

Abstract
RNA surveillance via the nuclear exosome requires cofactors such as the helicase SKIV2L2 to process and degrade certain noncoding RNAs. This research aimed to characterize the phenotype associated with RNAi knockdown of Skiv2l2 in two murine cancer cell lines: Neuro2A and P19. SKIV2L2 depletion in Neuro2A and P19 cells induced changes in gene expression indicative of cell differentiation and reduced cellular proliferation by 30%. Propidium iodide-based cell-cycle analysis of Skiv2l2 knockdown cells revealed defective progression through the G2/M phase and an accumulation of mitotic cells, suggesting SKIV2L2 contributes to mitotic progression. Since SKIV2L2 targets RNAs to the nuclear exosome for processing and degradation, we identified RNA targets elevated in cells depleted of SKIV2L2 that could account for the observed twofold increase in mitotic cells. Skiv2l2 knockdown cells accumulated replication-dependent histone mRNAs, among other RNAs, that could impede mitotic progression and indirectly trigger differentiation.
Keywords: histones, RNA turnover, RNA surveillance, TRAMP complex
INTRODUCTION
During the course of transcription, nuclear RNA surveillance orchestrates the proper processing and turnover of certain RNAs. The nuclear exosome mediates much of this RNA surveillance to prevent aberrant RNAs from either accumulating in the nucleus or being exported to the cytoplasm. Highly conserved, the eukaryotic nuclear exosome consists of a nine-subunit catalytically inactive core, a trimeric cap that binds RNA, and accessory proteins consisting of both endonucleases and exonucleases, such as Dis3 and Rrp6 (Lykke-Andersen et al. 2009).
The nuclear exosome requires a series of cofactors for preparation and delivery of RNA substrates (Houseley et al. 2006), and one such cofactor, the Trf4/Air2/Mtr4 polyadenylation (TRAMP) complex, was first discovered in Saccharomyces cerevisiae (LaCava et al. 2005; Houseley et al. 2006). The TRAMP complex, consisting of the helicase Mtr4p, the RNA binding protein Air1/2p, and the noncanonical poly(A) polymerase Trf4/5p, tags nuclear RNAs to be degraded with a short adenosine tail, guides those RNAs to the exosome, and then activates the exosome's exonucleolytic activity (LaCava et al. 2005; Houseley et al. 2006; Lykke-Andersen et al. 2009). Trf4/5p adds 3–5 adenosines to RNA substrates bound by Air1/2p (Schmidt and Butler 2013). The resulting A tail not only distinguishes exosome substrates from RNAs stabilized by a longer poly(A) tail (Andersen et al. 2008), but also generates the 3′ overhang necessary for the helicase Mtr4p to bind the RNA and unwind any secondary structure that would impede entry into the nuclear exosome (LaCava et al. 2005; Houseley et al. 2006; Andersen et al. 2008; Lykke-Andersen et al. 2009; Jia et al. 2012). The yeast TRAMP complex has been found to be essential in RNA surveillance of certain transcripts, including the turnover of hypomodified tRNAiMET (Wang et al. 2007a,b), 3′-end trimming of the 5.8S rRNA (de la Cruz et al. 1998), processing of the 7S rRNA precursor (Callahan and Butler 2009), degradation of cryptic unstable transcripts (CUTs) arising from heterochromatic regions (Thiebaut et al. 2006), and efficient splicing and intron degradation (Nag and Steitz 2012; Kong et al. 2013).
Recently, the homologs of the yeast TRAMP complex were found to be highly conserved in mammals, with the poly(A) polymerase PAPD5 having 37% amino acid sequence identity to Trf4p in its catalytic domain, ZCCHC7 showing similarity to Air1p, and SKIV2L2 retaining 51% amino acid identity with the helicase Mtr4p (Norbury 2011; Schmidt and Butler 2013). Mtr4p, the yeast homolog of SKIV2L2, has been shown to function as a DExH-box helicase with 3′ → 5′ helicase activity and ATPase activity (Weir et al. 2010; Norbury 2011). In addition to the TRAMP complex, mammalian SKIV2L2 interacts with the zinc knuckle protein ZCCHC8 and the putative RNA-binding protein RBM7 to form the nuclear exosome targeting (NEXT) complex (Lubas et al. 2011; Norbury 2011). Studies in mammals suggest that the TRAMP complex's location and RNA targets are primarily nucleolar, while the NEXT complex is located in the nucleoplasm and target nuclear RNAs (Lubas et al. 2011). Research focused on identifying substrates of the mammalian nuclear exosome has demonstrated that SKIV2L2 is necessary for the maturation of ribosomal RNA precursors (Schilders et al. 2007; Lubas et al. 2011) and facilitates the degradation of PROMPTs (promoter upstream transcripts) in concert with the NEXT complex (Tiedje et al. 2014). Furthermore, PAPD5 has been shown to bind RNA independent of the TRAMP complex (Rammelt et al. 2011) and modify miRNAs in vivo to alter their stability (Boele et al. 2014).
Because the helicase SKIV2L2 plays a central role in both the TRAMP and the NEXT complex, it acts as a key player in the study of mammalian nuclear RNA surveillance. In animals, SKIV2L2 has been implicated in development. Forward genetic screens in zebrafish identified SKIV2L2 as a possible factor in stem cell maintenance, as mutant zebrafish have reduced brain size and lack the ability to regenerate melanocytes or regrow tails (Yang et al. 2007; Hultman and Johnson 2010; Cox et al. 2011). In teleost fishes, Skiv2l2 expression colocalizes with the cell proliferation marker PCNA (Yang et al. 2007), indicating that Skiv2l2 expression is enhanced in actively dividing populations of stem cells. Taken together, the phenotypes associated with loss of SKIV2L2 in fish indicate that SKIV2L2 may help maintain stem cell populations. In mammals, preliminary screens suggest siRNA knockdown of Skiv2l2 results in impaired cell division and death of embryonic stem cells (Fazzio et al. 2008; Neumann et al. 2010). Additionally, Skiv2l2 is down-regulated during differentiation, with Skiv2l2 falling within the top 2% of genes whose expression is positively correlated with pluripotent embryonic stem cells and negatively correlated with differentiated cells (Cinghu et al. 2014). This trend has been confirmed in the testes where SKIV2L2 levels decline in response to testosterone during spermatogenesis (Osman et al. 2011). Because Skiv2l2 expression and function appear to be important in development, we focused our studies on how SKIV2L2, and therefore RNA surveillance, might affect pluripotency and proliferation in mammalian cancer cell lines.
By using Neuro2A (N2A, ATCC #CCL-131) cells derived from a murine neuroblastoma (Nilbratt et al. 2009; Tremblay et al. 2010; Zhi et al. 2012) and P19 cells (ATCC #CRL-1825) derived from a murine embryonic teratocarcinoma (Osman et al. 2011), we investigated the role of Skiv2l2 (Gene ID: 72198) in differentiation and proliferation. These mammalian cancer cell lines provide a system to investigate cell proliferation and model differentiation, as chemicals induce the differentiation of N2A cells into neurons (Nilbratt et al. 2009; Tremblay et al. 2010) and P19 cells into either neurons or myocytes (Jasmin et al. 2010; Dong et al. 2012). This research sought to characterize the phenotype associated with SKIV2L2 depletion via RNAi and to identify RNA substrates of SKIV2L2, particularly those that could affect proliferation or differentiation. Skiv2l2 knockdown cells exhibited changes in cell morphology and gene expression associated with cellular differentiation, including an increase in cell-fate-specific gene expression and a decrease in the expression of pluripotency genes. Skiv2l2 knockdown via RNAi also reduced cellular proliferation, particularly attributed to a slowed progression through the G2/M phase.
While previous research has uncovered a plethora of noncoding RNAs targeted for processing and degradation by the TRAMP and NEXT complexes, we sought to expand our studies beyond noncoding RNAs. With current known targets consisting of miRNA 5′ leader sequences (Dorweiler et al. 2014), snoRNAs (Lubas et al. 2015), rRNAs (Berndt et al. 2012), PROMPTs (Tiedje et al. 2014), and lncRNAs (Beaulieu et al. 2012), our research investigated the possibility of protein-coding mRNA turnover via the TRAMP and NEXT complex. In accomplishment of this, RNA-seq identified SKIV2L2-target RNAs that could be responsible for perturbing mitotic progression. Most notably, replication-dependent histone mRNAs accumulated following SKIV2L2 depletion. By demonstrating that SKIV2L2 directly binds histone mRNAs in vivo and that SKIV2L2 depletion doubles histone mRNA half-life, these results establish SKIV2L2-mediated RNA surveillance as essential for replication-dependent histone mRNA turnover.
RESULTS
RNAi-mediated knockdown of Skiv2l2 increases cell differentiation
To discern the phenotype associated with SKIV2L2 depletion in Mus musculus cell lines, RNAi was utilized to reduce Skiv2l2 expression in both N2A and P19 cells. Three siRNAs were initially tested (siRNA ID #177474, 177475, 177476) for Skiv2l2 knockdown efficiency, and siRNA ID #177475 was selected for subsequent studies based on knockdown efficiency at low concentrations and consistency (data not shown, Dorweiler et al. 2014). To address the possibility of off-target effects, experiments were replicated with siRNA ID #177476 (Supplemental Fig. S1). Following transfection with siRNA ID #177475 (Skiv2l2 siRNA), Western blotting and qRT-PCR confirmed knockdown of Skiv2l2 levels. When transfected with Skiv2l2 siRNA, N2A cells expressed on average 6.3-fold less SKIV2L2 protein than N2A cells transfected with nontargeting control siRNA (n = 3, P-value <0.0004, Fig. 1A), and Skiv2l2 mRNA levels decreased by 7.9-fold (n = 3, P-value <0.008) as measured via quantitative RT-PCR (qRT-PCR) (Fig. 1B). Initial observations revealed that following treatment with siRNAs directed against Skiv2l2, N2A cells failed to grow to 100% confluency, unlike N2A cells treated with the nontargeting control siRNA (data not shown). Further inspection revealed increased extension of axons and dendrites following SKIV2L2 depletion (Fig. 1C), hinting that SKIV2L2 may play a role in maintaining cancer cell proliferation or preventing differentiation. These observations led to experiments to determine whether the phenotype observed with Skiv2l2 knockdown might be due to increased N2A cell differentiation or reduced proliferation.
FIGURE 1.

Knockdown of Skiv2l2 in N2A cells enhances differentiation into neurons. (A) Western blots to detect SKIV2L2 protein in whole-cell extract from cells transfected with nontargeting control siRNA or Skiv2l2 siRNA. N2A cells transfected with control siRNA were grown in either 10% fetal bovine serum or 2% fetal bovine serum with 20 µM ATRA to induce differentiation. SKIV2L2 protein levels (118 kD) were normalized to β-actin protein levels (42 kD). (B) qRT-PCR of Skiv2l2 mRNA from N2A cells treated with control siRNA, ATRA, or Skiv2l2 siRNA. Skiv2l2 mRNA levels measured via ΔCq values were normalized to β-actin mRNA. Error bars represent ±SD for n = 3. (C) Neurite outgrowth imaging. Scale is 250 µM. Neuronal processes (marked by arrowheads) were observed among N2A cells and counted. (D) qRT-PCR of neuronal differentiation markers. Tubb3 and ChAT mRNA levels were calculated from ΔCq values and normalized to β-actin mRNA levels in control and Skiv2l2 siRNA-treated N2A cells (error bars represent ±SD, n = 3). (E) qRT-PCR of pluripotency markers Nestin and Sox2. Nestin and Sox2 mRNA levels were calculated as stated above (error bars represent ±SD for n = 3).
