

Abstract
Sphingosine-1-phosphate (S1P) is a lipid mediator that can activate five cell membrane G protein-coupled receptors (GPCRs) which carry a variety of essential functions and are promising drug targets. S1P is composed of a polar zwitterionic head-group and a hydrophobic alkyl chain. This implies an activation mechanism of its cognate receptor that must be significantly different from what is known for prototypical GPCRs (ie receptor to small hydrophilic ligands). Here we aim to identify the structural features responsible for S1P agonism by combining molecular dynamics simulations and functional assays using S1P analogs of different alkyl chain lengths. We propose that high affinity binding involves polar interactions between the lipid head-group and receptor side chains while activation is due to hydrophobic interactions between the lipid tail and residues in a distinct binding site. We observe that ligand efficacy is directly related to alkyl chain length but also varies with receptor subtypes in correlation with the size of this binding pocket. Integrating experimental and computational data, we propose an activation mechanism for the S1P receptors involving agonist-induced conformational events that are conserved throughout class A GPCRs.
Introduction
By ensuring the conversion of extracellular stimuli into cellular responses, G protein-coupled receptors (GPCRs) modulate signaling pathways in a wide variety of biological processes. The diversity of GPCRs functions is associated with a remarkable variety in their cognate ligands, from both structural and chemical standpoints. Indeed, these receptors bind entities as different as calcium ions, small organic molecules (amines, steroids), nucleotides, peptides, proteins or lipids1. GPCRs are classified into five main families or classes based on sequence similarity2, the class A, also known as rhodopsin family, being the largest and most studied. High-resolution structures of receptors from different families have confirmed that GPCRs share a similar architecture of seven transmembrane (TM) α-helices forming a bundle and that the TM domains are structurally conserved3, 4. Class A GPCRs exhibit a distinctive feature that most of their ligands bind to a cavity inside the TM helices. While most of them recognize small polar agonists, GPCRs for lipid mediators are activated by hormone-like signaling molecules derived from lipid species, which possess long hydrophobic moieties5. This subfamily is mostly composed of the sphingosine-1-phosphate (S1P) and lysophosphatidic acid (LPA) receptors (formerly grouped in the endothelial differentiation gene or EDG family) as well as the cannabinoid receptors. Sphingosine-1-phosphate (S1P) regulates a plethora of biological functions in the central nervous system, immune and cardiovascular systems as well as numerous pathophysiological processes6. This lysophospholipid is produced intracellularly from sphingolipid catabolism and exerts its functions mainly via activation of five cell membrane specific GPCRs, S1P1-5 (initially edg-1, 3, 5, 6, and 8)7. The diversity of S1P signaling and its regulation stems from synthesis and degradation balance, differential expression of S1P receptors in various cell types as well as from their distinct coupling to G proteins subtypes8. S1P receptors display a variety of essential functions while activated by a unique agonist, conferring them a major therapeutic potential, as underlined by the approval by the US FDA of the S1P1-mediated immune modulator FTY720 (fingolimod; GilenyaTM, Novartis) for the treatment of relapsing-remitting multiple sclerosis9, 10. Fingolimod is a prodrug that is phosphorylated to form fingolimod-phosphate (Fig. 1), which activates lymphocyte S1P1 and prevents lymphocyte egress from lymphoid tissues11.
Figure 1.
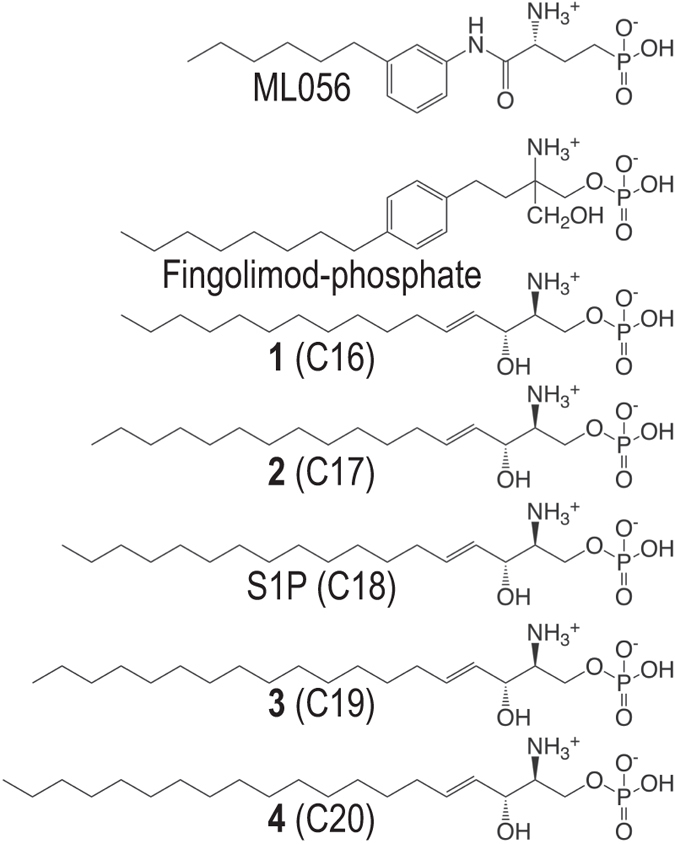
Structure of ML056, Fingolimod-phosphate, S1P and synthetic derivatives 1–4 of variable chain length.
S1P is an amphipathic molecule composed of a polar zwitteronic head-group and a hydrophobic alkyl chain (Fig. 1). Several studies combining molecular modeling and mutagenesis investigating the binding of S1P to S1P receptors show that the agonist head-group interacts with charged residues at one site of the receptor and its alkyl chain lies at a hydrophobic binding pocket6. The antagonist-bound S1P1 receptor structure12 and molecular dynamics (MD) simulations of the process of ligand entry from the membrane bilayer to the binding pocket13 have confirmed the role of the ligand head-group in both the process of ligand entry and receptor recognition. The S1P1 antagonist ML056 polar moiety possesses a primary amine and phosphonate group, features that are closely related to those of S1P (Fig. 1). Though the role of ligand polar moiety has been investigated for the native agonist14 as well as for synthetic molecules15, the implication of the alkyl chain in receptors activation remains elusive. While the S1P head-group is required for its high affinity binding to the receptor, acting as an anchor, elements suggest that it is not key in receptor activation, but rather that the alkyl chain may be involved in agonist activity16. This assumption is supported by the fact that an antagonist head-group can share closely related chemical functions with S1P and interact via the same residues, as exemplified by ML05612. The hypothesis that agonism could depend on the deformation of the TM pocket induced by a hydrophobic volume is underpinned by the investigation of S1P1 interactions with a series of synthetic agonists17, as well as structure-activity relationships work on FTY720 analogs18, 19. The latter studies reveal that the change of alkyl substituent position from para- to meta- on the aromatic ring converts the agonist to an antagonist, whereas changing chain length from 6 to 10 carbons on the meta- analog restores agonist activity.
In this work, by combining computational and experimental approaches, we identify the structural and chemical features that are responsible for S1P agonism, leading to a comprehensive activation mechanism for the S1P receptor family.
