 that was not mediated by T3. Interestingly, we observed a significant decrease in the level of TRβ1 in HFD fed rats and this effect was not rescued by either T2 or T3 (Suppl Fig. 5). These findings raise the possibility that some of the metabolic effects of both T2 and T3 during the adaptive phase may not require TRβ1.
Figure 4.

T2 but not T3 affects insulin/growth factor signaling in livers of HFD fed rats. Representative cropped Immunoblot and densitometry of ERK and AKT protein levels showing THs influence on the correlated signaling pathway. Values are means ± SEM (n = 4). *P < 0.05 in NCD Vs HFD; #P < 0.05 in HFD Vs HFD+T2/HFD+T3.
Discussion
Iodothyronines have become attractive pharmacological compounds to treat NAFLD. Presently, two iodothyronines, T2 and T3, both have shown efficacy in reducing the severity of NAFLD in both cell culture and animal models of NAFLD7, 25–28. It has been proposed that T2 and T3 employ different mechanisms for their effects, with T3 acting at a genomic level and T2 via non-genomic signaling12, 16. Most previous studies in the literature have not made a side-by-side comparison of the activities of these two iodothyronines; thus, our understanding of the relative differences between both their biological and metabolic effects is limited. In this study, we used a state-of-the-art metabolomics approach to compare the effects of T2 and T3 on hepatic adaptation to short-term HFD feeding in rats. The salient findings of our study were: i) Both T2 and T3 prevented hepatic fat accumulation which was associated with increased autophagy, lipolysis and FAO; ii) T2 and T3 decreased the synthesis of hepatic sphingolipids such as ceramides in response to HFD; and iii) T2, but not T3, rescued the impairment of hepatic AKT & ERK signaling pathways in rats fed HFD.
Our results were consistent with earlier reports showing lipid-lowering effects of T2 and T3 in animal models of NAFLD7, 25–28. However, in this study, we employed a more comprehensive metabolomics approach to understand the effects of these iodothyronines on the metabolic processes that control hepatic FAO in rats fed HFD. The increase in acylcarnitines (AC) is representative of increased lipolysis (long-chain AC) as well as increased fatty acid oxidation in the mitochondria (short-chain AC). Hepatic cells exhibit increased AC when fed HFD, due to increased uptake and utilization of fatty acids from HFD. However, the further increase in AC species by both T2 and T3 beyond that observed during HFD feeding alone suggested the induction of lipolysis of hepatic fat stores and fatty acid oxidation, and accounted for the decreased hepatic TG content observed in Fig. 1A. Consistently, we found that SCACs, which serve as proxy markers of FAO, were increased during HFD due to free fatty acids from lipolysis and diet, and further increased after T2 and T3 treatment due to induction of autophagy/lipophagy, CPT1α expression, and FAO. However, the effects of T2 and T3 were not uniform across the different SCACs as T2 effects were more pronounced on C:2 and C:4 whereas T3 caused significant increases in the C:5 and C4-OH (ketogenesis markers) species. These results suggested that although iodothyronines in general increased FAO, T2 and T3 affected different enzymatic pathways of downstream fatty acid metabolism. Furthermore, since some SCACs are derived from both organic and amino acid oxidation, our data implied that there may be more complex effects of T2 and T3 on cellular metabolism. Branched-chain amino acids (BCAA) including leucine, isoleucine and valine have been shown to be increased in human NAFLD and positively correlated with hepatic insulin resistance34. In this connection, our metabolomic analysis also showed that even short-term feeding on HFD caused a significant increase in the BCAA, valine. Interestingly, both T2 and T3 were able to rescue this effect. Other amino acids such as phenylalanine, tryptophan, tyrosine and leucine, which are ketogenic in nature, also were decreased by T2, and to a lesser extent by T3. This down-regulation may be a reflection of the increased oxidation of these amino acids since SCAC levels were increased.
Surprisingly, the levels of MCACs, for the most part, were either suppressed or unchanged by T2 and T3 treatment when compared with hepatic levels of MCACs from livers of rats fed only HFD. Furthermore, levels of most LCACs and VLCACs were increased in livers from rats fed HFD with T2 or T3 treatment than those from rats fed HFD alone. This scenario resembles a block in LCAC oxidation; however, when observed in the context of decreased levels of hepatic SCACs from livers from T2- and T3-treated rats, it is more likely that the reduced MCAC levels were due to increased oxidation of MCACs to SCACs. Furthermore, proteins that were positively-associated with increased FAO such as CPT1α and UCP2 also were increased by T3, and to a lesser extent, by T2. Finally, since LCACs and VLACs are derived from TG hydrolysis, our results implied that both T2 and T3 increased lipolysis.