To examine the correlation between SKIV2L2 levels and differentiation, N2A cells were chemically induced to differentiate. Serum starvation and treatment with 20 µM all-trans retinoic acid (ATRA) initiate the differentiation of N2A cells into cholinergic neurons (Nilbratt et al. 2009), visualized as the extension of neuronal processes. This differentiation can also be quantified through the expression of genes indicative of cholinergic neurons, such as acetylcholine transferase (ChAT), which is necessary for the biogenesis of acetylcholine (Sidiropoulou et al. 2008), and the axonal-specific β-tubulin-III (Tubb3) (Supplemental Fig. S2A; Zhi et al. 2012). Western blotting revealed that chemically differentiated N2A cells express ∼50% less SKIV2L2 protein when compared to N2A cells grown in standard growth media containing 10% fetal bovine serum (P-value <0.0001, Fig. 1A). As measured via qRT-PCR, Skiv2l2 mRNA levels were reduced by 4.3-fold in chemically differentiated N2A cells (P-value <0.003, Fig. 1B). Further studies have demonstrated that this reduction is not due to post-transcriptional control via the 3′ UTR of Skiv2l2 (Supplemental Fig. S3), suggesting that this down-regulation might be due to developmental changes in transcription.
Upon observation that differentiation in N2A cells down-regulates Skiv2l2, we hypothesized that SKIV2L2 depletion enhances differentiation. Both the quantification of N2A cells extending neuronal processes (axons and dendrites) and the measurement of differentiation marker expression were utilized to determine whether Skiv2l2 knockdown enhances the differentiation of N2A cells into neurons. N2A cells extending processes were counted in both control siRNA- and Skiv2l2 siRNA-transfected cells (Fig. 1C). Skiv2l2 knockdown resulted in a 20% increase in cells extending long neuronal processes over control cells (Supplemental Fig. S4, P-value <0.05).
As N2A cells differentiate, genes specific to neurons, such as ChAT and Tubb3, are transcribed, while pluripotency genes such as Sox2 and Nestin are silenced (Zhi et al. 2012; Supplemental Fig. S2A). Measured via qRT-PCR, Tubb3 expression increased 2.0-fold (P-value <0.048) and ChAT expression increased 3.2-fold following SKIV2L2 depletion (P-value <0.0001) (Fig. 1D). This increase in Tubb3 and ChAT expression, representative of axon structure and function, is consistent with the enhanced neuronal cell morphology in Skiv2l2 knockdown cells. Furthermore, the expression of pluripotency markers Nestin and Sox2 was found to exhibit a 2.97-fold decrease (P-value <0.03) and a 2.98-fold decrease (P-value <0.01), respectively, in Skiv2l2 knockdown cells when compared to control siRNA-treated cells (Fig. 1E). The increase in neuronal cell morphology, elevated Tubb3 and ChAT expression, and loss of pluripotency markers suggest that decreased levels of SKIV2L2 result in loss of multipotency and enhanced differentiation in N2A cells. Depletion of TRAMP component Papd5 via siRNA (siRNA ID #s103146) also increased extension of neuronal processes and higher Tubb3 and ChAT mRNA levels (Supplemental Fig. S5), suggesting that the observed phenotype relates to loss of RNA surveillance as opposed to off-target effects.
We next explored the possibility that Skiv2l2 knockdown triggers cell differentiation in multiple cell types by repeating RNAi knockdown of Skiv2l2 in the mouse embryonic teratocarcinoma line, P19. Skiv2l2 knockdown in P19 cells resulted in a 70% reduction in SKIV2L2 protein levels compared to control siRNA-treated cells (P-value <0.0001, Fig. 2A) and an eightfold decrease in Skiv2l2 mRNA levels (P-value <0.01, n = 4). Differentiation of P19 cells into cardiac myocytes with DMSO resulted in a significant 86% decrease in SKIV2L2 protein levels (P-value <0.04, Fig. 2A) and a threefold decrease in Skiv2l2 mRNA levels (P-value <0.03, Fig. 2B). This demonstrates that differentiation into multiple cell types, specifically cholinergic neurons and cardiac myocytes, correlates with a down-regulation of SKIV2L2. Due to the ability of P19 cells to form cardiac muscle, skeletal muscle, and various neurons, the phenotype of P19 cells following Skiv2l2 knockdown was examined through qRT-PCR of cardiac myocyte markers and pluripotency markers, as cell morphology was not a reliable measurement of differentiation. Gene transcripts selected to screen for differentiation included the cardiac muscle transcription factors GATA4 and Nkx2.5 (Choi et al. 2004), fatty-acid binding protein 3 (Fabp3) specific to cardiac muscle (Zhu et al. 2011), and the intermediate filament Desmin expressed in all muscle cell types (Supplemental Fig. S2B; Choi et al. 2004; Dey et al. 2010). High levels of the transcription factors Nanog (Zhu et al. 2011) and Sox2 (Dong et al. 2012) characterize P19 cell pluripotency (Supplemental Fig. S2B). Following Skiv2l2 knockdown, both GATA4 and Nkx2.5 mRNA levels increased 3.9-fold above those found in P19 cells treated with control siRNA (Fig. 2C, P-values <0.0008, 0.008). Fabp3 expression was elevated 1.8-fold (P-value <0.02), and Desmin expression increased 2.6-fold (Fig. 2D, P-value <0.04) in Skiv2l2 knockdown cells. Overall, the expression pattern following SKIV2L2 depletion in P19 cells indicates enhanced differentiation into cardiac myocytes. Down-regulation of the pluripotency transcription factors Sox2 and Nanog by 50-fold (P-value <0.0001) and 2.3-fold (P-value <0.04), respectively, denote a shift in P19 cells from a pluripotent to a more differentiated state following Skiv2l2 knockdown (Fig. 2E; Dey et al. 2010; Zhu et al. 2011; Dong et al. 2012). Therefore, RNAi against Skiv2l2 promotes differentiation into both neurons in N2A cells and cardiac myocytes in P19 cells.
FIGURE 2.

Knockdown of Skiv2l2 in P19 cells enhances differentiation into cardiomyocytes. (A) Western blot to detect SKIV2L2 protein in P19 whole-cell extract from cells transfected with control siRNA, Skiv2l2 siRNA, or grown in 0.1% DMSO. SKIV2L2 protein levels (118 kD) were normalized to β-actin (42 kD). (B) qRT-PCR of Skiv2l2 mRNA in P19 cells. Skiv2l2 mRNA levels in control siRNA-treated cells, 0.1% DMSO-treated cells, and Skiv2l2 siRNA-treated cells were measured via ΔCq values and normalized to β-actin (error bars represent ±SD for n = 4). (C) qRT-PCR of myocyte differentiation transcription factors in P19 cells. GATA4 and Nkx2.5 mRNA levels calculated as in B (error bars represent ±SD for n = 3). (D) qRT-PCR of myocyte differentiation markers in P19 cells. Fabp3 and Desmin mRNA levels calculated as in B (error bars represent ±SD for n = 3). (E) qRT-PCR of pluripotency markers Sox2 and Nanog in P19 cells. Sox2 and Nanog mRNA levels calculated as in B (error bars represent ±SD for n = 3).
SKIV2L2 depletion impairs cellular proliferation
Since SKIV2L2 depletion enhanced differentiation into two cell types in two different murine cell lines, we hypothesized that SKIV2L2 might affect cellular proliferation, consequently influencing the differentiation phenotype observed. Differentiation and proliferation are often viewed as antagonistic processes because cell-cycle arrest in the G1 phase usually precedes cell differentiation (Liu et al. 2013). The majority of terminally differentiated cells lose the ability to divide, with new cells coming only from a stem cell pool. Additionally, many cancer cell lines, including N2A cells, differentiate following serum starvation, indicating that the growth factors that keep cancer cell lines proliferating are necessary to prevent their differentiation (Phillips et al. 2006). Since slowed cellular proliferation due to cell-cycle arrest could result in the increased differentiation seen with Skiv2l2 knockdown, experiments were utilized to determine whether reduced SKIV2L2 levels impair progression through the cell cycle.
MTT assays were used to quantify the proliferation of both N2A and P19 cells transfected with either control or Skiv2l2 siRNA. MTT assays estimate the relative number of proliferating cells, because only proliferating cells are capable of reducing MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to purple formazan crystal (Freimoser et al. 1999; Xie et al. 2010; Gui et al. 2014). Lower levels of MTT reduction correspond to a decrease in the number of proliferating cells, which could be due to cell death, cell differentiation, or slower progression through the cell cycle (Freimoser et al. 1999). In both N2A cells and P19 cells, SKIV2L2 depletion resulted in decreased formazan production. Skiv2l2 knockdown in N2A cells resulted in a 38% (P-value <0.0001) overall decrease in formazan production (Supplemental Fig. S6). Time course MTT assays revealed that the proliferation of both ATRA-treated cells and Skiv2l2 knockdown cells slowed similarly over time (Fig. 3A), with Skiv2l2 siRNA resulting in a 22% reduction in proliferation at 28 h (P-value <0.0007) and a 27% reduction at 32 h when compared to control siRNA-treated cells (P-value <0.002). In P19 cells, Skiv2l2 knockdown cells showed a similar 30% decrease in formazan production when compared to cells transfected with control siRNA (Fig. 3B, P-value <0.04). This result demonstrates a 22%–38% reduction in the number of proliferating cells following SKIV2L2 depletion.
FIGURE 3.
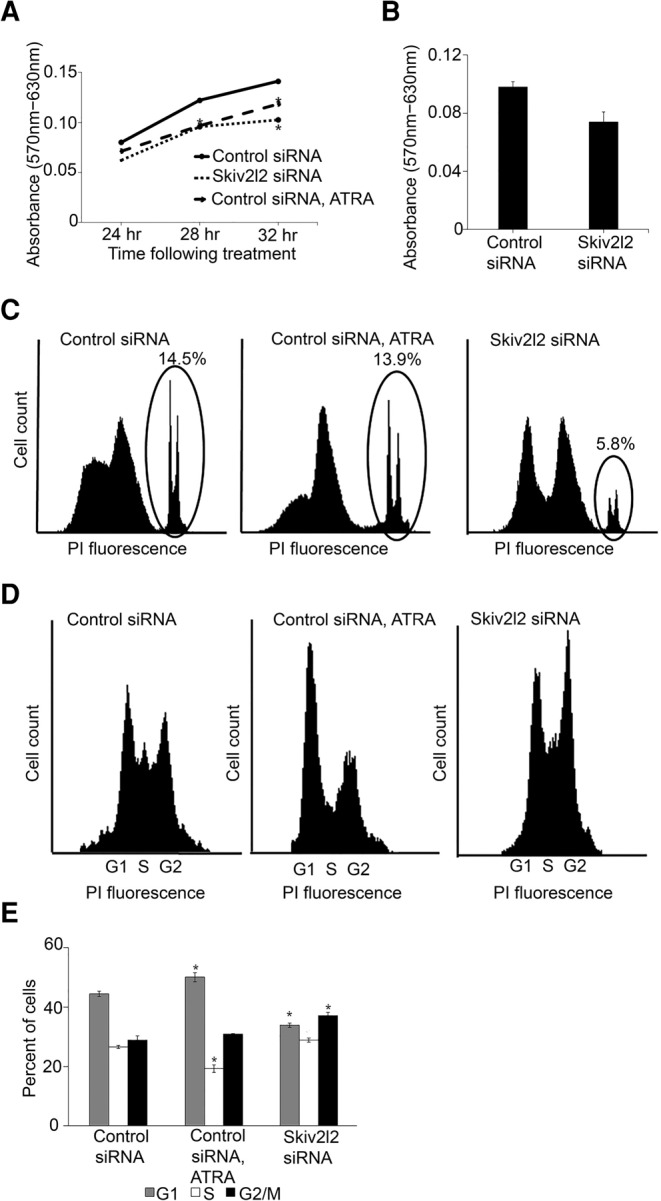
Skiv2l2 knockdown results in delayed proliferation attributed to slowed G2/M phase progression. (A) MTT time point assay in N2A cells. Absorbance of formazan (570 nm) at 24, 28, and 32 h following treatment with control siRNA, ATRA, or Skiv2l2 siRNA (n = 4 for each time point, [*] represents P-value <0.05). (B) MTT assay in P19 cells. Absorbance of formazan was measured after 48 h of treatment with control siRNA, ATRA, or Skiv2l2 siRNA (error bars represent ±SD for n = 9). (C) Propidium iodide viability staining of N2A cells. Fluorescing cells (dead or necrotic) detected by the FL-2 filter are circled, and the Accuri C6 software was used to quantify the percentage of control, ATRA treated, and Skiv2l2 knockdown cells staining with propidium iodide. (D) Cell-cycle analysis with propidium iodide staining of fixed N2A cells. Cells treated with control siRNA, Skiv2l2 siRNA, or ATRA were excited at 488 nm and sorted using FL-2 on the Accuri C6 based on propidium iodide fluorescence. The sorted cells corresponded to G1, S, and G2/M phase as denoted. (E) Quantification of cell-cycle profile. Based on the cell-cycle landscapes generated in D, the Accuri C6 software was used to calculate the percentage of cells in G1, S, and G2/M phase, where n = 3 sets of 50,000 cells and statistically significant differences (P-value <0.05) are denoted with an asterisk (*).