Results and Discussion
Molecular dynamics simulations of S1P1 in complex with the natural S1P agonist
We first aimed at identifying the molecular interactions between the natural agonist S1P and its cognate receptor, using the crystal structure of the S1P1 receptor in complex with the antagonist ML05612 (Supplementary Fig. S1A), which bears significant chemical and structural similarities with S1P (Fig. 1). We performed unbiased 2 μs MD simulation of S1P1, in the inactive conformation as determined in the crystal structure, with the natural S1P agonist bound to the receptor (see Experimental Procedures). The zwitterionic head-group of S1P was docked into the receptor analogously to the binding mode of the co-crystallized ML056 antagonist, interacting with N1012.60, R1203.28 and E1213.29 as previously determined by site-directed mutagenesis14. The alkyl chain inserts into a hydrophobic/aromatic pocket between TMs 3, 5 and 6 (Supplementary Fig. S1B), similarly to previous proposed models12, 20. This docking pose suggests that the end-terminal tail of the ligand points either toward TMs 3 and 5 (mainly F1253.33, L1283.36, C2065.43, F2105.47 shown in light green spheres) or toward TMs 5 and 6 (T2075.44, F2105.47, F2736.52, L2766.55 shown in dark green spheres). However, as shown in Supplementary Fig. S1B, these side chains are highly packed together in the crystal structure of inactive S1P1. This suggests that the additional methylene units of the S1P requires modification of the conformation of these side chains and the packing of TMs 3, 5, or/and 6 for agonist binding and thus receptor activation (see below). This assumption seems likely since the side chains at positions 3.3621, 22, 5.4323, 5.4724, 25, 6.5226 and 6.5527 have been proposed to be involved in receptor activation in other members of the class A GPCR family. We have monitored the influence of S1P on dihedral χ1 angles of these side chains (red dots) during the MD simulation (Supplementary Fig. S1E). Importantly, no significant changes in the conformation of the side chains are observed. Thus, these results suggest that the additional length of the alkyl chain of the S1P agonist, relative to the ML056 antagonist, has not been capable to modify the packing of TMs 3, 5, and 6 in the MD simulation of inactive S1P1. However a plot of the root-mean-square-deviation (rmsd) of S1P’s zwitterionic head-group relative to the initial, ML056-like binding pose (Supplementary Fig. S1E, red line) clearly shows the head-group shifts out from the initial binding position as revealed by rmsd values >5 Å (see Supplementary Fig. S1C). In contrast, rmsd values of the TM domain of the receptor (broken red line in Supplementary Fig. S1E) remain low, indicating no significant conformational changes of the S1P1 structure. These results suggest that S1P cannot be accommodated by an inactive structure of S1P1 while maintaining the key interactions with N1012.60, R1203.28 and E1213.29.
GPCRs are dynamic proteins that permit rapid small-scale structural fluctuations and pass through an energy landscape to adopt a number of conformations ranging from inactive to active28. However, pioneering work by the Kobilka group has shown that an agonist alone is not capable to stabilize the fully active conformation of the receptor in the absence of the G protein29. Similarly, MD simulations of an agonist bound to the inactive state of the receptor are not capable to reach active-like conformations in the absence of the G protein. Thus, to study S1P1 activation by S1P, we built an active-like model of S1P1 using the crystal structure of a nanobody-stabilized active state of the β2-adrenergic receptor23 as a template (see Experimental Procedures). In this model the intracellular part of S1P1 has been constructed as in the active conformation of β2-receptor (see Supplementary Fig. S2), in which the intracellular cavity for G protein binding is opened through the movement of the cytoplasmic end of TM 6 away from TM 3 and towards TM 5. Analogously to the structure of β2-receptor, the active-like model of S1P1 includes the G-protein-mimetic nanobody30, 31. It is important to remark that the extracellular part, including receptor side chain conformations, of the active-like model of S1P1 is similar to the inactive model (Supplementary Fig. S2D). For the active-like model, two independent replicas of 500 ns each were simulated, starting at different docking poses of the alkyl chain of S1P (Fig. 2A). Analysis of the trajectories revealed that these predicted complexes were highly stable, both the S1P ligand and the nanobody bound to the receptor (Supplementary Fig. S3). Thus, in contrast to the simulation of inactive S1P1, ligand rmsd of the zwitterionic head-group of S1P remained low (Supplementary Fig. S1E), indicating that this part of the ligand remained bound to N1012.60, R1203.28 and E1213.29. More important, in the two simulations, the terminal part of the alkyl chain of the ligand orients toward a small cavity that is opened between TMs 3 and 5 (Fig. 2B). Figure 2B shows that, independently of the starting position, the final conformational of the alkyl chain of S1P is similar in the two MD simulations.
Figure 2.
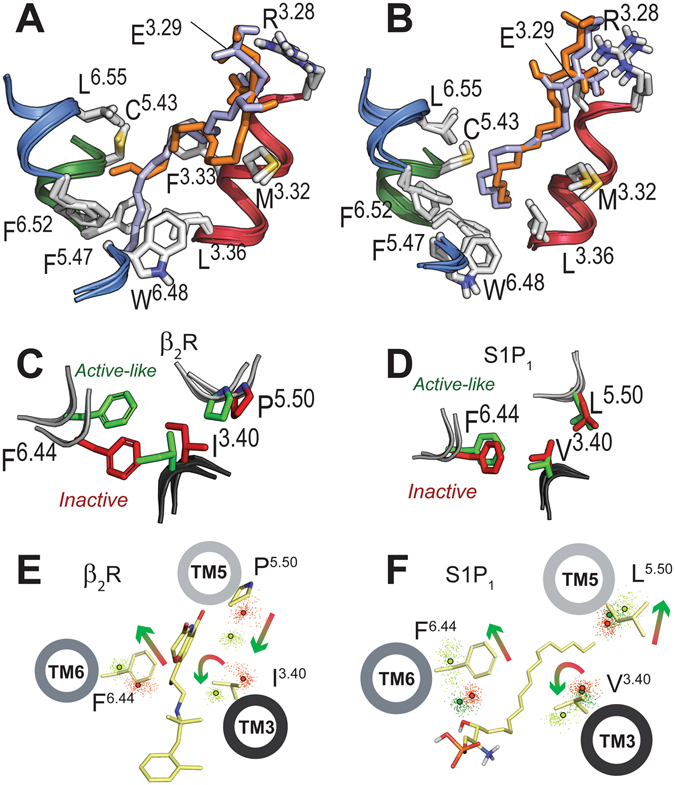
Mechanism of S1P-induced activation of S1P1. (A) Detailed view of the extracellular domain of the energy-minimized inactive-like and active-like models of S1P1. The initial binding pose of S1P used both in the inactive- and active-like MD simulation is shown in orange. A different conformation of the alkyl chain of S1P (in light blue) was also simulated for the active-like model of S1P1. (B) The final conformation, at 0.5 μs, obtained in the MD simulations of both starting conformations of S1P in complex with the active-like model of S1P1. (C,D) The starting positions of I1213.40, P2115.50 and F2826.44 (C) and V1323.40, L2135.50 and F2656.44 (D) (‘transmission switch’) used in the MD simulations of inactive- (in red) and active- (in green) conformations of the β2-adrenergic receptor (C) and S1P1 (D). Because the extracellular part of the active-like model of S1P1 is comparable to the inactive model (see Fig. 2A and Supplementary Fig. S2D), initial positions of these side chains, in the simulations, are similar and correspond to the inactive conformation. (E,F) Evolution of the Cβ atoms (dots) of I1213.40, P2115.50 and F2826.44 of β2- (E) and V1323.40, L2135.50 and F2656.44 of S1P1 (F) during the MD simulations (0.5 μs) of inactive- (in red) and active- (in light and dark green) like conformation of β2- in complex with the BI167107 agonist (light green, panel E) and S1P1 in complex with S1P (light green, panel F) and ligand-free (dark green, panel F). Centroids (calculated from 250 snapshots) of the Cβ atoms of these side chains are shown in red (inactive), dark green (ligand-free active conformation) and light green (agonist-bound active conformation) circles. Displayed helices, side chains, and agonists are shown for clarity. Arrows represent the observed movement of the helices during the MD simulations.