Hepatic lipolysis is mediated by either the classical lipases (e. g., ATGL) or autophagy31, 32. ATGL activity is inhibited by AMPK, which is a negative regulator of ATGL-mediated lipolysis35. Our results showed that neither T2 nor T3 significantly affected the AMPK–mediated phosphorylation of ATGL so it is likely that the increase in hepatic lipolysis induced by these iodothyronines did not involve stimulation of ATGL activity. Autophagy also has been implicated in the degradation and metabolism of lipids in the liver. In this connection autophagy-deficient mice developed fatty liver disease32. Moreover, the transcriptional master regulators of autophagy and lysosomal biogenesis, TFEB and TFE3, also have been linked to lipid catabolism in liver36. We previously showed that autophagy-mediated hepatic lipolysis or “lipophagy” was induced by T3 26. In this study, we examined the short-term effects of T2 and T3 in rats fed HFD and found that autophagic markers such as LC3-II and TFE3 were up-regulated in treated rats when compared to rats fed HFD alone. These findings suggested that lipophagy, rather than stimulation of ATGL, was the major mechanism employed for hepatic TG lipolysis and lipid clearance during the hepatic adaptive phase by these two THs.
Increased rate of FAO often generates cytotoxic free radicals known as reactive oxygen species (ROS) that can damage mitochondria. To sustain mitochondrial function there needs to be efficient mitochondrial quality control that involves co-ordinated mitochondrial biogenesis to maintain a healthy pool of mitochondria as well as anti-oxidant enzymes to get rid of mitochondria damaging free radicals. T3 can increase mitochondrial biogenesis37, most likely in conjunction with mitophagy38, 39. Similarly, we found that T3 significantly increased PGC1α, NRF-1, and TFAM in the livers of rats red HFD. In contrast, T2 did not increase expression of these proteins. Therefore, although both iodothyronines increased mitochondrial function and FAO, only T3 increased mitochondrial biogenesis. In contrast, we found that T2 maintained SOD1 protein expression whereas T3 decreased its expression in livers from rats fed HFD. Therefore, these results suggest that T3 may rely upon mitochondrial turnover whereas T2 may relay more on the induction of anti-oxidant proteins such as SOD1 to counteract oxidative stress and maintain quality control of mitochondria.
Sphingolipids are components of membrane bilayers that also serve as regulators for apoptosis, cellular senescence, stress response, inflammation, and metabolism5. Ceramides contribute to the cellular damage caused by inflammation from insulin resistance, mitochondrial dysfunction, and oxidative stress in NASH3. Currently, little is known about TH effects on ceramide synthesis in hepatic cells. Our results showed that both T2 and T3, in general, prevented induction of ceramide synthesis across different chain lengths in livers from rats fed HFD. Similarly, sphingomyelin which is associated with fatty liver and insulin resistance40, was significantly reduced by both the iodothyronines. Since saturated fats are the precursors of ceramide biosynthesis, the increased FAO in the mitochondria induced by T2 and T3 may explain the decrease in ceramide and sphingomyelins by these two iodothyronines in livers from rats fed HFD. Our findings have demonstrated a novel role for THs in regulating sphingolipid metabolism and hepatic toxicity through their induction of FAO.
Previously, it was shown that while both T2 and T3 decreased hepatic steatosis in rats, only T2 rescued the insulin resistance associated with fatty liver13. HFD inhibits AKT activation to decrease cell survival and increase hepatic metabolic processes such as gluconeogenesis that are negatively regulated by insulin41. MAPK/ERK signaling also is critical for insulin signaling in hepatic cells and is down-regulated during NAFLD progression42. Since insulin resistance frequently is associated with NAFLD progression, we examined the effects of T2 and T3 on these two cellular signaling pathways in rats fed short-term with HFD. Interestingly, only T2 rescued the negative effects of HFD on AKT and ERK signaling. The inability of T3 to increase insulin signaling despite decreasing hepatic fat content shows that there was a dissociation between its lipid-lowering and insulin-sensitizing effects. In agreement with our findings, Vatner et al. found that T3 paradoxically inhibited insulin signaling in liver despite rescuing fatty liver development in animals fed HFD43. In contrast, T2 was reported to rescue insulin resistance in NAFLD associated with obesity13. The dual effects of T3 on lipid metabolism and insulin resistance suggest that the positive effects of T3 in reducing liver TG, ceramides, and BCAA are counter-balanced by its potential negative effects on insulin resistance such as generation of diacylglycerol43, inhibition of MTORC244, and increased hepatic glucose production. It will be important to determine whether there are dose-dependent effects of T3 that preferentially enhance the positive effects on lipid metabolism over the negative ones on gluconeogenesis and diacylglycerol production. In this regard, T2 may offer more therapeutic potential than T3 since it can increase autophagy and FAO while improving insulin resistance caused by HFD. Our data showing the effects of T2 on MAPK/ERK and PI3K pathways also are consistent with those reported by Rochira A et al.45. According to this study, T2 most likely induced these pathways via a rapid non-genomic pathway that did not involve TRs45. Interestingly we found that that hepatic TRs levels were decreased in animals treated with HFD alone, and with HFD and T2 or T3. These results support the notion that some of the of the short-term metabolic actions of HFD, T2, and T3 may not require TRβ1.