Because the MTT assay measures overall proliferation, it was possible that reduced proliferation of Skiv2l2 knockdown cells could be due to cell death. Therefore, a two-pronged approach was utilized to measure cell proliferation: MTT assays to directly measure the relative amount of proliferative cells and propidium iodide staining to measure cell death. N2A cells were stained with propidium iodide, which passes through the compromised membranes of dead cells and binds to DNA (Lima et al. 2008). Quantifying the percentage of dead and necrotic cells staining with propidium iodide through FACs analysis revealed that transfection of N2A cells with control siRNA and differentiation with ATRA resulted in a 14% cell death rate (P-value <0.72, n = 3). In contrast, Skiv2l2 knockdown did not result in significant cell death, as only 5% of Skiv2l2 knockdown cells stained with propidium iodide (P-value <0.0001, n = 3) (Fig. 3C). There was no statistically significant change in the profile of cells staining with low levels of propidium iodide, possibly representing apoptotic cells (P-values all >0.05). In addition, granularity was measured via side-scattering versus forward-scattering. Cells demonstrating increased side-scattering with low forward-scattering are associated with apoptosis, and the percentage of cells meeting this criterion were quantified for each sample (Darzynkiewicz et al. 1992). This calculation showed that while ATRA treatment increased the number of cells with high side-scattering (P-value <0.0096), there was no difference between the control siRNA and Skiv2l2 siRNA-treated samples (P-value <0.2). This denotes that increased cell death could not account for the reduced levels of proliferation seen in the MTT assay following SKIV2L2 depletion.
Since Skiv2l2 knockdown results in increased differentiation, we hypothesized that loss of SKIV2L2 might cause G1 arrest followed by differentiation. The cell cycle was examined through fluorescent-activated cell sorting of fixed N2A cells stained with propidium iodide. Cells were sorted based on the intensity of propidium iodide fluorescence, which correlates to DNA content (Fig. 3D,E; Krishan 1975; Zhi et al. 2012; Wesley et al. 2015). For N2A cells transfected with the control siRNA, 44.5% of cells were found to be in G1 phase, as they possess half the DNA content of cells in G2 phase and therefore half the intensity of propidium iodide fluorescence. Smaller proportions of cells were observed in S phase (26.6%) and G2/M phase (28.8%), both denoted by increased DNA content and propidium iodide fluorescence. As expected, differentiation of N2A cells with ATRA induced a small but significant 11% increase in the proportion of cells in G1 phase compared to control cells (P-value <0.0025). Surprisingly, SKIV2L2 depletion did not cause G1 arrest like chemically induced differentiation. Instead, 37.2% of Skiv2l2 knockdown cells exhibited double the DNA content, which is indicative of G2/M phase arrest. This modest but statistically significant 23% increase (P-value <0.000001) in G2/M phase cells implies that loss of SKIV2L2 hinders the progression of cells through G2/M phase, which is distinct from G1 phase arrest seen in chemically differentiating N2A cells. Overall, both the reduced proliferation and G2/M phase arrest observed following SKIV2L2 depletion suggest SKIV2L2 helps maintain cell proliferation.
Skiv2l2 knockdown impairs mitotic progression
Following observance of delayed G2/M phase progression after Skiv2l2 knockdown, further experiments aimed to determine whether SKIV2L2 depletion impaired the G2 or mitotic phases of the cell cycle. Phosphorylation of serine 10 on histone H3 is a well-accepted marker of mitosis, as H3 phospho-S10 increases with chromosome condensation during mitosis (Crosio et al. 2002). To examine whether SKIV2L2 depletion impedes mitotic progression, Western blots were performed to detect phosphorylated H3 (H3 phospho-S10) in control and knockdown N2A cells. Whole-cell extracts were fractionated into cytoplasmic and nuclear fractions. Histone proteins were acid-extracted from the nuclear fraction for analysis, while the β-actin protein was detected in the cytoplasmic fraction. Skiv2l2 knockdown N2A cells exhibited an average 2.5-fold increase in overall H3 phospho-S10 levels compared to control cells (Fig. 4A, n = 8, P-value <0.0071). The experiment was replicated in P19 cells, where Skiv2l2 knockdown resulted in a fourfold increase in H3 phospho-S10 over control levels (Fig. 4B, n = 8, P-value <0.04).
FIGURE 4.
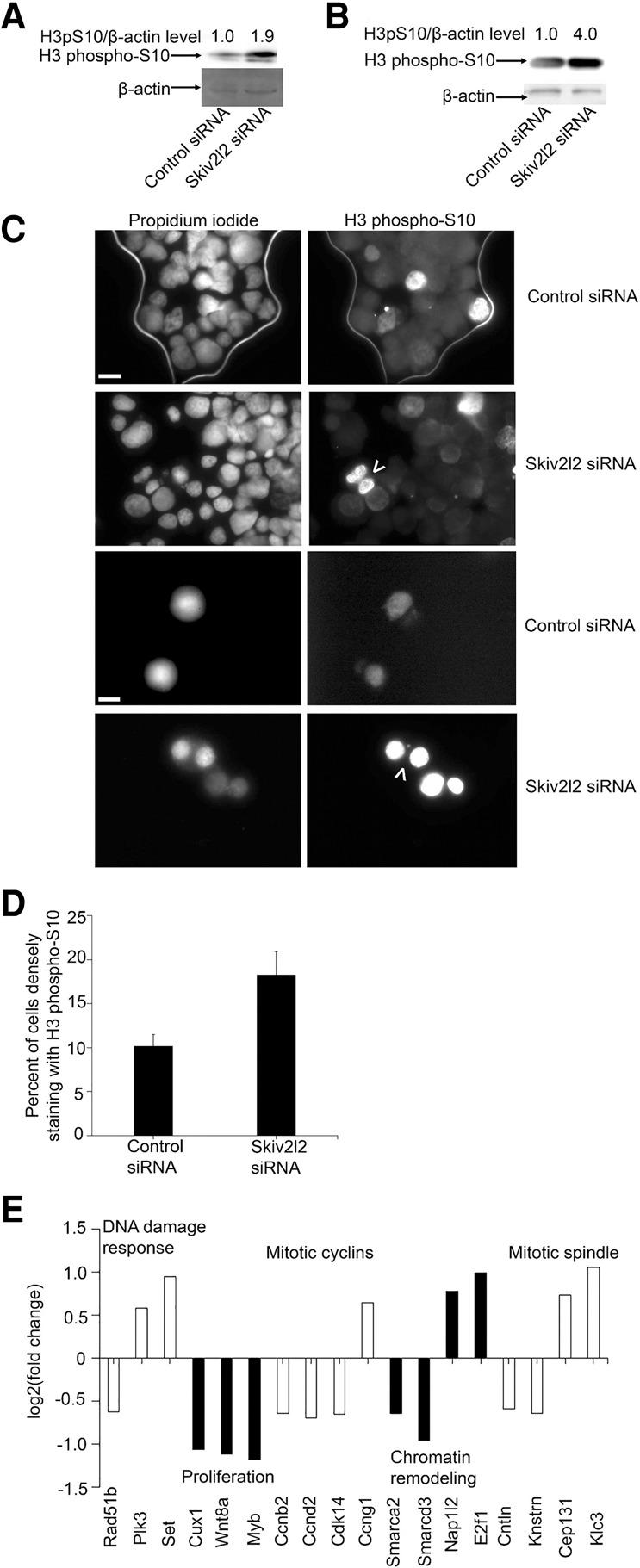
Skiv2l2 knockdown leads to delayed progression through mitosis. (A) Western blot to detect histone H3 phosphorylated on serine 10 (H3 phospho-S10) in N2A cells. H3 phospho-S10 protein (17 kD) levels from nuclear fractions following acid extraction were normalized to β-actin detected in the cytoplasmic fraction from whole-cell extracts from control and Skiv2l2 knockdown cells. (B) Western blot to detect H3 phospho-S10 normalized to β-actin protein levels in P19 cells. Performed as stated previously on acid-extracted nuclear and cytoplasmic fractions from control or Skiv2l2 siRNA-treated P19 cells. (C) Indirect immunofluorescent staining of H3 phospho-S10 in N2A cells. Propidium iodide and H3 phospho-S10 antibody, followed by detection with anti-rabbit AlexaFluor488, stained mitotic cells, 15 µM scale bar for top six panels, 20 µM for bottom two panels. (D) Quantification of H3 phospho-S10 staining nuclei. Nuclei densely staining with H3 phospho-S10 antibody were divided by the number of nuclei staining with propidium iodide to obtain the percentage of nuclei undergoing mitosis (n = 4, P-value <0.02). (E) Graph of cell-cycle control genes with differential expression in Skiv2l2 knockdown cells detected via RNA-seq in P19 cells. Up-regulated and down-regulated genes, shown respectively as above and below zero, in Skiv2l2 knockdown cells found to be involved in cell-cycle control. Statistically significant changes in RNA levels have a q-value of <0.05.
However, this increase in H3 phospho-S10 could correlate with increased overall histone levels in G2/M phase. Therefore, indirect immunofluorescence microscopy was used to count the number of cells positive for the H3 phospho-S10 marker (Fig. 4C). This would confirm that the overall increase in H3 phospho-S10 correlates to an increase in cells in mitosis. Using this method, 10.7% (±4.3% SD) of cell nuclei staining with propidium iodide also expressed H3 phospho-S10 in control siRNA-treated N2a cells. Comparatively, treatment with Skiv2l2 siRNA resulted in 20.1% (±4.3% SD) of cell nuclei staining with the mitotic marker H3 phospho-S10 antibody (Fig. 4C). This represents nearly a twofold increase in cells expressing H3 phospho-S10 following Skiv2l2 knockdown (n = 4, P-value <0.02), which is similar to the average 2.5-fold increase in overall H3 phospho-S10 levels measured via Western blotting. FACs analysis based on the detection of H3 phospho-S10 replicated this observed doubling in mitotic cells with SKIV2L2 depletion (Supplemental Fig. S7). Interestingly, binucleated cells were observed in Skiv2l2 knockdown cells at a 5% incidence rate (Fig. 4B), denoted by arrows, while no binucleated cells were observed in control cells (0% incidence rate). These observed binucleated cells depict a delay or failure in cytokinesis following SKIV2L2 depletion. Together, reduced SKIV2L2 levels increased the number of mitotic cells, suggesting impaired mitotic progression, and resulted in binucleation in a subset of cells expressing the mitotic marker H3 phospho-S10.
We reasoned that the inhibition of RNA surveillance via Skiv2l2 knockdown might result in the accumulation of RNAs that could account for the mitotic arrest observed. Therefore, RNA-seq data obtained from P19 cells treated with control or Skiv2l2 siRNA were interrogated to identify changes in the transcriptome that might be indicative of proliferation defects. GO-ontology analysis indicated SKIV2L2 depletion correlates with misregulation of genes involved with the DNA damage response, signaling-induced cell proliferation, mitotic cyclins, chromatin remodeling, and mitotic spindle assembly. Figure 4E depicts a subset of these misregulated genes. Of particular interest, cells depleted of SKIV2L2 showed higher expression of cyclin-G1 (Kimura et al. 2001), which accumulates in G2/M phase arrested cells, and lower expression of both the G1–S phase transition cyclin-D1 (Matsuura et al. 2004) and the mitotic progression cyclin-B2 (Wu et al. 2010). This cyclin expression pattern is indicative of cells arrested in G2/M phase. Additionally, GO-ontology analysis of misregulated genes in Skiv2l2 knockdown cells revealed certain genes that positively influence cell proliferation were down-regulated (P-value <0.03), while specific anti-apoptotic genes were up-regulated (P-value <0.001). Together, this affirms that the observed defects in cell proliferation correlates with changes in gene expression.