Mechanism of S1P-induced activation of S1P1
We assume, as a working hypothesis, that activation of S1P1 involves rearrangement of the conserved ‘transmission switch’ made of the amino acids at positions 3.40, 5.50, and 6.4432. This conserved ‘transmission switch’ has been proposed for other GPCRs based on the fact that, for instance, a hydrogen bond interaction between agonists and TM 5 in β1-33 and β2-23 or the conformational change of inactive 11-cis retinal to the active 11-trans retinal in rhodopsin34 or agonist-binding to A2A35, stabilizes a receptor conformation that includes an inward movement of TM 5 at the highly conserved P5.50, relative to inactive structures. For comparison purposes with S1P1, we simulated this ‘transmission switch’ in the β2-adrenergic receptor, a prototypical GPCR, for which experimental structures of the inactive and active conformation of the receptor are available (Fig. 2C). Clearly, the inward movement of TM 5 is sterically competing with a bulky hydrophobic side chain at position 3.40, triggering a small counterclockwise rotation, viewed from the extracellular part, of TM 336 (Fig. 2E). Finally, the rotation of TM 3 repositions the side chain of F6.44, facilitating the outward movement of TM 6 for receptor activation23 (Fig. 2E). Next we analyzed in the MD simulations of inactive- and active-like S1P1 receptor this ‘transmission switch’ by monitoring the rotation and displacement of TMs 3, 5 and 6 (Fig. 2F). It is important to note that, in contrast to the β2-adrenergic receptor (Fig. 2C), the initial position of the TMs and the key V1323.40, L2135.50 and F2656.44 side chains of S1P1 were similar in inactive- and active-like conformations (Fig. 2D). However, the MD simulations reveal different final position of the helices (Fig. 2F). Relative to the inactive simulation, in the active simulation we observe an extracellular outward movement of TM 5, while TM 3 performs the proposed counterclockwise rotation of 10° (Fig. 2F and Supplementary Fig. S1D) positioning V1323.40 towards TM 6, which leads to a steric exclusion of V1323.40 with the side chain of F2656.44 and the outward movement of TM 6 for receptor activation. Clearly, the extracellular outward movement of TM 5, away from TM 3, during S1P1 activation is different to the inward movement observed in the process of activation of “typical” GPCRs such as the β2-receptor (compare Fig. 2E,F). Conformational divergence is expected in TM 5 as GPCRs activated by lipid mediators (S1P, LPA and cannabinoid receptors) lack the highly conserved P5.50 present in other GPCRs, thus suggesting a local conformational specificity. Importantly, the V1323.40L, L2135.50G or F2656.44G mutations, performed by others14, decreased the S1P-induced Emax by 55%, 46%, or 28%, respectively, relative to wt. Thus, we can conclude that activation of S1P1 involves several structural elements of the ‘transmission switch’ present in the other members of the GPCR family.
It is important to discriminate the effect of the ligand at the extracellular domain and of the nanobody at the intracellular domain in triggering these conformational changes of receptor activation. Thus, we performed an additional 0.5 μs of MD simulations of the active-like model of S1P1 bound to the nanobody but in the absence of the ligand for comparison purposes (see Experimental Procedures). Clearly, the ‘transmission switch’ elements of receptor activation do not occur in the absence of the ligand (Fig. 2F, light green vs. dark green). Thus, our simulations indicate that the S1P ligand or the nanobody alone cannot activate S1P1 and the ‘transmission switch’ elements of receptor activation are only observed in the presence of both the ligand and the G protein-mimetic.
What are the initial structural changes of S1P-induced receptor activation responsible for the rearrangement of the ‘transmission switch’? Analysis of the MD simulations shows that the terminal part of the alkyl chain of the ligand (the additional methylene units present in the agonist) inserts into a small cavity between TMs 3 and 5 (Fig. 2B). This cavity is created following conformational changes of L1283.36 from trans to gauche- and F2105.47 from gauche- to trans (Supplementary Fig. S1E). We propose that the conformational changes of the L1283.36 and F2105.47 side chains, opening the cavity for the ligand tail, initiate activation of the ‘transmission switch’ via rigid body motions of TMs 3 and 5. F2105.47 is pointing toward TM 3 in the inactive gauche- conformation and toward TM 6 in the active trans conformation to interact with W2696.48, whereas L1283.36 is pointing toward TM 6 in the inactive trans conformation and toward TM 7 in the active gauche- conformation (Fig. 2A,B). Fujiwara et al. showed that the L1283.36G, F2105.47G or W2696.48A mutations impede S1P-induced receptor activation without modifying ligand binding14. Moreover, the role of these side chains at position 3.3621, 22, 5.4724, 25 or 6.4821, 22, 24 in receptor activation has previously been described for other GPCRs. Importantly, the MD simulation of S1P1 bound to the nanobody but in the absence of the ligand does not trigger the conformational changes of L1283.36 from trans to gauche- and F2105.47 from gauche- to trans (dark green data points distribute like red data points in Supplementary Fig. S1E). These findings reinforce our proposal that both the ligand and the intracellular nanobody are required for receptor activation.
Therefore our simulations suggest that the ability of the ligand to stabilize the active state of the S1P1 receptor is coupled to the insertion of the lipid tail inside the hydrophobic pocket between TMs 3, 5 and 6. We have subsequently tested this model by measuring activation of the receptor using S1P analogs of different chain length.
Synthesis of S1P analogs
We synthesized S1P analogs 1–4 with decreasing and increasing number of methylene units (n = 10, 11, 13, 14) in the hydrophobic chain with respect to S1P (n = 12), while the head-group of the molecule was kept identical. The compounds were prepared as depicted in Fig. S4. Thus, the corresponding terminal alkenes were coupled to phosphorylated fragment 5 via an olefin cross metathesis reaction to obtain intermediates 6–9. Then, treatment of these intermediates with bromotrimethylsilane afforded the desired sphingosine derivatives 1–4 by simultaneous deprotection of the amino and phosphate groups. S1P, with a total number of 18 carbon atoms (C18), and the synthesized derivatives of 16 (1 or C16), 17 (2 or C17), 19 (3 or C19) and 20 (4 or C20) carbon atoms are represented in Fig. 1.
Functional characterization of S1P analogs at S1P receptors
In order to assess the influence of ligands chain length on the activation of S1P receptors, we compared the functional response induced by the synthesized S1P derivatives on mammalian cell lines expressing the receptor of interest (see Experimental Procedures). Figure 3A shows the dose-dependent curves corresponding to S1P1 functional responses. We observe that all the S1P analogs tested are agonists of the S1P1 receptor, but to different extents, both in terms of potency and efficacy. Mean S1P-normalized Emax (maximal efficacy) and pEC50 values of the different compounds towards S1P1 obtained from dose-response curves are presented in Tables 1 and 2, respectively. Clearly, the rank order of receptor activation (Emax) is C19 (116% of S1P maximal activation) ≈ C17 ≈ C18 (S1P) > C16 (78%). For C20, due to the lower potency, Emax determination was no reliable within the range of concentrations tested and is thus not taken into account in the activation efficacy interpretation. The mean Emax values on 4 independent experiments for each compound are represented on the corresponding histogram of Fig. 3A, illustrating the reproducibility of the observed trend in maximal efficacy. The fact that at saturating concentration the different S1P analogs exhibit different maximal response clearly indicates that the alkyl chain is involved in stabilizing the active conformation of the receptor. Supplementary Fig. S6 shows the position of V1323.40, L2135.50 and F2656.44 (‘transmission switch’) obtained during MD simulations of S1P1 bound to these ligands. The rank order of Emax correlates with the movements of the ‘transmission switch’ amino acids. Clearly, the partial agonist C16 triggers the least outward movement of TM 5, rotation of TM 3, and the outward movement of TM 6 relative to the other full agonist ligands.
Figure 3.