In summary, we used a metabolomics approach to obtain a comprehensive and comparative view of the metabolic actions of T2 and T3 on the livers of rats fed short-term HFD. Both compounds had lipolytic effects in the liver mediated by autophagy and increased FAO although the metabolic profiles suggested that there may be some differences in the mechanism(s) and magnitude of their metabolic effects. It is noteworthy that T3 induced mitophagy and mitochondrial biogenesis, whereas T2 did not appear to do so despite its ability to induce FAO. Additionally, the increased FAO by both iodothyronines was able to reduce intrahepatic ceramide levels, and thus may protect hepatocytes against lipotoxicity due to increased intracellular saturated fatty acids from intrahepatic hydrolysis of triglycerides and imported FFAs generated by lipolysis of adipose tissue. Last, although there have been several studies showing beneficial effects of both T2 and T3 for reducing hepatosteatosis7, 46, T2 may offer potential therapeutic advantages by activating insulin-signaling pathways instead of inducing metabolic and cell signaling effects that counteract insulin action. Although several issues such as the role of TH receptors in T2 action and the assessment of the metabolomic changes in chronic HFD models still need to be addressed, our results have provided novel insights into the metabolomic actions of T2 and T3 during the early hepatic metabolic adaptation to lipid challenge.
Methods
Animals and Drugs Treatment
The studies were performed in male Wistar rats (250–300 g) purchased from Charles River Laboratories. They were maintained and used in accordance with the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. All animals were kept one per cage in a temperature-controlled room at 28 °C under a 12-h light/12-h dark cycle and water was available ad libitum. Rats were divided into four groups and treated for 1 week. The first group (group NCD) received a standard diet (total metabolizable percentage of energy: 60.4 carbohydrates, 29 proteins, 10.6 fat J/J; 15.88 KJ gross energy/g; Muscedola, Milan, Italy). The second (group HFD) received an HFD (consisting of 280 g diet supplemented with 395 g lyophilized lamb meat [Liomellin, Milan, Italy], 120 g cellulose [Sigma-Aldrich, St. Louis,MO], 20 g mineral mix [ICN Biomedical, Solon, OH], 7 g vitamin mix [ICN], and 200 g low-salt butter [Lurpak, Denmark]) (total metabolizable percentage of energy: 21 carbohydrates, 29 proteins, 50 fat J/J; 19.85 KJ gross energy/g). The third group (group HFD-T2) received the same HFD together with a daily intraperitoneal injection of T2 (25 µg/100 g body wt) (Sigma-Aldrich). The fourth group (group HFD-T3) received the same HFD together with a daily intraperitoneal injection of T3 (2.5 µg/100 g body wt) (Sigma-Aldrich). After 1 week of treatment, rats were anesthetized by an intraperitoneal injection of chloral hydrate (40 mg/100 g body wt) and then killed by decapitation. Liver was excised and immediately frozen in liquid nitrogen for subsequent analysis. The authors also confirm that all experiments were performed in accordance with relevant guidelines and regulations. The authors confirm that the experimental protocols were approved by Seconda Università degli Studi di Napoli, Caserta, Italy and Duke-NUS Medical School, Singapore institutional committee.