Various classes of RNAs accumulate in Skiv2l2 knockdown cells
Those RNAs that accumulate in cells depleted of SKIV2L2 potentially represent two classes of RNAs: direct RNA targets of SKIV2L2-mediated processing or turnover and RNAs whose levels change indirectly due to loss of nuclear RNA surveillance. The RNA-seq study in P19 cells sought to identify RNAs that change abundance following SKIV2L2 depletion, and effort focused on discovering potential direct targets of SKIV2L2 that might perturb mitosis upon their accumulation. RNA-seq was performed on total RNA isolated from control siRNA and Skiv2l2 siRNA-treated P19 cells. RNA was treated with DNase and RiboZero to eliminate DNA and rRNA prior to sequencing via Illumina Hi-Seq 2000 paired end sequencing. Sequenced reads were mapped to the mm10 genome build via TopHat, and differential changes in RNA levels were detected and calculated via cufflinks and cuffdif. In this manner, 1473 RefSeq genes were identified as dysregulated in Skiv2l2 knockdown cells (Supplemental Tables S1, S2). Of these, 585 genes were elevated following Skiv2l2 knockdown, while 887 genes were reduced, with <5% potentially being false discoveries (q-value <0.05). Elevated genes (337) and reduced genes (428) showing greater than a 1.5-fold change were probed in further detail (Supplemental Table S6). Of these, genes elevated in the Skiv2l2 knockdown samples included 36 replication-dependent histone mRNA genes, 47 noncoding RNAs, and 254 other mRNAs (Fig. 5A). Genes showing decreased steady-state levels in cells depleted of SKIV2L2 included 416 mRNAs, 10 noncoding RNAs, and only two noncanonical histone mRNA transcripts (Fig. 5B). Additionally, 772 intergenic regions of the mouse genome were significantly elevated, while only 99 intergenic regions were down-regulated, suggesting that SKIV2L2 depletion causes a substantial increase in intergenic transcripts (Supplemental Tables S1, S3). A fraction of these intergenic regions contain retrogenes and pseudogenes annotated on the UCSC Genome Browser, while some of these intergenic regions fall upstream of promoters, possibly representing PROMPTs (promoter upstream transcripts), a known target of nuclear RNA surveillance (Tiedje et al. 2014; Lubas et al. 2015). Since noncoding RNAs represent the majority of RNAs previously discovered as targets of the TRAMP complex, these noncoding RNAs were closely inspected. Long noncoding RNAs (lncRNAs) represented the majority of accumulating noncoding RNAs. Other up-regulated noncoding RNAs included both new and previously reported RNA surveillance targets: miRNAs, snoRNAs, and small nuclear RNAs such as MRP RNA and ribonuclease P RNA (Supplemental Fig. S8; Schilders et al. 2007; Dorweiler et al. 2014; Tiedje et al. 2014; Lubas et al. 2015).
FIGURE 5.

Replication-dependent histone mRNAs accumulate in Skiv2l2 knockdown cells. (A) Transcripts up-regulated in P19 cells following Skiv2l2 knockdown as measured by RNA-seq. Transcripts up-regulated in Skiv2l2 knockdown cells were categorized based on coding and noncoding annotations (q-value <0.05, >1.5-fold change). (B) Transcripts down-regulated in P19 cells following Skiv2l2 knockdown as measured by RNA-seq. Transcripts down-regulated in Skiv2l2 knockdown cells categorized based on coding and noncoding annotations (q-value <0.05, >1.5-fold change). (C) qRT-PCR of histone mRNAs in N2A cells. Histone mRNA levels were calculated using ΔCq values and normalized to β-actin mRNA (error bars represent ±SD for n = 3). (D) qRT-PCR of histone mRNAs in P19 cells. Histone mRNA levels calculated as in C (error bars represent ±SD for n = 3).
Interestingly, certain trends emerged in the RNA-seq data. While previous research focused on noncoding RNA targets of nuclear RNA surveillance, at least three types of coding RNAs noticeably accumulate in Skiv2l2 knockdown cells. First, imprinted genes were overrepresented among genes dysregulated following SKIV2L2 depletion. Out of the 129 imprinted genes in the mouse genome, 16 were found to have changed expression in Skiv2l2 knockdown cells, which is a statistically significant overrepresentation of imprinted genes among those dysregulated (P-value <2.52 × 10−6, Supplemental Table S4). In addition to imprinted genes, a statistically significant change in ribosomal protein mRNAs emerged in Skiv2l2 knockdown cells. Approximately one-third of all ribosomal protein mRNAs accumulated with SKIV2L2 depletion, with no ribosomal protein mRNAs being down-regulated, which is a statistically significant link between SKIV2L2 and ribosomal protein mRNA regulation (26 out of 39, P-value <4.59 × 10−20). Finally, histone mRNAs were found to be significantly overrepresented in the data (P-value <1.85 × 10−57). Forty-eight out of 60 histone genes (80%) were dysregulated in cells depleted of SKIV2L2. Replication-dependent histone mRNAs consistently accumulated in Skiv2l2 knockdown cells, while two noncanonical histone mRNA transcripts, H2A.x and H2A.z, were down-regulated (Supplemental Table S5). Analysis of the RNA-seq reads suggests that these accumulating RNAs are not 3′ extended or misprocessed mRNAs, with reads from control and Skiv2l2 siRNA cells mapping to identical positions (Supplemental Fig. S9). These findings confirm the involvement of SKIV2L2-dependent RNA surveillance in the turnover of noncoding RNAs (Schilders et al. 2007; Dorweiler et al. 2014; Tiedje et al. 2014; Lubas et al. 2015), and, more importantly, they suggest that SKIV2L2-dependent RNA surveillance helps facilitate the degradation of certain coding RNAs, specifically replication-dependent histone mRNAs.
SKIV2L2 depletion impairs replication-dependent histone mRNA turnover
The sequencing data from P19 cells insinuated that SKIV2L2 is necessary for efficient turnover of replication-dependent histone mRNAs. To our knowledge, nuclear turnover of histone mRNAs has not been reported in mammalian cells. Studies have decisively shown that cytoplasmic histone mRNA turnover occurs following histone mRNA uridylation, but these reports conflict on the involvement of PAPD5 in this process (Kaygun and Marzluff 2005a,b; Marzluff et al. 2008; Mullen and Marzulff 2008; Schmidt et al. 2010). However, the fact that Trf4p, the yeast homolog of PAPD5, and the TRAMP complex have been implicated in histone mRNA turnover in yeast (Reis and Campbell 2006; Canavan and Bond 2007) prompted further investigation into the possibility that SKIV2L2 mediates the degradation of histone mRNAs in Mus musculus. Moreover, accumulation of histone mRNAs and proteins have been shown to induce mitotic arrest in eukaryotes (Singh et al. 2010), making histone mRNAs a potential candidate responsible for the observed delayed mitotic progression in Skiv2l2 knockdown cells.
Quantitative RT-PCR verified that the replication-dependent histone transcripts hist1h1b (H1), hist1h2an (H2A), hist1h2bc (H2B), hist1h3a (H3), and hist1h4d (H4) were significantly elevated in both N2a cells and P19 cells following Skiv2l2 knockdown (Fig. 5C,D, P-values <0.05). Attempts to detect histones following reverse transcription with oligo(dT) were unsuccessful, suggesting that the accumulating histones lack long poly(A) tails. However, this does not eliminate the possibility that these histone mRNAs are uridylated or possess short A tails.
In order to support the assertion that replication-dependent histone mRNA turnover requires SKIV2L2, experiments sought to demonstrate that SKIV2L2 binds histone mRNAs in vivo, thereby establishing a direct, physical link between SKIV2L2 and histone mRNA. In addition, evidence that loss of SKIV2L2 leads to a reduction in histone mRNA turnover and an increase in histone mRNA half-life would further support the notion of SKIV2L2-mediated turnover of replication-dependent histone mRNAs. To demonstrate that histone mRNAs directly associate with SKIV2L2, cross-linked immunoprecipitation was performed to detect histone mRNAs bound to SKIV2L2. Both N2A and P19 cells were irradiated with UV light to crosslink RNA to protein, and proteins were bound to beads coated with antibodies directed against either SKIV2L2 or a negative control goat IgG. Western blots demonstrated that anti-SKIV2L2 successfully pulled down ∼10% of cellular SKIV2L2, while no SKIV2L2 was detected in the eluate from control anti-goat IgG (Fig. 6A,B). RNAs immunoprecipitated with anti-SKIV2L2 were compared to those RNAs immunoprecipitated with the control antibody using quantitative RT-PCR. Histone mRNA levels in immunoprecipitates were normalized against Skiv2l2 mRNA, which showed no binding to SKIV2L2. Compared to RNA immunoprecipitated with anti-goat IgG, replication-dependent histone mRNAs (H1, H2A, H2B, H3, and H4) were elevated among RNAs immunoprecipitated with anti-SKIV2L2, as measured via qRT-PCR (Fig. 6C,D, all P-values <0.05). In N2A cells, H1, H2A, and H4 were enriched among those RNAs pulled down with anti-SKIV2L2 by more than fivefold, while H2B and H3 was found at levels fourfold and twofold above the control, respectively (Fig. 6C). Among those RNAs that immunoprecipitated with SKIV2L2, retro-Phgdh mRNA levels were not above those seen in the control immunoprecipitate, suggesting that the protocol sufficiently distinguished between RNAs bound to SKIV2L2 and unbound RNA. P19 cells proved to yield a more dramatic result, with H1, H2A, and H3 being pulled down with anti-SKIV2L2 at levels 20-fold or greater above those seen in control cells (Fig. 6D).
FIGURE 6.
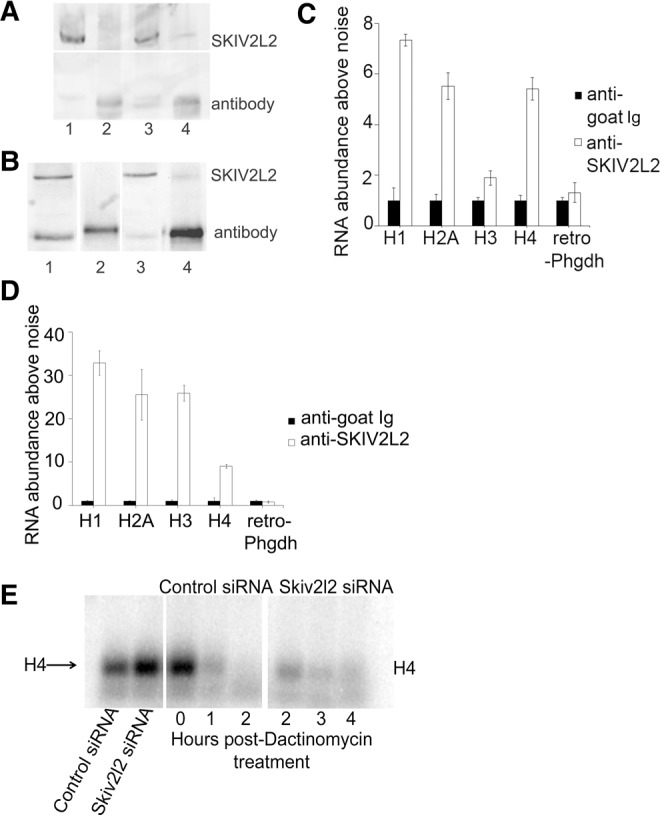
SKIV2L2 binds to and modulates the half-life of replication dependent histone mRNAs. (A) Western blot to detect immunoprecipitated SKIV2L2 in N2A cells. Western blots to detect SKIV2L2 were performed on the following samples from N2A cells. (Lane 1) Whole-cell extract incubated with anti-goat IgG. (Lane 2) Immunoprecipitate with anti-goat IgG. (Lane 3) Whole-cell extract incubated with anti-SKIV2L2. (Lane 4) Immunoprecipitate with SKIV2L2. The top band indicates the position of SKIV2L2, and the lower band represents the heavy chain from the antibody used for immunoprecipitation. (B) Western blot to detect immunoprecipitated SKIV2L2 in P19 cells. Western blots to detect SKIV2L2 were performed in P19 cells as stated above. (Lane 1) Whole-cell extract incubated with anti-goat IgG. (Lane 2) Immunoprecipitate with anti-goat IgG. (Lane 3) Whole-cell extract incubated with anti-SKIV2L2. (Lane 4) Immunoprecipitate with SKIV2L2. (C) qRT-PCR of histone RNAs bound to immunoprecipitated SKIV2L2 in N2A cells. Proteins were extracted from N2A cells irradiated with UV light. SKIV2L2 was immunoprecipitated with its cross-linked RNAs using anti-SKIV2L2, with anti-goat used as a negative control. RNA was isolated from both anti-SKIV2L2 and anti-goat immunoprecipitates. Using qRT-PCR, RNA abundance levels of histone mRNAs immunoprecipitated in either sample were calculated using ΔCq values, and normalized to the amplification of Skiv2l2 mRNA, which was unbound in both samples (error bars represent ±SD for n = 3). Immunoprecipitated retro-Phgdh represents an RNA precipitated equally by anti-goat and anti-SKIV2L2. (D) qRT-PCR of histone RNAs bound to immunoprecipitation SKIV2L2 in P19 cells. Performed as stated in C on immunoprecipitate from P19 cells. (E) Northern blot demonstrating H4 mRNA turnover following Dactinomycin treatment. Following transfection with control or Skiv2l2 siRNA, RNA was extracted at 0, 1, 2, 3, and 4 h following application of Dactinomycin. Northern blotting detected H4 mRNA, which was quantified and normalized to ribosomal RNA levels to calculate half-life.