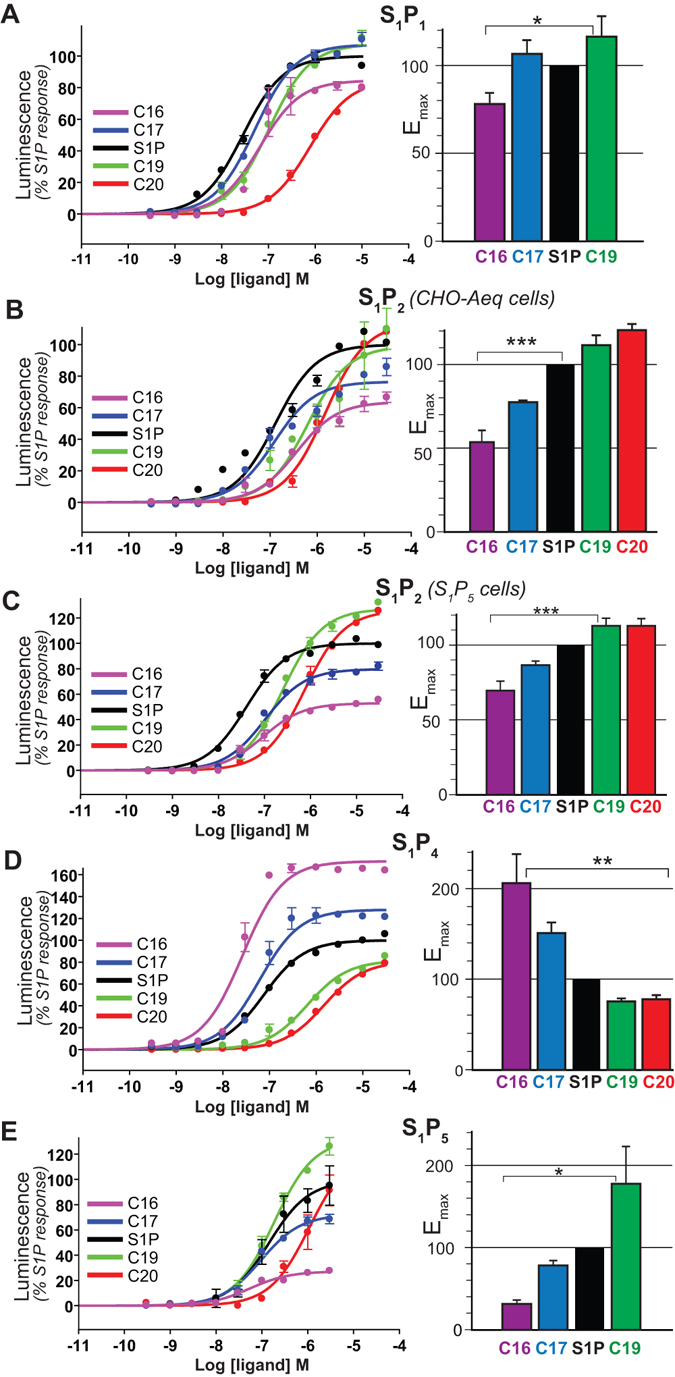
Functional response of S1P receptors to S1P synthetic analogs. Aequoscreen CHO-K1 cells expressing the different S1P receptors were subjected to stimulation with increasing concentrations of S1P-derived ligands and the resulting luminescence was measured. (A) S1P1, n = 4 (B) S1P2 from CHO-Aeq cells, n = 3 (C) S1P2 from S1P5 cells, n = 4 (D) S1P4, n = 4 (E) S1P5, n = 2. The corresponding dose-response curves for one representative experiment are shown, where each curve represents the mean ± S.E.M of duplicate data points. Luminescence intensities are normalized on maximal response to the natural agonist S1P. Histograms on the right show maximal efficacy (Emax) values calculated for each S1P analog at S1P receptors, based on the sigmoidal fitting of aequorin functional response. The data plotted represent the mean ± S.E.M and are expressed in % of maximal efficacy in response to stimulation by S1P. Statistical significance was assessed using one-way ANOVA with a Scheffe’s post hoc test: *p < 0.05, **p < 0.01, ***p < 0.001.
Table 1.
Emax values of S1P derivatives at S1P receptors.
| C16 | C17 | S1P | C19 | C20 | |
|---|---|---|---|---|---|
| S1P1 (n = 4) | 78 ± 6 | 107 ± 8 | 100 | 117 ± 12 | n.r. |
| S1P2 1 (n = 4) | 70 ± 6 | 87 ± 3 | 100 | 113 ± 1 | 113 ± 5 |
| S1P2 2 (n = 3) | 54 ± 7 | 78 ± 1 | 100 | 112 ± 6 | 121 ± 4 |
| S1P4 (n = 4) | 206 ± 32 | 151 ± 12 | 100 | 76 ± 3 | 79 ± 4 |
| S1P5 (n = 2) | 32 ± 5 | 79 ± 6 | 100 | 178 ± 45 | n.r. |
The maximal efficacy for each compound at each S1P subtype was determined from the dose-response curves of the aequorin-based functional assay. Mean values on 2 to 4 independent experiments are normalized on S1P maximal response and expressed in %, along with the corresponding S.E.M. values.
1S1P5 cells, 2CHO-Aeq cells, n.r. = not reliable.
Table 2.
pEC50 values of S1P derivatives at S1P receptors.
| C16 | C17 | S1P | C19 | C20 | |
|---|---|---|---|---|---|
| S1P1 (n = 4) | 6.5 ± 0.3 | 6.9 ± 0.2 | 7.1 ± 0.3 | 6.6 ± 0.2 | n.r. |
| S1P2 1 (n = 4) | 6.9 ± 0.1 | 7.0 ± 0.1 | 7.5 ± 0.0 | 6.7 ± 0.1 | 6.3 ± 0.1 |
| S1P2 2 (n = 3) | 6.5 ± 0.2 | 7.1 ± 0.1 | 7.3 ± 0.2 | 6.5 ± 0.2 | 6.0 ± 0.2 |
| S1P4 (n = 4) | 7.3 ± 0.2 | 7.1 ± 0.1 | 7.0 ± 0.1 | 6.1 ± 0.1 | 5.6 ± 0.1 |
| S1P5 (n = 2) | 7.4 ± 0.2 | 6.9 ± 0.1 | 7.3 ± 0.5 | 6.7 ± 0.1 | n.r. |
The experimental EC50 for all S1P derivatives were determined from the dose-response curves of the aequorin-based functional assays. Mean values of pEC50 (negative logarithm of EC50) on 2 to 4 independent experiments are presented along with the corresponding S.E.M. values.
1S1P5 cells, 2CHO-Aeq cells, n.r. = not reliable.
In order to further test this hypothesis, we extended our investigation to other receptors of the S1P family, by performing functional assays with the synthetic ligands on S1P2, S1P4, or S1P5 receptors. Figure 3 shows the dose-response curves obtained for the other sphingosine-1-phosphate receptors (S1P2, S1P4 and S1P5) upon stimulation by S1P and its synthetic derivatives. All compounds display positive efficacies for all the S1P receptor subtypes tested and thus behave like agonists at these receptors. The corresponding Emax and pEC50 values are shown in Tables 1 and 2. For S1P2 coming from CHO-Aeq cells (Fig. 3B), C20 is the more efficacious with 120% of S1P maximal response, followed by C19 at 112%, whereas C17 and C16 display a lower efficacy than S1P with respectively 78% and 54%. The reproducible trend of Emax values shows that the longer the alkyl chain, the higher the efficacy of the compound. For S1P2 in the CHO-Aeq system, we observed a non-specific contribution at high concentrations (unlikely due to another S1P receptor) that lowers the quality of fitting. Therefore we confirmed our S1P2 data using S1P5 cell line, knowing that about 90% of the functional response detected in these cells is inhibited by JTE013, a specific S1P2 antagonist, and thus is due to S1P2 receptors. In this system (Fig. 3C), C19 displays a higher Emax value than S1P of about 113%, whereas C17 displays lower Emax than S1P around 87%. Finally, C16 has the lowest efficacy with 70% of maximal S1P activation. Functional responses obtained on S1P5 cell line are exempt of non-specific contribution at high concentrations of ligands, within the range tested. As the two cell lines are transfected with distinct promiscuous G proteins, we can expect that the apparent potencies and efficacies can vary slightly from one system to another, even when looking at identical receptors. Nevertheless, we observe the same behavior in both systems: compounds with longer hydrophobic chain activate S1P2 receptor more efficiently (Fig. 3C). This agrees with the overall behavior observed with S1P1.