Metabolomics
Metabolomic analysis was performed as described previously47. Amino-acids were extracted from 100 μL of liver homogenate using methanol and then derivatized to form butyl esters using 3 M HCl in butanol. Samples were then reconstituted in 80% aqueous methanol and 4 μL of this solution was injected into an Agilent SB-C8 column (12 × 50 vmm with 1.8 um particle size) for analysis. Mobile phase used was 80% methanol and 20% water, and flow rate was maintained at 0.4 ml/min for 2 min. Isocratic flow of 0.6 ml/min of 30% acetonitrile and 70% water with 0.1% formic acid was maintained for 5.5 min. For Sphingolipid analysis tissue homogenate was resuspended in 900 μl of ice-cold chloroform-methanol (1:2) and incubated in ice for 15 min with vortexing every 5 min. Three hundred microliters of ice-cold distilled water (dH2O) and 300 μl of ice-cold chloroform were added to the samples, which were then vortexed and centrifuged at 8,000 × g for 2 min at 4 °C. The lower organic phase was transferred into a clean microcentrifuge tube. A second extraction was performed by adding 300 μl of ice-cold chloroform, and the lower organic phase was pooled with that of the first extraction. For acylcarnitine extraction, 100 µL of tissue homogenate was extracted using methanol. The acylcarnitine extracts were derivatised with 3 M Hydrochloric acid in methanol, dried and reconstituted in methanol for analysis in LC-MS.The collected samples were dried under a stream of nitrogen and stored at −80 °C until ready for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. Data acquisition and analysis were performed on Agilent MassHunter Workstation B.06.00 Software.
Western blotting
Tissue samples were lysed using CelLytic™ M Cell Lysis Reagent (Sigma, C2978) and immunobloting was performed as described previously.25 Image acquisition was done using ChemiDoc (Bio-Rad ChemiDoc™ MP System, 1708280). Densitometry analysis was performed using ImageJ software (NIH, Bethesda, MD, USA).
Reagents
Antibody details are as follows: TFAM (Cell Signaling Technology, 7495); PGC-1α (Santa Cruz Biotechnology, sc-13067); NRF1 (Cell Signaling Technology, 12381); UCP2 (Santa Cruz Biotechnology, sc-6525); CPT1α (Abcam, ab128568); phoshpo-Acetyl-CoA Carboxylase (Cell Signaling Technology, 11818); Acetyl-CoA Carboxylase (Cell Signaling Technology, 4190S); LC3B-II (Cell Signaling Technology, 2775); TFE3 (Cell Signaling Technology, 14779); TFEB (Abcam, ab2636); phospho-ATGL S406 (Abcam ab135093); ATGL (Abcam ab57562); Phospho-p44/42 MAPK (Erk1/2) Thr202/Tyr204 (Cell Signaling Technology, 9101); p44/42 MAPK (Erk1/2) (Cell Signaling Technology, 9102); Phospho-Akt S473 (Cell Signaling Technology, 4058); Akt (Cell Signaling Technology, 9272); SOD1 (Cell Signaling Technology, 4266); ACTB/β-Actin (Santa Cruz Biotechnology, sc-81178); GAPDH (Cell Signaling Technology, 2118).
Liver triglyceride estimation
Liver triglycerides were estimated using manufacturer’s guidelines (Cayman chemicals, Item No: 10010303)
Statistical analysis
Results are expressed as means (n = 4 ± SEM). The statistical significance of differences between groups was determined using one-way ANOVA followed by a Student-Newman-Keuls test. Differences were considered significant at P < 0.05.
Electronic supplementary material
Acknowledgements
This work was supported by NMRC/CSA/0054/2013 (PMY), and from Singapore Ministry of Health NMRC/BNIG/2025/2014 (RAS). This research was financially supported by grants from the University of Campania “L. Vanvitelli” and from the University of Sannio (AL & FG).We thank Dr. J-P Kovalik (CVMD, Duke-NUS Medical School) for helpful discussions of the metabolic data.
Author Contributions
L.F.I. performed the experiments, F.C. and R.S. helped in sample collection and processing, A.L., P.M.Y., F.G. and R.A.S. designed the study and wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02205-1
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Antonia Lanni, Email: antonia.lanni@unina2.it.