In an attempt to further support the hypothesis that SKIV2L2 is involved with replication-dependent histone mRNA turnover, the half-life of H4 mRNA was measured in both control and Skiv2l2 knockdown cells. N2A cells were treated with Dactinomycin to inhibit RNA polymerase II transcription, and Northern blots allowed for the visualization of H4 mRNA levels over time. Histone H4 mRNA was selected as all nine H4 mRNAs expressed from histone cluster one significantly accumulated with SKIV2L2 depletion as identified via RNA-seq, and there are no noncanonical H4 variants that could be detected. In N2a cells, qRT-PCR identified a threefold increase in histone H4 mRNA with Skiv2l2 knockdown. Northern blots detected a twofold increase in histone H4 mRNA (P-value <0.04, n = 3), which is similar to the accumulation seen via qRT-PCR, demonstrating that Northern blotting is sensitive enough to discriminate differences in H4 mRNA levels (Fig. 6E). Cells were then treated with Dactinomycin to observe histone H4 mRNA turnover in the absence of transcription. Histone H4 mRNA levels were monitored at five time points following Dactinomycin treatment, ranging from 0 to 4 h. H4 mRNA levels were detected via Northern blotting, quantified using ImageJ software, and normalized to ribosomal RNA levels to account for differences in load. In control cells, H4 mRNA was depleted to negligible levels 4 h after Dactinomycin treatment (Fig. 6E). However, at 4 h post-Dactinomycin treatment, Skiv2l2 knockdown cells retained higher levels of H4 mRNA (Fig. 6E; Supplemental Fig. S10), demonstrating that cells depleted of SKIV2L2 exhibit a decreased rate of H4 mRNA turnover. The mRNA half-life for histone H4 was calculated at each time point over 4 h for three separate trials. Based on the average of three independent trials, the half-life of H4 mRNA in control cells was 34 min, and this half-life increased to 72 min following Skiv2l2 knockdown (P-value <0.04). Given that Dactinomycin inhibited transcription in these samples, a slower rate of mRNA decay must be responsible for the increase in H4 mRNA half-life. Since replication-dependent histone mRNAs immunoprecipitated with SKIV2L2 at high levels, these results conclusively demonstrate SKIV2L2 directly binds to histone mRNAs in vivo. Coupled with the doubling of H4 mRNA half-life following SKIV2L2 depletion, we conclude that SKIV2L2 directly aids in the turnover of replication-dependent histone mRNAs.
DISCUSSION
Here, we have demonstrated that RNAi knockdown of Skiv2l2 increased differentiation of two murine cell lines into two cell fates. This differentiation correlates to a decrease in cellular proliferation, attributed to delayed mitotic progression following Skiv2l2 knockdown. The observed binucleated cells could arise via “mitotic slippage” where activation of the spindle assembly checkpoint temporarily arrests cells in mitosis, but the checkpoint cannot be maintained, causing cells to aberrantly continue through mitosis and undergo failed cytokinesis (Rieder and Maiato 2004; Stevens et al. 2007; Freije et al. 2014; Dikovskaya et al. 2015). Examining potential RNAs targeted by SKIV2L2 revealed that SKIV2L2 directly binds to replication-dependent histone mRNAs, which are elevated in cells depleted of SKIV2L2 due to a slower decay rate. Taken together, these results are highly suggestive of the involvement of RNA surveillance factor SKIV2L2 in histone mRNA turnover, where SKIV2L2 binds histone mRNAs to mediate their degradation.
Because differentiation reduces Skiv2l2 protein and mRNA levels, we conclude that Skiv2l2 regulation occurs at the transcriptional level. Additionally, the presence of a CpG island promoter that Skiv2l2 shares with another RNA helicase, Dhx29, implies that DNA methyltransferases may developmentally regulate Skiv2l2 transcription. ChIP data suggest that multiple transcription factors such as STAT3 (Chen et al. 2008; Hutchins et al. 2012), CREB (Lesiak et al. 2013), E2F1 (Chen et al. 2008), and Myc (Chen et al. 2008; Sabò et al. 2014) bind to the Skiv2l2 promoter, and microarray studies have demonstrated that Skiv2l2 mRNA increases with cAMP/PKA activation (Guo et al. 2012). Additionally, the SKIV2L2 protein has been previously found to be a target of the MAPK–ERK signaling cascade in mice (Sundaram 2013), and the transcription factor Smad4 has been shown to regulate Skiv2l2 transcription (Fei et al. 2009). Our research has also demonstrated that chemically differentiating cancer cell lines down-regulate Skiv2l2 and that decreased SKIV2L2 levels enhance cell differentiation. Similar to how autophagy pathways are up-regulated in stem cells due to increased translation (Phadwal et al. 2012), stem cells may also up-regulate RNA surveillance pathways in response to the high levels of transcription and protein synthesis that stem cells require to efficiently divide and maintain pluripotency (Boisvert et al. 2007). One such gene whose transcription increases in proliferating cells, Rn45s (45S ribosomal RNA precursor), undergoes processing dependent on SKIV2L2-mediated RNA surveillance (Schilders et al. 2007). In this paper, we have identified replication-dependent histone mRNAs as another potential SKIV2L2 target whose mRNA levels increase in proliferating cells. With the switch from proliferation to post-mitotic differentiation, high levels of SKIV2L2 and RNA surveillance may no longer be necessary, resulting in the reduced levels of SKIV2L2 observed in chemically differentiated cells.
Since loss of SKIV2L2 enhances both the differentiation of N2A cells into cholinergic neurons and P19 cells into cardiac myocytes, we conclude that SKIV2L2 functions in mouse cells to maintain pluripotency as opposed to preventing differentiation into a certain cell type. Additionally, the delay in G2/M phase progression in Skiv2l2 knockdown cells differs from the canonical G1 arrest seen in chemically or developmentally differentiated cells, suggesting that the differentiation following loss of SKIV2L2 occurs indirectly as opposed to through direct induction of differentiation pathways. While the results of mitotic arrest and increased differentiation appear to conflict, there are possible explanations for the phenotype seen with SKIV2L2 depletion. The differentiation observed in N2A and P19 cells following Skiv2l2 knockdown may result from mitotic stress that generates polyploid cells unable to undergo apoptosis. Failed cytokinesis following mitotic slippage generates one 4N G1 cell that can undergo apoptosis, enter S phase with significant genomic damage, or arrest in G1 to either senesce or differentiate (Rieder and Maiato 2004). This phenomenon of mitotic slippage could potentially account for the phenotype observed following SKIV2L2 depletion, as mitotic stress can induce differentiation either naturally or following p53 inhibition (Smith et al. 1998; Freije et al. 2014). The p53 signaling pathway has been shown to be inactivated in P19 cell lines (Chang et al. 2010), lending support to the idea that differentiation could result from a failure to trigger apoptosis following mitotic slippage in cells depleted of SKIV2L2. We have also demonstrated that SKIV2L2 depletion results in decreased cellular proliferation, specifically attributed to perturbed progression through G2/M phase and seen as an increase in G2/M phase cells, elevated levels of the mitotic marker H3 phospho-S10, and the observance of binuclear cells. In light of this, SKIV2L2 may promote efficient progression through the cell cycle to allow for self-renewal in stem cells. This hypothesis would explain studies in zebrafish, where Skiv2l2 mutants fail to regenerate melanocytes or tails, presumably due to a decrease in the stem cell population (Yang et al. 2007).
This research has illuminated SKIV2L2 target RNAs that could be responsible for perturbed mitotic progression. In particular, SKIV2L2 depletion in N2A and P19 cells results in the accumulation of the replication-dependent histone mRNAs. Histone mRNA levels depend on cell-cycle regulation, with high levels of histone mRNAs necessary in proliferating cells. During S phase, histone mRNA transcription increases three- to fivefold (Marzluff et al. 2008), but at the end of S phase, histone mRNA half-life decreases substantially. Unlike other coding mRNAs, histone mRNAs lack poly(A) tails, with the processing and stability of the mRNA depending on the binding of SLBP (stem–loop-binding protein) (Lanzotti et al. 2002; Zhao et al. 2004; Zhang et al. 2012). The down-regulation of both SLBP and histone mRNAs at the end of S phase is necessary for cell-cycle progression because the expression of histone proteins outside of S phase negatively impacts chromosome segregation during mitosis and may titrate histone modifiers away from chromatin (Meeks-Wagner and Hartwell 1986; Marzluff et al. 2008; Singh et al. 2010). This research implicates SKIV2L2 in the turnover of replication-dependent histone mRNAs. In yeast, loss of Mtr4p results in M phase arrest due to impaired microtubule spindle assembly (Smith et al. 2011), but it is unclear if this is due to a histone imbalance. Additionally, loss of Trf4p and the exosome component Rrp6p lead to histone accumulation in yeast (Reis and Campbell 2006; Canavan and Bond 2007). Similarly, knockdown of Rrp6 in Drosophila results in the accumulation of histone mRNAs and inhibits progression into M phase (Graham et al. 2009). However, previous research failed to establish a definitive link between histone turnover and nuclear RNA surveillance components in mammals. The results presented in this paper demonstrate that SKIV2L2 not only binds histone mRNAs, but also that SKIV2L2 depletion causes histone mRNAs to accumulate due to impaired histone mRNA turnover. Together, this offers highly suggestive evidence that at least one component of nuclear RNA surveillance, the helicase SKIV2L2, mediates the turnover of replication-dependent histone mRNAs. In humans, the discovery that h3′exo, an exonuclease responsible for histone mRNA turnover, is not sufficient to displace SLBP and degrade the stem–loop of histone mRNA (Dominski et al. 2003) lends promise to the idea that the RNA helicase SKIV2L2 may be necessary to unwind histone mRNAs prior to degradation. The connection between SKIV2L2 and histone accumulation implies that SKIV2L2 directs histone mRNAs to the exosome for degradation. In addition, human PM/Scl-100 (Rrp6) has been shown to be necessary for the degradation of cytoplasmic histone mRNAs (Slevin et al. 2014). The cellular localization of PM/Scl-100 is primarily nuclear, as it associates with the nuclear exosome (Allmang et al. 1999; Brouwer et al. 2000). However, certain studies have detected a very small proportion of PM/Scl-100 in the cytoplasm (Lejeune et al. 2003), although this finding is not definitive. Localization studies detect SKIV2L2 exclusively in the nucleus, with a higher concentration in the nucleolus (Schilders et al. 2007; Lubas et al. 2011, 2015; Osman et al. 2011). To our knowledge, SKIV2L2 has not been detected in the cytoplasm. Since the nuclear RNA surveillance components PM/Scl-100 and SKIV2L2 have been implicated in the replication-dependent histone turnover, it is possible that these proteins play a role in cytoplasmic RNA surveillance or a more complicated relationship exists between histone mRNA turnover and nuclear RNA surveillance.