S1P4 activation profiles (Fig. 3D) display obvious differences with the previous S1P receptor subtypes tested. Strikingly, efficacy at the S1P4 receptor is 2 times higher for C16 than for S1P and 1.5 time for C17, while compounds C19 and C20 display Emax values between 75 and 80% of S1P maximal activation. Therefore, the activation trend observed is the opposite from the other subtypes with respect to S1P showing that shorter chains clearly favor S1P4 activation.
The S1P5 functional response was determined in the presence of the S1P2 specific antagonist JTE013, which allows to entirely remove the contribution of endogenous S1P2 receptor (Fig. 3E). The efficacies show that the longer chains activate better the receptor than the shorter ones, with C19 Emax estimation (~180%) higher than S1P, whereas C17 show 78% and C16 only 32% efficacy. Note that C19 Emax value could be underestimated given that the plateau was not clearly reached. As in the case of S1P1, the C20 response curve did not allow the determination of a reliable Emax, due to its low EC50.
Tables 1 and 2 summarize the S1P-normalized Emax and pEC50 values obtained for S1P and all the synthetic derivatives at the four S1P receptor subtypes tested. As explained above, Emax determination for C20 on S1P1 and S1P5 receptors was not considered as reliable. By comparing efficacies, we can conclude that S1P2 and S1P5 are activated better by longer hydrophobic chains. S1P1 follows a similar trend, but with lower distinction between the compounds triggering the maximal activity C19, S1P and C17, while significantly lower efficacy is observed for C16. Interestingly, S1P4 presents the opposite trend, meaning that the shorter chains activate the system to the higher level than the longer ones.
Taken together, our results on G protein-triggered calcium mobilization support that the alkyl chain length is a key factor of agonism, an observation that can be applied to almost the whole S1P receptors family. In addition, functional properties are subtype-dependent within S1P receptors family, suggesting a specific behavior in response to hydrophobic chain length modulation.
In order to verify that the measured response was not influenced by the coupling system (i.e. co-expression of apoaequorin along with a promiscuous G protein subtype) we then tested if the influence of the alkyl chain length can be observed at the metabolic level. Specifically, we measured NF-kB activation following stimulation of C16, S1P (C18) and C19 compounds analogs in BEAS-2B where such metabolic response to S1P is solely due to endogenous S1P2 activation37. When we stimulated BEAS-2B cells with various concentrations and measured NF-kB activity (see Experimental Procedures) we observe concentration-dependent response to the S1P and its analogs and the maximal level of activation reached differs from one compound to another (Fig. 4).
Figure 4.
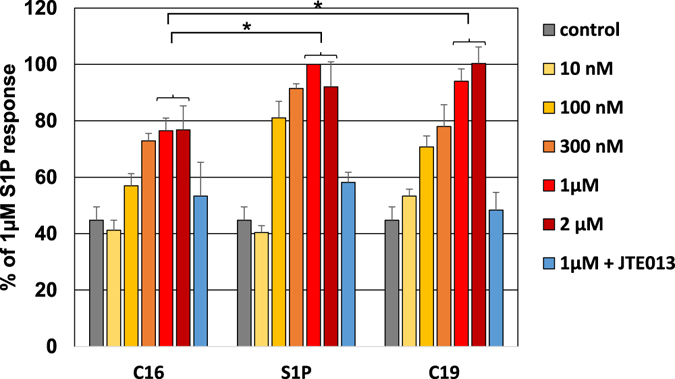
NF-κB activation via S1P2 in response to S1P analogs. Human BEAS-2B cells naturally expressing S1P2 receptor were subjected to stimulation with increasing concentrations (10 nM to 2 µM, from light yellow to dark red) of C16, S1P and C19 and resulting NF-κB activation was followed by luminescence (luciferase reporter). Untreated cells were used as a control (in grey) and 1 µM condition was also performed after pre-incubation with the selective S1P2 antagonist JTE013 (in blue). Histograms represent the mean values ± S.E.M. on 3 independent experiments for each condition, expressed as a percentage of 1 µM S1P response. Statistical significance between maximal activation levels (plateau at 1–2 µM) for different compounds was assessed using one-way ANOVA with a Scheffe’s post hoc test: *p < 0.05.
The efficacy of the alkyl chain variants are different, with C16 displaying the lowest efficacy, with about 77% of S1P activation, and C19 showing an equal to higher efficacy than S1P (note that Emax is not reached at 1 or 2 µM for S1P2 in the aequorin assay, see Fig. 3B). The differences between the maximal activation level induced by the different analogs are statistically significant (p < 0.05) (Fig. 4). The trend observed using an S1P2-mediated physiological metabolic response is thus consistent with the activation trend deciphered from our aequorin-based assay for S1P2 (Fig. 3 and Table 1).
As the differences observed are independent of the functional assay, we can therefore conclude that the alkyl chain is a determinant of receptor activation in terms of G protein-coupling but also of biological outcome.
Structure-activity relationships at the sphingosine receptor family
The above results clearly show that ligand chain length influences the activation of sphingosine-1-phosphate receptors in a subtype-dependent manner, with S1P4 behaving quite differently. This seems due to the narrow channel (binding cavity), formed by the hydrophobic and bulky side chains of the amino acids at positions 3.32–3.33, 3.36–3.37, 4.56, 5.42–5.43, 5.46–5.47, 6.51–6.52, 6.55 and 7.39, where the alkyl tail must expand (Fig. 5A,B). The end of this channel is delimited by the amino acids at positions 4.56, 5.42 and 5.46, whereas the other side chains define the shape of the channel. We explored the volume of this channel from the conformational ensembles obtained in the MD trajectories of the natural S1P agonist in complex with the different sphingosine-1-phosphate receptor subtypes. Clearly, the amino acid side chains of S1P4 delineate the smallest channel, the side chains of S1P5 the largest, whereas the channels formed by the side chains of S1P1 and S1P2 are in between (Fig. 5C). This rank order follows our experimental results in which short alkyl chains (suitable for fitting a small size channel) favor S1P4 activation, long alkyl chains (appropriate for fitting a large channel) activate S1P5 more efficiently, and S1P1 and S1P2 favor long alkyl chains but less markedly than S1P5. Analysis of the amino acid sequence forming this channel between S1P4 (the smallest) and S1P5 (the largest) shows differences only at positions 3.32, 3.37 and 5.46 (Fig. 5D). Specifically, S1P4 contains the bulky and β-branched side chain of Ile at position 5.46 while S1P5 contains the small Ala amino acid at the end of the channel. Notably, mutation of the β-branched V2095.46 to Leu in S1P1, increasing side chain flexibility due to the lack of the β-branched character of Leu, augments S1P-induced Emax to 147%14.
Figure 5.
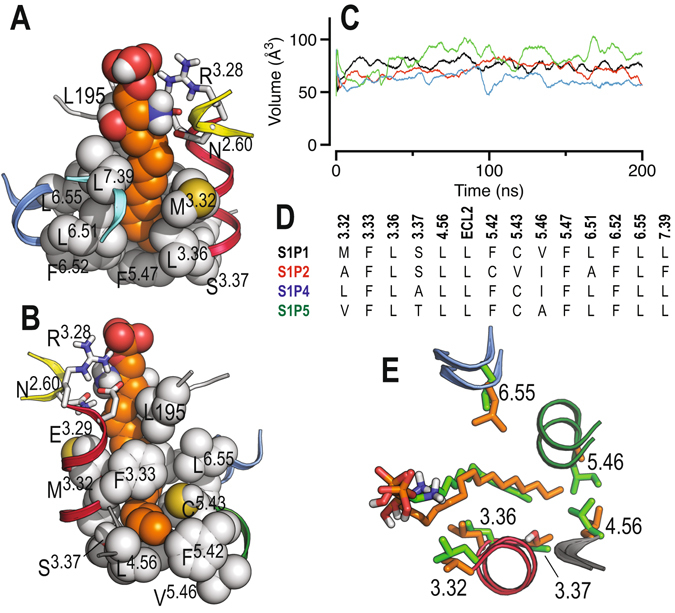
Molecular dynamics simulations of sphingosine-1-phosphate receptors. (A) Detailed view of the narrow channel of S1P1 where the alkyl tail of S1P (in orange) must expand. The end of the channel is delimited by the amino acids at positions 4.56, 5.42 and 5.46. The structure depicts the final conformation at 0.2 μs. (B) Same as in panel A but rotated 180°. (C) Volume of the channel in S1P1 (black), S1P2 (red), S1P4 (blue) and S1P5 (green) along the MD trajectories with the natural S1P agonist as calculated with POVME49. (D) Sequence alignment, among sphingosine receptors, of the amino acids forming this channel. (E) The final conformation, at 0.2 μs, obtained in the MD simulations of S1P4 in complex with C16 (in green) and S1P5 in complex with C20 (in orange).