Paul M. Yen, Email: paul.yen@duke-nus.edu.sg
Rohit A. Sinha, Email: anthony.rohit@gmail.com
References
- 1.Khan RS, Newsome PN. Non-alcoholic fatty liver disease and liver transplantation. Metabolism: clinical and experimental. 2016;65:1208–1223. doi: 10.1016/j.metabol.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Pais R, et al. NAFLD and liver transplantation: Current burden and expected challenges. Journal of hepatology. 2016;65:1245–1257. doi: 10.1016/j.jhep.2016.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology (Baltimore. Md.) 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 4.Cazanave SC, Gores GJ. Mechanisms and clinical implications of hepatocyte lipoapoptosis. Clinical lipidology. 2010;5:71–85. doi: 10.2217/clp.09.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaurasia B, Summers SA. Ceramides - Lipotoxic Inducers of Metabolic Disorders. Trends in endocrinology and metabolism: TEM. 2015;26:538–550. doi: 10.1016/j.tem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Sunny NE, et al. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. American journal of physiology. Endocrinology and metabolism. 2015;309:E311–319. doi: 10.1152/ajpendo.00161.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cable EE, et al. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology (Baltimore, Md.) 2009;49:407–417. doi: 10.1002/hep.22572. [DOI] [PubMed] [Google Scholar]
- 8.Yehuda-Shnaidman E, Kalderon B, Bar-Tana J. Thyroid hormone, thyromimetics, and metabolic efficiency. Endocrine reviews. 2014;35:35–58. doi: 10.1210/er.2013-1006. [DOI] [PubMed] [Google Scholar]
- 9.Senese R, Lasala P, Leanza C, de Lange P. New avenues for regulation of lipid metabolism by thyroid hormones and analogs. Frontiers in physiology. 2014;5:475. doi: 10.3389/fphys.2014.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silvestri E, Coppola M, Cioffi F, Goglia F. Proteomic approaches for the study of tissue specific effects of 3,5,3′-triiodo-L-thyronine and 3,5-diiodo-L-thyronine in conditions of altered energy metabolism. Frontiers in physiology. 2014;5:491. doi: 10.3389/fphys.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha, R. & Yen, P. M. In Endotext (eds L. J. De Groot et al.) (MDText.com, Inc., 2000).
- 12.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocrine reviews. 2010;31:139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Lange P, et al. Nonthyrotoxic prevention of diet-induced insulin resistance by 3,5-diiodo-L-thyronine in rats. Diabetes. 2011;60:2730–2739. doi: 10.2337/db11-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goglia F, Lanni A, Barth J, Kadenbach B. Interaction of diiodothyronines with isolated cytochrome c oxidase. FEBS letters. 1994;346:295–298. doi: 10.1016/0014-5793(94)00476-5. [DOI] [PubMed] [Google Scholar]
- 15.Arnold S, Goglia F, Kadenbach B. 3,5-Diiodothyronine binds to subunit Va of cytochrome-c oxidase and abolishes the allosteric inhibition of respiration by ATP. European journal of biochemistry. 1998;252:325–330. doi: 10.1046/j.1432-1327.1998.2520325.x. [DOI] [PubMed] [Google Scholar]
- 16.Senese R, Cioffi F, de Lange P, Goglia F, Lanni A. Thyroid: biological actions of ‘nonclassical’ thyroid hormones. The Journal of endocrinology. 2014;221:R1–12. doi: 10.1530/JOE-13-0573. [DOI] [PubMed] [Google Scholar]
- 17.Cano A, Alonso C. Deciphering non-alcoholic fatty liver disease through metabolomics. Biochemical Society transactions. 2014;42:1447–1452. doi: 10.1042/BST20140138. [DOI] [PubMed] [Google Scholar]
- 18.Dumas ME, Kinross J, Nicholson JK. Metabolic phenotyping and systems biology approaches to understanding metabolic syndrome and fatty liver disease. Gastroenterology. 2014;146:46–62. doi: 10.1053/j.gastro.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Kaikkonen, J. E. et al. Metabolic profiling of fatty liver in young and middle-aged adults - cross-sectional and prospective analyses of the Young Finns Study. Hepatology (Baltimore, Md.), doi:10.1002/hep.28899 (2016). [DOI] [PMC free article] [PubMed]
- 20.Lai YS, et al. Mass-Spectrometry-Based Serum Metabolomics of a C57BL/6J Mouse Model of High-Fat-Diet-Induced Non-alcoholic Fatty Liver Disease Development. Journal of agricultural and food chemistry. 2015;63:7873–7884. doi: 10.1021/acs.jafc.5b02830. [DOI] [PubMed] [Google Scholar]
- 21.Safaei A, et al. Metabolomic analysis of human cirrhosis, hepatocellular carcinoma, non-alcoholic fatty liver disease and non-alcoholic steatohepatitis diseases. Gastroenterology and hepatology from bed to bench. 2016;9:158–173. [PMC free article] [PubMed] [Google Scholar]
- 22.Terashima Y, et al. Metabolomics-based search for therapeutic agents for non-alcoholic steatohepatitis. Archives of biochemistry and biophysics. 2014;555–556:55–65. doi: 10.1016/j.abb.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 23.von Schonfels W, et al. Metabolomic tissue signature in human non-alcoholic fatty liver disease identifies protective candidate metabolites. Liver international: official journal of the International Association for the Study of the Liver. 2015;35:207–214. doi: 10.1111/liv.12476. [DOI] [PubMed] [Google Scholar]