In addition to regulating histone mRNA levels, our data have implicated SKIV2L2-mediated RNA surveillance in snoRNA and miRNA biogenesis, lncRNA turnover, and degradation of transcripts originating from intergenic regions, such as retrotransposed mRNAs, pseudogenes, and PROMPTs. These targets could also influence proliferation and pluripotency through various mechanisms. Verifying and exploring these RNA surveillance targets will drive forward a better understanding of how RNA surveillance can influence RNA transcription and RNA biogenesis pathways on a more global scale and lead to greater insight on the regulation of mammalian gene expression.
Our studies on SKIV2L2 in mammalian cells establish a connection between RNA surveillance and cell-cycle progression. While there is a correlation between histone mRNA accumulation and mitotic arrest in Skiv2l2 knockdown cells, the definitive cause of this phenotype remains elusive. Since SKIV2L2 functions in nucleolar RNA surveillance, and the regulation of cell growth and division largely depends on the nucleolus (Tsai 2014), we hypothesize that the turnover of histone mRNAs or the processing and turnover of nucleolar RNAs are necessary for mitotic progression. Alternatively, because SKIV2L2 facilitates the processing of mammalian pre-rRNAs, SKIV2L2 may enhance the efficiency of rRNA processing and ribosome assembly, which are necessary to maintain cell proliferation in actively dividing cells (Boisvert et al. 2007). Indeed, our data also hint at a central role for SKIV2L2 in regulating ribosome biogenesis, as cells depleted of SKIV2L2 accumulate ribosomal protein mRNAs and show perturbed ribosomal RNA processing (Berndt et al. 2012). Similar to the results seen in this study for SKIV2L2, the methyltransferase fibrillarin has also drawn a connection between stem cell maintenance and ribosome biogenesis (Watanabe-Susaki et al. 2014). Like SKIV2L2, fibrillarin is necessary for rRNA maturation and highly expressed in embryonic stem cells, with knockdown of fibrillarin inducing differentiation and inhibiting cell growth (Watanabe-Susaki et al. 2014). In support of the hypothesis SKIV2L2 may induce differentiation due to impaired ribosome biogenesis, the down-regulation of rRNAs has been shown to trigger differentiation in human HL-60 and THP-1 cell lines (Hayashi et al. 2014), and ribosomal protein mRNA expression changes during differentiation in human NTERA2 cells (Bevort and Leffers 2000). While the exact function of SKIV2L2 in the cell cycle is unclear, an imbalance of at least two SKIV2L2 substrates, histone mRNAs and ribosomal protein mRNAs, could be responsible for the defects in mitotic progression observed following Skiv2l2 knockdown. Deeper investigation into the role of SKIV2L2 and nuclear RNA surveillance in cell-cycle regulation may prove important in understanding diseases marked by changes in SKIV2L2, such as cancer.
While noncoding RNAs represent the vast majority of discovered nuclear RNA surveillance targets, it was of great interest that replication-dependent histone mRNAs accumulate following loss of SKIV2L2-mediated RNA surveillance. Since histone mRNAs lack a poly(A) tail like many noncoding RNAs (Marzluff et al. 2008), it reasons that the nuclear turnover of histone mRNAs may be similar to that seen for noncoding RNAs. Replication-dependent histones are transcribed at high levels during S phase but are rapidly turned over upon entry into G2 phase (Marzluff et al. 2008). While the turnover of histones following uridylation in the cytoplasm has been extensively studied (Kaygun and Marzluff 2005a,b; Mullen and Marzulff 2008), our research suggests that SKIV2L2 also mediates the turnover of histone mRNAs, as SKIV2L2 directly binds histone mRNAs in vivo and SKIV2L2 depletion increases the half-life of histone H4 mRNA. This histone mRNA turnover could potentially occur in the nucleus, where SKIV2l2 is localized, or in the cytoplasm following breakdown of the nuclear envelope. Since SKIV2l2 is necessary for replication-dependent histone mRNA turnover, it is hypothesized that with SKIV2L2 depletion, histone mRNAs fail to be properly turned over at the end of S phase, leading to a histone imbalance, which has previously been shown to disrupt mitosis (Meeks-Wagner and Hartwell 1986; Singh et al. 2010; Günesdogan et al. 2014).
MATERIALS AND METHODS
Cell culture
Neuro2A (N2A, ATCC #CCL-131) cells were grown at 37°C, 5% CO2, and 95% humidity in DMEM (Sigma-Aldrich #D6429), 10% fetal bovine serum (FBS, Sigma-Aldrich #12306C). Cells were plated on Greiner Bio-One Advanced TC 35- or 60-mm plates. Cells were split at 1:5 or 1:10 every 48 h. Differentiation was initiated by plating the cells in DMEM, 2% FBS, 20 µM all-trans retinoic acid (ATRA, Sigma-Aldrich #R2625). Cells were harvested after 48 h by simple pipetting.
P19 cells (ATCC #CRL-1825) were cultured in α-MEM (Sigma-Aldrich # M8042), 10% FBS (Sigma-Aldrich #12306C) at 37°C, 5% CO2, and 95% humidity. P19 cells were passaged every 48 h at 1:5 or 1:10. Cells were differentiated using α-MEM growth medium, 10% FBS, 0.1% dimethyl sulfoxide (Sigma-Aldrich #D2438). Cells were harvested after 48 h by simple pipetting.
RNAi
Before RNAi knockdown, 3 × 105 N2A or P19 cells were plated on 35-mm plates. When cells were at ∼60% confluency, cells were transfected with the appropriate siRNA. Skiv2l2 knockdowns were performed by transfecting cells with either 67 nM Skiv2l2 Silencer siRNA ID #177475 or #177476 (Thermo Fisher #AM16704, #AM16708) or 67 nM Negative Control Silencer siRNA by Ambion (Thermo Fisher #AM4635). Delivery of siRNAs was accomplished with 1 mL of Opti-MEM (Thermo Fisher #31985) and 0.1% Lipofectamine RNAiMax by Invitrogen (Thermo Fisher #13778) for N2A cells or 0.1% Lipofectamine 3000 by Invitrogen (Thermo Fisher #L300008) for P19 cells according to the manufacturer's protocol.
Cells treated with the siRNAs were grown for 48 h at 37°C, 5% CO2, and 95% humidity. The cells were then harvested following RNAi using simple pipetting.
Neuronal processes quantification
Following RNAi, N2A cells were counted following treatment with the Neural Outgrowth Staining kit (Thermo Fisher #A15001) according to the manufacturer's protocol. Cells were viewed on the Leica DMI6000 B inverted microscope with excitation at 495 nM and 555 nM for visualization of the cell viability stain (green) and the cell membrane stain (orange). Random fields were selected, and 900 cells were scored for Skiv2l2 and negative control siRNA-treated cells. Cells staining both orange and green (yellow overlay) with processes extending longer than twice the diameter of the cell body were counted as differentiated. The number of differentiated cells was then divided by the total number of cells counted to obtain the percentage of dead and differentiated cells. Samples were compared using a binomial comparison of proportions and χ2 test.
Protein extraction
Harvested N2A and P19 cells were lysed on ice for 15 min with protein lysis buffer containing 1% Triton-X, 150 mM sodium chloride, 50 mM Tris-HCl pH 8, and cOmplete EDTA-free proteinase inhibitor tablet (04693132001 Roche). Cells were then spun down at 16,000g at 4°C for 15 min, and the supernatant containing the protein was collected. Bradford assays were performed to quantify the total protein using Bradford Reagent (Sigma-Aldrich #B6916). Histone proteins were extracted by incubating the nuclear pellet with 0.2 N HCl overnight at 4°C.
RNA isolation
Cells were lysed in 1 mL TRI Reagent (Sigma-Aldrich #T9424), and RNA was isolated according to the manufacturer's protocol. RNA was DNase treated according to the TURBO DNA-free kit protocol (Ambion #AM1907) and quantified using the NanoDrop 2000 (Thermo Scientific). RNA isolated from P19 cells for sequencing was tested for RNA integrity using the RNA 6000 Nano Kit (Agilent #5067-1511) on the Agilent 2100 Bioanalyzer according to the manufacturer's protocol.
Western blotting
Protein extracts in Laemmli buffer were run at 100V on a 10% acrylamide (37.5:1) SDS-PAGE gel. Acid-extracted nuclear fractions and cytoplasmic fractions were run on separate gels on the same day. Proteins were then transferred onto BioTrace nitrocellulose membrane, 0.2 µm pore size (Pall Corporation #66485) at 4°C and 15 V for 12 h. The membrane was incubated in blotting milk containing polyclonal antibodies raised in rabbit against mouse SKIV2L2 (1:1000 dilution, Abcam ab187884), β-actin (1:50 dilution, Thermo Fisher #PA5-16914), or H3 phospho-S10 (1:500 dilution, Abcam ab5176) for 16 h at 4°C, followed by goat anti-rabbit antibody conjugated to horseradish peroxidase (1:5,000 dilution, Abcam ab6721) Protein was detected using luminol, ρ-coumaric acid, and hydrogen peroxide and capturing chemiluminescence with the UVP Biospectrum System. SKIV2L2 and H3 phospho-S10 protein levels were quantified by normalizing to ACTB using the ImageJ software provided by the NIH. A Student's t-test measured significance between control and Skiv2l2 siRNA samples, while a one-way ANOVA with Tukey's HSD test was used with comparison to chemically induced differentiation.
Quantitative RT-PCR
DNase-treated RNA isolated from N2A and P19 cells was reversed transcribed using oligo-dT or gene-specific primers (Supplemental Table S6) according to M-MLV reverse transcriptase protocol (Promega #M1705). PCR cleanup (IBI Scientific, VWR #95039) was then carried out according to the manufacturer's protocol on the reverse transcription reactions, and the reactions were diluted with DEPC-treated water by a factor of 10. Targets (primers listed in Supplemental Table S6) were amplified from the reverse transcription reactions using iQ SYBR Green Supermix (BioRad #1708880). Amplification was detected using the CFX Connect Real-Time PCR detection system. Cycles 1–35 were analyzed using regression threshold analysis for samples with no amplification in control reactions without reverse transcriptase. Target “expression” (steady-state mRNA level) was calculated using CFX Manager Software, where expression equals 2−(Cqtarget−CqActB) with targets normalized to β-actin and control treatment. Statistics were calculated using the Student's t-test for comparison between control and Skiv2l2 knockdown. For comparison with ATRA treatment as well, a one-way ANOVA followed by the Tukey's HSD test was used.
MTT assay
Immediately following transfection of N2A or P19 cells with negative control siRNA or Skiv2l2 siRNA, cells were harvested and transferred to a 96-well plate. 100 µL of cells were plated in each well, with 10 µL of 5 mg/mL Thiazolyl blue tetrazolium bromide (MTT, Sigma-Aldrich #M5655) according to the manufacturer's protocol at various time points during 37°C incubation. Absorbance of formazan was measured according to the manufacturer's protocol after 3 h of incubation with MTT. Formazan absorbance was calculated by subtracting the absorbance at 630 nm from the absorbance at 570 nm for 10 wells per time point. Formazan absorbance was then used to calculate cell proliferation by dividing any given formazan absorbance by the initial formazan absorbance at the time of transfection. Overall proliferation at 36 h was calculated for both N2a and P19 cells, and results were compared using the Student's t-test. For comparison between control siRNA, ATRA treatment, and Skiv2l2 siRNA over time, a one-way ANOVA was used in conjunction with the Tukey's HSD test at each time point.
Fluorescent activated cell sorting
Following RNAi, knockdown cells were harvested and washed in PBS supplemented with 2% FBS and resuspended in 1 mL of PBS.