In addition, the side chain at position 3.37 (Ala in S1P4 and Thr in S1P5) influences the conformation of L4.56. In transmembrane α-helices side chain conformations of Thr are mostly limited to gauche+38 in which the hydroxyl group forms hydrogen bonds with the backbone carbonyl oxygen at position i-3 and the methyl group points toward L4.56. The methyl group of Thr forces L4.56 to adopt the gauche+ conformation pointing away from the bundle and expanding the length of the channel in S1P5, in contrast to the small Ala that permits L4.56, in the trans conformation, to pack against TM 3 making the channel shorter in S1P4 (Fig. 5E).
A major question is to understand at the molecular level the different activation trends observed based on Emax values, among different ligand chain lengths and receptor subtypes. Thus, we explored the initial structural changes triggered by the C16, C17, C18, C19 and C20 ligands in the S1P1, S1P2, S1P4 and S1P5 receptor subtypes by MD simulations (see Experimental Procedures). Although ligand efficacy is a function of multiple factors, we found a clear correlation between Emax values and the conformation of L3.36 (no significant differences in the side chain conformation of F2105.47 are observed), specifically the amount of time the L3.36 side chain spends in the inactive trans or active gauche- conformation (Supplementary Fig. S7). Short alkyl chains (C16-C18) stabilize the active gauche- conformation of L3.36 better than long alkyl chains (C19-C20) in S1P4, whereas this trend is the opposite for S1P5 in which long alkyl chains (C19-C20) are necessary for achieving the active conformation in agreement with the experimental rank order of Emax obtained for these receptor subtypes (Table 1). In addition, the rank order of Emax in S1P1 and S1P2 (Table 1) also correlates with the amount of time L3.36 spends in the active gauche- conformation (C16 < C17 ≈ C18 ≈ C19 for S1P1 and C16 < C17 < C18 < C19 ≈ C20 for S1P2).
Conclusions
Our study demonstrates that the alkyl chain length of the lipidic agonist is a key feature in activation of S1P receptors. This is achieved by hydrophobic interactions between the lipid tail and residues in a binding pocket located between TMs 3–6. We can also extrapolate these findings to fingolimod-phosphate. The additional methylene units of the fingolimod-phosphate agonist, relative to the ML056 antagonist (Fig. 1), also seems a key feature for their different functional activity. Moreover, subtype-specific effects were observed within S1P receptors family, especially marked in the case of S1P4, can be rationalized based on the different volumes of the binding cavities. Our data suggest a model where one region of the ligand (the head group of S1P) is responsible for high-affinity binding, while a distinct part (the alkyl chain) is responsible for triggering activation. Therefore that high affinity binding and receptor activation can be partially uncoupled. This can be illustrated by comparing the profiles of S1P4R, where Emax and EC50 correlate, with that of S1P2 where it is not the case, as S1P has a better efficacy but a lower potency then C19 or C20.
A key question in GPCR biology is how structurally and chemically diverse ligands can activate receptors with the same overall structure and common signaling effectors, i.e. how the binding of epinephrine on β2- and of sphingosine-1-phosphate on S1P1 receptor both lead to an outward movement of TM6 at the intracellular side. Obviously, the binding site is specific to the cognate ligand, but what are the conserved structural elements that allow convergence of the conformational pathways? Residues at TM3-5-6 interface interacting with ligand’s alkyl chain have been identified in the activation mechanism of other GPCRs. This suggests the existence of common structural events following agonist-induced structural changes that are conserved throughout class A and where the different conformational pathways converge. Still, these convergence points have required specific adaptation of each receptor to accommodate the structural specificities of their cognate ligand, as illustrated by the lack of the crucial P5.50 residue in TM5, a characteristic feature of receptors for lipid mediators.
The model proposed for activation of S1P receptors is likely to be applicable to some extent to the other lipid-binding GPCRs, such as related LPA receptors, but also opens perspectives to understanding the transmission switch in other receptors families. In particular, it would be interesting to investigate whether a dual-site model may apply to other types of GPCRs, as already shown for chemokine receptors39.
Methods
Molecular dynamics simulations of sphingosine-1-phosphate receptors in complex with ligands
The crystal structure of inactive S1P1 (PDB code 3V2Y)12 was used for the construction of the S1P1 model. The T4-lysozyme used to stabilize the structure of S1P1 40 was removed and ICLs 2 (Met149-Ser155) and 3 (Ser232-Lys243) were modeled using ICL 2 of dopamine D3 (PDB code 3PBL) and ICL 3 of squid rhodopsin (PDB code 2Z73) as templates with Modeller 9.1241. The natural S1P agonist was docked into the S1P1 receptor using the Autodock Vina tool42. All docking solutions were visually inspected and the poses in which the zwitterionic head-group interacts with N1012.60, R1203.28 and E1213.29 in a similar way as the co-crystalized ML056 antagonist were energy minimized. In these docking poses the alkyl chain of S1P expands towards TMs 3, 5 and 6, similarly to previous proposed docking models12, 20. An alternative conformation of S1P in which the alkyl chain is pointing towards the intracellular part was also considered (Fig. 2A). In parallel, an “active” conformation of S1P1 was modeled from the crystal structure of a nanobody-stabilized active state of the β2-adrenergic receptor (PDB id 3P0G)23, by changing the conformation of the intracellular part of the TM helices of S1P1 (Ala1303.38 – Trp1684.50, Ser2165.53 – Ile2666.45, Ser3047.46 – Leu330CTerm) for the active conformation of β2- (Ala1193.38 – Trp1584.50, Ile2145.53 – Thr2836.45, Ser3197.46 – Leu342CTerm) (see Supplementary Fig. S2 for details). Modeller 9.12 was used to build homology models of “active” S1P2 (33% of sequence identity and 47% of sequence similarity), S1P4 (26% and 41%) and S1P5 (29% and 43%) subtypes using the constructed structure of “active” S1P1 as a template. These “active-like” models include the G-protein-mimetic nanobody23. Autodock Vina was also used to dock C16, C17, C18 (S1P), C19, and C20 ligands into the different “active” models of receptor subtypes. For comparison purposes the crystal structures of the inactive, carazolol-bound, β2-adrenergic receptor (PDB id 2RH1)43 and the nanobody-stabilized active β2-adrenergic receptor bound to the BI167107 agonist (PDB id 3P0G)23 were also studied. VMD and its membrane and solvate plugins were used to embed these computational models into a constructed POPC bilayer44. All molecules closer than 2 Å to any receptor atom were removed from the system. The resulting models, which consist in a rectangular box containing a lipid bilayer (~170 molecules of POPC) with explicit solvent (>16,000 water molecules) and a 0.15 M concentration of Na+ and Cl− ions (~120 and 140 ions, respectively), were energy-minimized and subsequently subjected to a 10 ns MD equilibration with positional restraints on protein coordinates to remove possible voids present in protein/lipids or proteins/water interfaces. After 10 ns, these restraints were released and MD trajectories were produced at constant pressure and temperature: 2 μs for inactive S1P1 + C18 ligand (S1P), two different replicas of 0.5 μs for “active” S1P1 + C18 ligand + nanobody, 0.5 μs for “active” S1P1+ no ligand + nanobody, 0.2 μs for “active” receptor subtypes (S1P1-S1P5) with each of the ligands (compounds C16-C20) + nanobody, 0.5 μs for inactive β2- + carazolol and 0.5 μs for active β2- + BI167107 + nanobody (see Supplementary Table S1). Computer simulations were performed with the GROMACS 4.6 simulation package45, using the AMBER99SB-ILDN force field as implemented in GROMACS, Berger parameters for POPC lipids, and the general Amber force field (GAFF) and HF/6-31 G*-derived RESP atomic charges for the ligands. This procedure has been previously validated46.