- 24.Lanni A, et al. 3,5-diiodo-L-thyronine powerfully reduces adiposity in rats by increasing the burning of fats. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1552–1554. doi: 10.1096/fj.05-3977fje. [DOI] [PubMed] [Google Scholar]
- 25.Cavallo A, et al. 3,5-Diiodo-L-thyronine administration to hypothyroid rats rapidly enhances fatty acid oxidation rate and bioenergetic parameters in liver cells. PloS one. 2013;8:e52328. doi: 10.1371/journal.pone.0052328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha RA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. The Journal of clinical investigation. 2012;122:2428–2438. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grasselli E, et al. 3,5-Diiodo-L-thyronine modulates the expression of genes of lipid metabolism in a rat model of fatty liver. The Journal of endocrinology. 2012;212:149–158. doi: 10.1530/JOE-11-0288. [DOI] [PubMed] [Google Scholar]
- 28.Grasselli E, et al. Direct effects of iodothyronines on excess fat storage in rat hepatocytes. Journal of hepatology. 2011;54:1230–1236. doi: 10.1016/j.jhep.2010.09.027. [DOI] [PubMed] [Google Scholar]
- 29.Sinha RA, Singh BK, Yen PM. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends in endocrinology and metabolism: TEM. 2014;25:538–545. doi: 10.1016/j.tem.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Schooneman MG, Vaz FM, Houten SM, Soeters MR. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes. 2013;62:1–8. doi: 10.2337/db12-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology (Baltimore, Md.) 2011;53:116–126. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buettner R, Scholmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity. 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- 34.Lake AD, et al. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino acids. 2015;47:603–615. doi: 10.1007/s00726-014-1894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmadian M, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell metabolism. 2011;13:739–748. doi: 10.1016/j.cmet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Settembre C, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature cell biology. 2013;15:647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weitzel JM, Iwen KA. Coordination of mitochondrial biogenesis by thyroid hormone. Molecular and cellular endocrinology. 2011;342:1–7. doi: 10.1016/j.mce.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Sinha RA, et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy. 2015;11:1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesmana R, et al. Thyroid Hormone Stimulation of Autophagy Is Essential for Mitochondrial Biogenesis and Activity in Skeletal Muscle. Endocrinology. 2016;157:23–38. doi: 10.1210/en.2015-1632. [DOI] [PubMed] [Google Scholar]
- 40.Chakraborty M, Jiang XC. Sphingomyelin and its role in cellular signaling. Advances in experimental medicine and biology. 2013;991:1–14. doi: 10.1007/978-94-007-6331-9_1. [DOI] [PubMed] [Google Scholar]
- 41.Samuel VT, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. The Journal of biological chemistry. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Doronin S, Malbon CC. Insulin activation of mitogen-activated protein kinases Erk1,2 is amplified via beta-adrenergic receptor expression and requires the integrity of the Tyr350 of the receptor. The Journal of biological chemistry. 2000;275:36086–36093. doi: 10.1074/jbc.M004404200. [DOI] [PubMed] [Google Scholar]
- 43.Vatner DF, et al. Thyroid hormone receptor-beta agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. American journal of physiology. Endocrinology and metabolism. 2013;305:E89–100. doi: 10.1152/ajpendo.00573.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh BK, et al. Hepatic FOXO1 Target Genes Are Co-regulated by Thyroid Hormone via RICTOR Protein Deacetylation and MTORC2-AKT Protein Inhibition. The Journal of biological chemistry. 2016;291:198–214. doi: 10.1074/jbc.M115.668673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rochira A, Damiano F, Marsigliante S, Gnoni GV, Siculella L. 3,5-Diiodo-l-thyronine induces SREBP-1 proteolytic cleavage block and apoptosis in human hepatoma (Hepg2) cells. Biochimica et biophysica acta. 2013;1831:1679–1689. doi: 10.1016/j.bbalip.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Vergani L. Lipid lowering effects of iodothyronines: In vivo and in vitro studies on rat liver. World journal of hepatology. 2014;6:169–177. doi: 10.4254/wjh.v6.i4.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinha RA, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology (Baltimore, Md.) 2014;59:1366–1380. doi: 10.1002/hep.26667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.