For viability staining, cells were then incubated with 1 U RNaseA for 1 h at 37°C. 100 µL of 1 mg/mL propidium iodide (Thermo Fisher #P3566) was added to the cells 20 min before cell sorting on the BD Accuri C6 cell sorter. Cells were excited with the 488 nm blue laser, and the filter was set to FL-2 to detect propidium iodide fluorescence. 50,000 cells were sorted for each sample, debris was gated out, and the percentage of stained cells was calculated using the CFlow Plus software. The percentage of stained cells across the three treatments were compared with a one-way ANOVA in conjunction with the Tukey's HSD test. In addition, cells with increased side-scatter were counted using the CFlow Plus software and compared across the three treatments with a one-way ANOVA and Tukey's HSD test as well.
For cell-cycle analysis, cells were fixed in ice cold ethanol for 30 min, washed twice in PBS, and resuspended in 1 mL PBS. The fixed cells were then incubated with 1 U RNaseA for 1 h at 37°C. 100 µL of 1 mg/mL propidium iodide (Thermo Fisher #P3566) was added to the fixed cells 20 min before cell sorting on the BD Accuri C6 cell sorter as stated above. The percentage of stained cells in G1, S, and G2/M phase was calculated using the CFlow Plus software based on FL-2 detection (DNA content) and cell count. The percentage of cells in each cell-cycle phase were compared across the three treatments with a one-way ANOVA in conjunction with the Tukey's HSD test.
Indirect immunofluorescence cell imaging
Following RNAi treatment, harvested cells were washed in PBS, 2% FBS and fixed for 30 min in ice-cold ethanol. After washing in PBS, 0.1% NP-40, pH 7.5, cells were blocked for 60 min at 21°C in PBS, 2% FBS. Cells were then incubated with the primary antibody against H3 phospho-S10 (1:500 dilution, Abcam ab5176) for 14 h at 4°C. Cells were again washed before incubation with the secondary antibody anti-rabbit conjugated to AlexaFluor 488 (1:500 dilution, Thermo Fisher #A-11034) for 2 h at 21°C. Cells were washed and resuspended in 1 mL PBS before viewing 200 µL of cells on slides. Cells were excited at 488 nm and both brightfield images and FL-1 filtered images were captured on the Nikon Eclipse E600 microscope. 150 cells per sample were scored based on fluorescence intensity, with four biological replicates. A Student's t-test was performed to compare control to Skiv2l2 knockdown samples.
RNA-seq
RNA was harvested from P19 cells as described above on three biological replicates of control siRNA and Skiv2l2 siRNA treated cells. RNA was tested for quality and treated with DNase and RiboZero kits to remove DNA and rRNA. Illumina Hi-Seq 2000 paired-end sequencing was performed at University of Wisconsin-Madison. FASTQ reads were trimmed and filtered to improve mapping quality before mapping to mm10 genomic build using TopHat, allowing for no more than two nucleotide mismatches per read and resulting in 95% mapping efficiency. Galaxy web-based tools were used to perform cufflinks, and cuffdiff was run to determine differential transcripts. Transcripts with a q-value less than 0.05 were considered to be differentially expressed in the Skiv2l2 knockdown cells, and elevated or down-regulated genes showing a fold change >1.5-fold were probed in detail. Files were converted into BigWig format to view genome coverage on the IGV Genome Browser. GO-enrichment analysis was run on the GO-ontology consortium website. Classes of mRNAs were identified using the geneimprint: imprinted gene database and NCBI. Statistics were performed in R Studio using the phyper function.
Protein–RNA immunoprecipitation
After 48 h of cell growth, untreated P19 and N2A cells were irradiated at 254 nm (150 mJ/cm2) to crosslink proteins and RNA using the UVP Crosslinker CL-1000 model. Protein A conjugated Dynabeads were prepared according to the manufacturer's protocol using the Dynabeads Protein A Immunoprecipitation Kit (Thermo Fisher #10006D). Dynabeads were incubated with 3 µg of rabbit polyclonal antibody against SKIV2L2 (Abcam #ab70552). Cells were lysed as described previously. Cellular lysate prepared UV irradiated cells were loaded onto the Dynabeads and incubated according to kit protocol. Immunoprecipation of SKIV2L2 according to kit protocol was performed, and the protein–RNA complexes were eluted off the Dynabeads. Eluate was treated with DNase and proteinase K. Samples of whole cell extracts (8 µL) after application to beads and immunoprecipitate after application to beads, washing, and elution (1 µL) were taken for Western blotting to verify recovery of SKIV2L2 protein, while RNA was isolated from eluate using TRIzol extraction with chloroform as previously described. cDNA preparation and qPCR were performed on the isolated RNA as mentioned previously. As Skiv2l2 mRNA did not immunoprecipitate with anti-SKIV2L2, Skiv2l2 mRNA target Cq values were representative of fluctuations in background fluorescence. Experimental Cq values were normalized to Skiv2l2 mRNA Cq values. The Student's t-test was performed to compare mRNA levels for anti-goat-Ig and anti-SKIV2L2 samples for three trials.
RNA decay assay
Following 24 h of control and Skiv2l2 RNAi as previously described, N2A cells were treated with Dactinomycin (Sigma #A1410) to a concentration of 5 µg/mL. Cells were harvested for RNA extraction at 0, 1, 2, 3, and 4 h. RNA was quantified and 8 µL of identical amounts of RNA (approximately 8 µg) were run on a 1.4% agarose gel at 90V. RNA was transferred to a blotting membrane overnight in 20X SSC before being crosslinked to the membrane. The Northern blot was probed using primers against H4 and ActB. Primers were radiolabeled with ATP, [γ-32P]-3000Ci/mmol 10mCi/ml EasyTide Lead (PerkinElmer), using T4-polynucleotide kinase (NEB #M0201) according to the manufacturer's protocol. Radiolabeled probes were incubated with the blot at 42°C for 16 h and developed using phosphorimaging screens and the Storm PhosphorImager from GE Healthcare. Peak quantification for H4 mRNA and rRNA was performed on Image J software, and H4 mRNA levels were normalized to 5S rRNA. The decay constant was calculated using the equation ln(C/C0) = −kdecayt, where t = time, kdecay = decay constant, C = RNA level at t, and C0 = RNA level at t = 0. H4 half-life (t1/2) was found by solving for t at ln(0.5). This half-life was calculated at every hour, 2, 3, and 4 h. These values were averaged to determine the overall half-life for each trial. The Student's t-test was performed on the H4 half-life calculated for three distinct trials.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University of Wisconsin Biotechnology Center DNA Sequencing Facility for providing NEXT-generation sequencing facilities and services and Fengchao Wang for technical assistance.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060640.117.
REFERENCES
- Allmang C, Petfalski E, Podtelejnikov A, Mann M, Tollervey D, Mitchell P. 1999. The yeast exosome and human PM-Scl are related complexes of 3′ → 5′ exonucleases. Genes Dev 13: 2148–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen KR, Jensen TH, Brodersen DE. 2008. Take the “A” tail–quality control of ribosomal and transfer RNA. Biochim Biophys Acta 1779: 532–537. [DOI] [PubMed] [Google Scholar]
- Beaulieu YB, Kleinman CL, Landry-Voyer AM, Majewski J, Bachand F. 2012. Polyadenylation-dependent control of long noncoding RNA expression by the poly(A)-binding protein nuclear 1. PLoS Genet 8: e1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt H, Harnisch C, Rammelt C, Stöhr N, Zirkel A, Dohm JC, Himmelbauer H, Tavanez JP, Hüttelmaier S, Wahle E. 2012. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA 18: 958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevort M, Leffers H. 2000. Down regulation of ribosomal protein mRNAs during neuronal differentiation of human NTERA2 cells. Differentiation 66: 81–92. [DOI] [PubMed] [Google Scholar]
- Boele J, Persson H, Shin JW, Ishizu Y, Newie IS, Søkilde R, Hawkins SM, Coarfa C, Ikeda K, Takayama K, et al. 2014. PAPD5-mediated 3′ adenylation and subsequent degradation of miR-21 is disrupted in proliferative disease. Proc Natl Acad Sci 111: 11467–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert FM, van Koningsbruggen S, Navascués J, Lamond AI. 2007. The multifunctional nucleolus. Nat Rev Mol Cell Biol 8: 574–585. [DOI] [PubMed] [Google Scholar]
- Brouwer R, Allmang C, Raijmakers R, van Aarssen Y, Egberts WV, Petfalski E, van Venrooij WJ, Tollervey D, Pruijn GJ. 2000. Three novel components of the human exosome. J Biol Chem 276: 6177–6184. [DOI] [PubMed] [Google Scholar]
- Callahan KP, Butler JS. 2009. TRAMP complex enhances RNA degradation by the nuclear exosome component Rrp6. J Biol Chem 285: 3540–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canavan R, Bond U. 2007. Deletion of the nuclear exosome component RRP6 leads to continued accumulation of the histone mRNA HTB1 in S-phase of the cell cycle in Saccharomyces cerevisiae. Nucleic Acids Res 35: 6268–6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G, Miao YL, Zhang Y, Liu S, Kou Z, Ding J, Chen DY, Sun QY, Gao S. 2010. Linking incomplete reprogramming to the improved pluripotency of murine embryonal carcinoma cell-derived pluripotent stem cells. PLoS One 5: e10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117. [DOI] [PubMed] [Google Scholar]
- Choi SC, Yoon J, Shim WJ, Ro YM, Lim DS. 2004. 5-azacytidine induces cardiac differentiation of P19 embryonic stem cells. Exp Mol Med 36: 515–523. [DOI] [PubMed] [Google Scholar]
- Cinghu S, Yellaboina S, Freudenberg JM, Ghosh S, Zheng X, Oldfield AJ, Lackford BL, Zaykin DV, Hu G, Jothi R. 2014. Integrative framework for identification of key cell identity genes uncovers determinants of ES cell identity and homeostasis. Proc Natl Acad Sci 111: E1581–E1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JA, McAdow AR, Dinitz AE, McCallion AS, Johnson SL, Voigt MM. 2011. A zebrafish SKIV2L2-enhancer trap line provides a useful tool for the study of peripheral sensory circuit development. Gene Expr Patterns 11: 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. 2002. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 22: 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. 1992. Features of apoptotic cells measured by flow cytometry. Cytometry 13: 795–808. [DOI] [PubMed] [Google Scholar]
- de la Cruz J, Kressler D, Tollervey D, Linder P. 1998. Dob1p (Mtr4p) is a putative ATP-dependent RNA helicase required for the 3′ end formation of 5.8S rRNA in Saccharomyces cerevisiae. EMBO J 17: 1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey BK, Gagan J, Dutta A. 2010. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol 31: 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikovskaya D, Cole JJ, Mason SM, Nixon C, Karim SA, McGarry L, Clark W, Hewitt RN, Sammons MA, Zhu J, et al. 2015. Mitotic stress is an integral part of the oncogene-induced senescence program that promotes multinucleation and cell cycle arrest. Cell Rep 12: 1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z, Yang XC, Kaygun H, Dadlez M, Marzluff WF. 2003. A 3′ exonuclease that specifically interacts with the 3′ end of histone mRNA. Mol Cell 12: 295–305. [DOI] [PubMed] [Google Scholar]
- Dong D, Meng L, Yu Q, Tan G, Ding M, Tan Y. 2012. Stable expression of FoxA1 promotes pluripotent P19 embryonal carcinoma cells to be neural stem-like cells. Gene Expr 15: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorweiler JE, Ni T, Zhu J, Munroe SH, Anderson JT. 2014. Certain adenylated non-coding RNAs, including 5′ leader sequences of primary microRNA transcripts, accumulate in mouse cells following depletion of the RNA helicase MTR4. PLoS One 9: e99430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Huff JT, Panning B. 2008. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei T, Xia K, Li Z, Zhou B, Zhu S, Chen H, Zhang J, Chen Z, Xiao H, Han JD, et al. 2009. Genome-wide mapping of SMAD target genes reveals the role of BMP signaling in embryonic stem cell fate determination. Genome Res 20: 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freije A, Molinuevo R, Ceballos L, Cagigas M, Alonso-Lecue P, Rodriguez R, Menendez P, Aberdam D, De Diego E, Gandarillas A. 2014. Inactivation of p53 in human keratinocytes leads to squamous differentiation and shedding via replication stress and mitotic slippage. Cell Rep 9: 1349–1360. [DOI] [PubMed] [Google Scholar]