Chemistry
Full synthetic details as well as characterization data of final compounds 1–4, together with synthesis of all intermediates, are described in the Supporting Information.
Functional characterization of S1P analogs at S1P receptors
Cell lines
All cell culture work on S1P receptors was performed using AequoScreen® CHO-K1 cell lines provided by Perkin Elmer, which are modified to stably express the mitochondrially targeted apo-aequorin protein (transfection with a bicistronic expression plasmid). CHO-K1 Parental Aequorin cell line (No. ES-000-A24) is stably expressing the mitochondrially targeted apoaequorin and the promiscuous G protein subtype Gα1, redirecting the coupling towards the PLC-calcium signaling cascade. This cell line was used for activation assays on constitutively expressed S1P2 (EDG5) receptor as well as for transient transfection and activation assay on human S1P1 (EDG1) receptor (see below). Activation assays on human S1P4 and S1P5 receptors were performed using aequorin CHO-K1 cells lines stably expressing S1P4 (ref No. ES-592-A) and S1P5 (ref No. ES-593-A) respectively, as well as the apoaequorin and the promiscuous Gαq/i(5) protein.
S1P2 functional response was assessed using the parental cell line (CHO-Aeq) and S1P5 cell line. Indeed, CHO-Aeq cells display significant S1P activity that is solely due to S1P2 as we could fully inhibit it using the S1P2 specific antagonist JTE013 (Supplementary Fig. S5). S1P4 expression was adequate for our assays. In contrast, S1P5 levels were low but experiments performed in presence of the specific S1P2 antagonist JTE013 to inhibit endogenous S1P2 signal which allowed to properly characterize S1P5 response.
Cell culture
All AequoScreen® CHO-K1 cell lines were grown at 37 °C and 5% CO2, in HAM’s F12 Nutrient Mix medium (Gibco, Life Technologies), supplemented with 10% fetal bovine serum (Biowest), penicillin and streptomycin at 50 units/mL (BioWhittaker, Lonza), and 1 mM sodium pyruvate (BioWhittaker, Lonza). Depending on the cell line, the following selection antibiotics were added to the media: 500 μg/mL Geneticin® (G418 sulfate) (Gibco, Life technologies) for the S1P receptors expression selection, 250 μg/mL Zeocin (Invitrogen, Life technologies) for the apoaequorin expression selection, 5 μg/mL Puromycin (Invitrogen, Life technologies) for promiscuous Gαq/i(5) protein expression selection. Cells were passaged, after one wash with Ca2+ and Mg2+-free Dubelcco’s Phosphate Saline Buffer (PBS), using Trypsin-EDTA mixture at 170 U/mL–0.2 mg/mL (BioWhittaker, Lonza).
Transient transfection for S1P1 expression
AequoScreen® CHO-K1 parental cell line stably transfected with pCAEQG plasmid leading to the co-expression of apoaequorin along with the promiscuous G protein subtype Gα16 were grown as described in section B, except from the replacement of regular FBS by Ultra-low Endotoxin FBS (BioWest). CHO-K1 parental cells were seeded in 6-well pates (Greiner) in medium without antibiotics 24 h before transfection, to reach a 50–70% confluency. Transient transfection with a pcDNA3.1+ vector encoding the human gene S1PR1 with an N-terminal HA-tag sequence (Missouri S&T cDNA Resource center), purified with an EndoFree Plasmid Maxi kit (Qiagen). Transient transfection was performed using the cationic polymer JetPei® (Polyplus-transfection) transfection agent at a 2:1 agent:DNA ratio (4 μg DNA/well), that gave the best expression based on western-blotting analysis and S1P functional response. After removal of the transfection agent cells were subjected to a 12 days antibiotic selection (G418, Life technologies) before performing the intracellular calcium mobilization assays.
Aequorin-based calcium mobilization assay
Functional response was assessed by measuring the luminescence of mitochondrial aequorin after agonist-induced intracellular Ca2+ release47. Antibiotics were removed from the cell medium 24 hours before the assays. Cells at 70–80% confluency were collected from culture plates with Ca2+ and Mg2+-free Dulbecco’s phosphate-buffered saline (Lonza) containing 1 mM EDTA (Invitrogen, Life Technologies), centrifuged at 1000 g for 5 min and resuspended at a density of 5.106 cells/mL in Dulbecco’s modified Eagle’s medium (DMEM) w/o phenol red supplemented with 0, 1% BSA (Sigma). Cells were incubated for 4 hours in the dark in the presence of the Aequorin chromophore coelenterazine H (Promega) at 5 μM. Cells were diluted 10 times in DMEM - 0, 1% BSA prior to use. 25000 cells (50 μL) were injected to each well of white 96-well plates Lumitrac200 (Greiner bio-one) containing the prepared dilutions of the ligands (50 μL) in DMEM-0, 1% BSA, ranging from 0.1 nM to 30 μM final concentration. When indicated, specific S1P2 antagonist JTE013 was added at a concentration corresponding to 20 times the IC50 and incubated for 15 min before performing the measurements. For inhibition curve determination (“antagonist mode”), cells were pre-incubated in wells with increasing concentrations of antagonist for 15 min and luminescence is measured after addition of S1P at the EC80 (concentration at 80% of the effect). The emitted light was measured for 30 sec with a CentroXS3 LB960 Luminometer (Berthold Technologies). Analysis of kinetics indicates that the luminescence signal was always contained within the 30 second windows independently of the analog or receptor tested (not shown). Duplicate measurements were performed for each concentration and every experiment with analogs was performed between 2 and 4 times depending on the subtype. As a positive control ATP (Sigma) at 20 μM was used, which leads to full activation of the endogenous purinergic receptors, allowing signal normalization for agonist response. Data were analyzed with the PRISM3.0 software (Graph-Pad Prism Software, San Diego, CA) using nonlinear regression fittings with sigmoidal dose-response model parameters, to determine EC50 (half maximal effective concentration) or IC50 (half maximal inhibitory concentration) and Emax (maximal efficacy) values. To compare the different S1P analogs, Emax values were further normalized on maximal response to the cognate agonist, S1P.
NF-κB luciferase reporter assay
Human BEAS-2B NF-κB luciferase reporter cells (provided by Dr. Simon Rousseau, McGill University) were seeded into 24-well plates at a density of 50000 cells/well and grown for 24 hours in DMEM (Lonza) supplemented with 10% fetal bovine serum (Biowest), penicillin and streptomycin at 50 units/mL (BioWhittaker, Lonza) and hygromycin B selection antibiotic (Invivogen) at 100 µg/mL. Cells were serum-starved overnight in DMEM supplemented with 0.1% bovine serum albumin (Sigma) and stimulated for 4 hours at 37 °C with C16, S1P or C19, ranging from 10 nM to 2 µM final concentration. Other cells were pretreated with the S1P2 antagonist JTE013 at 3 µM and then stimulated with 1 µM of C16, S1P or C19. At the end of the incubation with the compounds, cells were lysed with Reporter Lysis Buffer (Promega), the lysates were collected and centrifuged at 12000 g, 4 °C for 3 min. 10 µL of each supernatant were used for luciferase activity measurement. Luminescence intensity after addition of 40 µL of D-luciferin (Biosynth) reagent solution at 235 µM was measured for 15 sec with a CentroXS3 LB960 Luminometer (Berthold Technologies). Results were normalized on supernatants total protein content and expressed as a percentage of 1 µM S1P response. Dose response-curves were limited to the 10 nM–2 µM range because we observed irregular or non-reproducible data for values above 2 µM. Such erratic behavior is most likely due to toxic effects caused by long exposure (4 h) to high concentration of S1P (and analogs) which have been previously described can be receptor independent48. For this concentration range, according to our calcium mobilization assays, Emax should be reached for C16, barely for S1P and not yet for C19 (Fig. 3).
Statistical analysis
Data were fitted into a sigmoidal concentration-response curve using the GraphPad Prism software (San Diego, California, US). Statistical analysis was performed using one-way ANOVA with Scheffe’s post-test.
Electronic supplementary material
Acknowledgements
This study was supported by grants from the Fonds de la Recherche Scientifique F.R.S.-FNRS (grants F.4523.12, 34553.08), MINECO (SAF2016-77830-R) and Comunidad de Madrid (S2011/BMD2353). Authors thankfully acknowledge the computer resources provided by the Barcelona Supercomputing Center - Centro Nacional de Supercomputación. L.P. participates in the European COST Action CM1207 (GLISTEN). We thank Charazet Debza for technical assistance, Marc Parmentier and Jean-Marie Ruysschaert for helpful discussions, Simon Rousseau and Julie Bérubé for providing us BEAS-2B cells and assistance with the assays and Mercedes Campillo for help with statistical analysis. A.T.T. was a doctoral fellow of the F.R.S.-FNRS. C.G. is a Chercheur Qualifié of the FRS-FNRS and a WELBIO investigator.
Author Contributions
A.T.T. performed all functional experiments and analyzed the experimental data. J.Z. performed and analyzed all molecular dynamics simulations with the help of A.G., I.G.G., S.O.G. and M.L.L.R. performed the organic synthesis of the compounds. L.P. and C.G. provided overall project supervision and wrote the paper together with A.T.T. and J.Z.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Anastassia Troupiotis-Tsaïlaki and Julian Zachmann contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02104-5
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Leonardo Pardo, Email: Leonardo.Pardo@uab.cat.
Cedric Govaerts, Email: Cedric.Govaerts@ulb.ac.be.
References
- 1.Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fredriksson R, Schiöth HB. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharmacol. 2005;67:1414–1425. doi: 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez A, Cordomí A, Caltabiano G, Pardo L. Impact of Helix Irregularities on Sequence Alignment and Homology Modeling of G Protein-Coupled Receptors. ChemBioChem. 2012;13:1393–1399. doi: 10.1002/cbic.201200189. [DOI] [PubMed] [Google Scholar]
- 4.Wu H, et al. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Im D-S. Discovery of new G protein-coupled receptors for lipid mediators. J. Lipid Res. 2004;45:410–418. doi: 10.1194/jlr.R300006-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.O’Sullivan C, Dev KK. The structure and function of the S1P1 receptor. Trends Pharmacol. Sci. 2013;34:401–412. doi: 10.1016/j.tips.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010;688:141–155. doi: 10.1007/978-1-4419-6741-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen H, Gonzalez-Cabrera PJ, Sanna MG, Brown S. Sphingosine 1-phosphate receptor signaling. Annu. Rev. Biochem. 2009;78:743–768. doi: 10.1146/annurev.biochem.78.072407.103733. [DOI] [PubMed] [Google Scholar]
- 9.Mandala S, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 10.Im DS. Intercellular lipid mediators and GPCR drug discovery. Biomol. Ther. 2013;21:411–422. doi: 10.4062/biomolther.2013.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanson Ma, et al. Crystal Structure of a Lipid G Protein-Coupled Receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanley N, Pardo L, Fabritiis GD. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016;6:22639. doi: 10.1038/srep22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parrill AL, et al. Identification of Edg1 receptor residues that recognize sphingosine 1-Phosphate. J. Biol. Chem. 2000;275:39379–39384. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- 15.Pham T-CT, et al. Molecular recognition in the sphingosine 1-phosphate receptor family. J. Mol. Graph. Model. 2008;26:1189–1201. doi: 10.1016/j.jmgm.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujiwara Y, et al. Identification of the hydrophobic ligand binding pocket of the S1P1 receptor. J. Biol. Chem. 2007;282:2374–2385. doi: 10.1074/jbc.M609648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jo E, et al. S1P1-selective in vivo-active agonists from high-throughput screening: Off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem. Biol. 2005;12:703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 18.Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. J. Biol. Chem. 2005;280:9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- 19.Foss FW, et al. Synthesis and biological evaluation of gamma-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg. Med. Chem. 2007;15:663–677. doi: 10.1016/j.bmc.2006.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan S, Wu R, Latek D, Trzaskowski B, Filipek S. Lipid Receptor S1P1 Activation Scheme Concluded from Microsecond All-Atom Molecular Dynamics Simulations. PLoS Comput. Biol. 2013;9:1–8. doi: 10.1371/journal.pcbi.1003261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jongejan A, et al. Linking agonist binding to histamine H1 receptor activation. Nat. Chem. Biol. 2005;1:98–103. doi: 10.1038/nchembio714. [DOI] [PubMed] [Google Scholar]
- 22.Pellissier LP, et al. Conformational toggle switches implicated in basal constitutive and agonist-induced activated states of 5-hydroxytryptamine-4 receptors. Mol. Pharmacol. 2009;75:982–990. doi: 10.1124/mol.108.053686. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen SGF, et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holst B, et al. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J. Biol. Chem. 2010;285:3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ersoy BA, et al. Mechanism of N-terminal modulation of activity at the melanocortin-4 receptor GPCR. Nature Chemical Biology. 2012;8:725–730. doi: 10.1038/nchembio.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi L, et al. Beta2 adrenergic receptor activation: Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J. Biol. Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 27.Kruse AC, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–6. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deupi X, Kobilka BK. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology (Bethesda). 2010;25:293–303. doi: 10.1152/physiol.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nygaard R, et al. The dynamic process of β2-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen SGF, et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ring AM, et al. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–9. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–94. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 33.Warne T, et al. The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choe H-W, et al. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 35.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sansuk K, et al. A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol. Pharmacol. 2011;79:262–269. doi: 10.1124/mol.110.066068. [DOI] [PubMed] [Google Scholar]
- 37.O’Sullivan MJ, Hirota N, Martin JG. Sphingosine 1-Phosphate (S1P) Induced Interleukin-8 (IL-8) Release Is Mediated by S1P Receptor 2 and Nuclear Factor κB in BEAS-2B Cells. PLoS One. 2014;9:e95566. doi: 10.1371/journal.pone.0095566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deupi X, et al. Influence of the g- conformation of Ser and Thr on the structure of transmembrane helices. J. Struct. Biol. 2010;169:116–123. doi: 10.1016/j.jsb.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Kufareva I, Salanga CL, Handel TM. Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol. 2015;93:372–383. doi: 10.1038/icb.2015.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Day PW, et al. A monoclonal antibody for G protein-coupled receptor crystallography. Nat. Methods. 2007;4:927–929. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 41.Martí-Renom MA, et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 42.Trott O, Olson AJ. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rasmussen SGF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 44.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 45.Hess B, Kutzner C, Van Der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 46.Cordomí A, Caltabiano G, Pardo L. Membrane protein simulations using AMBER force field and Berger lipid parameters. J. Chem. Theory Comput. 2012;8:948–958. doi: 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
- 47.Stables J, et al. A bioluminescent assay for agonist activity at potentially any G-protein-coupled receptor. Anal. Biochem. 1997;252:115–126. doi: 10.1006/abio.1997.2308. [DOI] [PubMed] [Google Scholar]
- 48.Hagen N, et al. Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 2009;284:11346–11353. doi: 10.1074/jbc.M807336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durrant JD, Votapka L, Sørensen J, Amaro RE. POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics. J. Chem. Theory Comput. 2014;10:5047–5056. doi: 10.1021/ct500381c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.