- Freimoser FM, Jakob CA, Aebi M, Tuor U. 1999. The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities. Appl Environ Microbiol 65: 3727–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham AC, Kiss DL, Andrulis ED. 2009. Core exosome-independent roles for Rrp6 in cell cycle progression. Mol Biol Cell 20: 2242–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui MC, Chen B, Yu SS, Bu BT. 2014. Effects of suppressed autophagy on mitochondrial dynamics and cell cycle of N2a cells. J Huazhong Univ Sci Technol Med Sci 34: 157–160. [DOI] [PubMed] [Google Scholar]
- Günesdogan U, Jäckle H, Herzig A. 2014. Histone supply regulates S phase timing and cell cycle progression. eLife 3: e02443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Wilderman A, Zhang L, Taylor SS, Insel PA. 2012. Quantitative proteomics analysis of the cAMP/protein kinase A signaling pathway. Biochemistry 51: 9323–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Kuroda T, Kishimoto H, Wang H, Iwama A, Kimura K. 2014. Downregulation of rRNA transcription triggers cell differentiation. PLoS One 9: e98586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J, LaCava J, Tollervey D. 2006. RNA-quality control by the exosome. Nat Rev Mol Cell Biol 7: 529–539. [DOI] [PubMed] [Google Scholar]
- Hultman KA, Johnson SL. 2010. Differential contribution of direct-developing and stem cell-derived melanocytes to the zebrafish larval pigment pattern. Dev Biol 337: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins AP, Poulain S, Miranda-Saavedra D. 2012. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood 119: e110–e119. [DOI] [PubMed] [Google Scholar]
- Jasmin DC, Spray DC, Campos de Carvalho AC, Mendez-Otero R. 2010. Chemical induction of cardiac differentiation in P19 embryonal carcinoma stem cells. Stem Cells Dev 19: 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Wang X, Anderson JT, Jankowsky E. 2012. RNA unwinding by the Trf4/Air2/Mtr4 polyadenylation (TRAMP) complex. Proc Natl Acad Sci 109: 7292–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaygun H, Marzluff WF. 2005a. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat Struct Mol Biol 12: 794–800. [DOI] [PubMed] [Google Scholar]
- Kaygun H, Marzluff WF. 2005b. Translation termination is involved in histone mRNA degradation when DNA replication is inhibited. Mol Cell Biol 25: 6879–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura SH, Ikawa M, Ito A, Okabe M, Nojima H. 2001. Cyclin G1 is involved in G2/M arrest in response to DNA damage and in growth control after damage recovery. Oncogene 20: 3290–3300. [DOI] [PubMed] [Google Scholar]
- Kong KYE, Tang HM, Pan K, Huang Z, Lee TH, Hinnebusch AG, Jin GY, Wong CM. 2013. Cotranscriptional recruitment of yeast TRAMP complex to intronic sequences promotes optimal pre-mRNA splicing. Nucleic Acids Res 42: 643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishan A. 1975. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol 66: 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D. 2005. RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121: 713–724. [DOI] [PubMed] [Google Scholar]
- Lanzotti DJ, Kaygun H, Yang X, Duronio RJ, Marzluff WF. 2002. Developmental control of histone mRNA and dSLBP synthesis during Drosophila embryogenesis and the role of dSLBP in histone mRNA 3′ end processing in vivo. Mol Cell Biol 22: 2267–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Li X, Maquat LE. 2003. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12: 675–687. [DOI] [PubMed] [Google Scholar]
- Lesiak A, Pelz C, Ando H, Zhu M, Davare M, Lambert TJ, Hansen KF, Obrietan K, Appleyard SM, Impey S, et al. 2013. A genome-wide screen of CREB occupancy identifies the RhoA inhibitors Par6C and Rnd3 as regulators of BDNF-induced synaptogenesis. PLoS One 8: e64658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima RM, Alvarez LD, Costa MF, Costa SL, Clarêncio J, El-Bachá RS. 2008. Cytotoxic effects of catechol to neuroblastoma N2a cells. Gen Physiol Biophys 27: 306–314. [PubMed] [Google Scholar]
- Liu K, Lin B, Zhao M, Yang X, Chen M, Gao A, Liu F, Que J, Lan X. 2013. The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell Signal 25: 1264–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Dziembowski A, Jensen TH. 2011. Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 43: 624–637. [DOI] [PubMed] [Google Scholar]
- Lubas M, Andersen PR, Schein A, Dzeimbowski A, Kudla G, Jensen TH. 2015. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep 10: 178–192. [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen S, Brodersen D, Jensen T. 2009. Origins and activities of the eukaryotic exosome. J Cell Sci 122: 1487–1494. [DOI] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, Duronio RJ. 2008. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet 9: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. 2004. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 430: 226–231. [DOI] [PubMed] [Google Scholar]
- Meeks-Wagner D, Hartwell L. 1986. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell 44: 43–52. [DOI] [PubMed] [Google Scholar]
- Mullen TE, Marzluff WF. 2008. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev 22: 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag A, Steitz JA. 2012. Tri-snRNP-associated proteins interact with subunits of the TRAMP and nuclear exosome complexes, linking RNA decay and pre-mRNA splicing. RNA Biol 9: 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Walter T, Hériché JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. 2010. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464: 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilbratt M, Porras O, Marutle A, Hovatta O, Nordberg A. 2009. Neurotrophic factors promote cholinergic differentiation in human embryonic stem cell-derived neurons. J Cell Mol Med 14: 1476–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ. 2011. Regional specialization: the NEXT big thing in nuclear RNA turnover. Mol Cell 43: 502–504. [DOI] [PubMed] [Google Scholar]
- Osman BAH, Kawashima A, Tamba M, Satoh E, Kato Y, Iki A, Konishi K, Matsuda M, Okamura N. 2011. Localization of a novel RNA-binding protein, SKIV2L2, to the nucleus in the round spermatids of mice. J Reprod Dev 57: 457–467. [DOI] [PubMed] [Google Scholar]
- Phadwal K, Watson AS, Simon AK. 2012. Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci 70: 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips A, Jones CJ, Blaydes JP. 2006. The mechanisms of regulation of Hdm2 protein level by serum growth factors. FEBS Lett 580: 300–304. [DOI] [PubMed] [Google Scholar]
- Rammelt C, Bilen B, Zavolan M, Keller W. 2011. PAPD5, a noncanonical poly(A) polymerase with an unusual RNA-binding motif. RNA 17: 1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis CC, Campbell JL. 2006. Contribution of Trf4/5 and the nuclear exosome to genome stability through regulation of histone mRNA levels in Saccharomyces cerevisiae. Genetics 175: 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Maiato H. 2004. Stuck in division or passing through. Dev Cell 7: 637–651. [DOI] [PubMed] [Google Scholar]
- Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. 2014. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511: 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilders G, van Dijk E, Pruijn GJM. 2007. C1D and hMtr4p associate with the human exosome subunit PM/Scl-100 and are involved in pre-rRNA processing. Nucleic Acids Res 35: 2564–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Butler JS. 2013. Nuclear RNA surveillance: role of TRAMP in controlling exosome specificity. Wiley Interdiscip Rev RNA 4: 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MJ, West S, Norbury CJ. 2010. The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 17: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidiropoulou E, Sachana M, Flaskos J, Harris W, Hargreaves AJ, Woldehiwet Z. 2008. Diazinon oxon affects the differentiation of mouse N2a neuroblastoma cells. Arch Toxicol 83: 373–380. [DOI] [PubMed] [Google Scholar]
- Singh RK, Liang D, Gajjalaiahvari UR, Kabbaj MH, Paik J, Gunjan A. 2010. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 9: 4236–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slevin M, Meaux S, Welch JD, Bigler R, Miliani de Marval PL, Su W. 2014. Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol Cell 53: 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TK, Nylander KD, Schor NF. 1998. The roles of mitotic arrest and protein synthesis in induction of apoptosis and differentiation in neuroblastoma cells in culture. Dev Brain Res 105: 175–180. [DOI] [PubMed] [Google Scholar]
- Smith SB, Kiss DL, Turk E, Tartakoff AM, Andrulis ED. 2011. Pronounced and extensive microtubule defects in a Saccharomyces cerevisiae DIS3 mutant. Yeast 28: 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens FE, Beamish H, Warrener R, Gabrielli B. 2007. Histone deacetylase inhibitors induce mitotic slippage. Oncogene 27: 1345–1354. [DOI] [PubMed] [Google Scholar]
- Sundaram MV. 2013. Canonical RTK-Ras-ERK signaling and related alternative pathways. WormBook, pp. 1–38. http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. 2006. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the Nrd1-Nab3 pathway in genome surveillance. Mol Cell 23: 853–864. [DOI] [PubMed] [Google Scholar]
- Tiedje C, Lubas M, Tehrani M, Menon MB, Ronkina N, Roussesau S, Cohen P, Kotlyarov A, Gaestel M. 2014. p38MAPK/MK2-mediated phosphorylation of RBM7 regulates the human nuclear exosome targeting complex. RNA 21: 262–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay RG, Sikorska M, Sandhu JK, Lantheir P, Ribecco-Lutkiewicz M, Bani-Yaghoub M. 2010. Differentiation of mouse Neuro 2A cells into dopamine neurons. J Neurosci Methods 186: 60–67. [DOI] [PubMed] [Google Scholar]
- Tsai RY. 2014. Turning a new page on nucleostemin and self-renewal. J Cell Sci 127: 3885–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jia H, Jankowsky E, Anderson JT. 2007a. Degradation of hypomodified tRNAiMet in vivo involves RNA-dependent ATPase activity of the DExH helicase Mtr4p. RNA 14: 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. 2007b. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet 39: 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Susaki K, Takada H, Enomoto K, Miwata K, Ishimine H, Intoh A, Ohtaka M, Nakanishi M, Sugino H, et al. 2014. Biosynthesis of ribosomal RNA in nucleoli regulates pluripotency and differentiation ability of pluripotent stem cells. Stem Cells 32: 3099–3111. [DOI] [PubMed] [Google Scholar]
- Weir JR, Bonneau F, Hentschel J, Conti E. 2010. Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc Natl Acad Sci 107: 12139–12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley UV, Hatcher J, Dempsey RJ. 2015. Sphingomyelin synthase 1 regulates Neuro-2a cell proliferation and cell cycle progression through modulation of p27 expression and Akt signaling. Mol Neurobiol 51: 1530–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Zhang X, Huang X, Yang Y, Hua X. 2010. Regulation of cyclin B2 expression and cell cycle G2/M transition by menin. J Biol Chem 285: 18291–18300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Tan G, Ding M, Dong D, Chen T, Meng X, Huang X, Tan Y. 2010. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res 38: 8027–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CT, Hindes AE, Hultman KA, Johnson SL. 2007. Mutations in gfpt1 and skiv2l2 cause distinct stage-specific defects in larval melanocyte regeneration in zebrafish. PLoS Genet 3: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Lam TT, Tonelli M, Marzluff WF, Thapar R. 2012. Interaction of the histone mRNA hairpin with stem–loop binding protein (SLBP) and regulation of the SLBP–RNA complex by phosphorylation and proline isomerization. Biochemistry 51: 3215–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, McKillop-Smith S, Müller B. 2004. The human histone gene expression regulator HBP/SLBP is required for histone and DNA synthesis, cell cycle progression and cell proliferation in mitotic cells. J Cell Sci 117: 6043–6051. [DOI] [PubMed] [Google Scholar]
- Zhi F, Gong G, Xu Y, Zhu Y, Hu D, Yang Y, Hu Y. 2012. Activated β-catenin forces N2A cell-derived neurons back to tumor-like neuroblasts and positively correlates with a risk for human neuroblastoma. Int J Biol Sci 8: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Hu DL, Liu YQ, Zhang QJ, Chen FK, Kong XQ, Cao KJ, Zhang JS, Qian LM. 2011. Fabp3 inhibits proliferation and promotes apoptosis of embryonic myocardial cells. Cell Biochem Biophys 60: 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.