

Abstract
The absence of α2* nicotinic acetylcholine receptors (nAChRs) in oriens lacunosum moleculare (OLM) GABAergic interneurons ablate the facilitation of nicotine-induced hippocampal CA1 long-term potentiation and impair memory. The current study delineated whether genetic mutations of α2* nAChRs (Chrna2L9′S/L9′S and Chrna2KO) influence hippocampus-dependent learning and memory and CA1 synaptic plasticity. We substituted a serine for a leucine (L9′S) in the α2 subunit (encoded by the Chrna2 gene) to make a hypersensitive nAChR. Using a dorsal hippocampus-dependent task of preexposure-dependent contextual fear conditioning, adolescent hypersensitive Chrna2L9′S/L9′S male mice exhibited impaired learning and memory. The deficit was rescued by low-dose nicotine exposure. Electrophysiological studies demonstrated that hypersensitive α2 nAChRs potentiate acetylcholine-induced ion channel flux in oocytes and acute nicotine-induced facilitation of dorsal/intermediate CA1 hippocampal long-term potentiation in Chrna2L9′S/L9′S mice. Adolescent male mice null for the α2 nAChR subunit exhibited a baseline deficit in learning that was not reversed by an acute dose of nicotine. These effects were not influenced by locomotor, sensory or anxiety-related measures. Our results demonstrated that α2* nAChRs influenced hippocampus-dependent learning and memory, as well as nicotine-facilitated CA1 hippocampal synaptic plasticity.
Acetylcholine and nicotine activate nicotinic acetylcholine receptors (nAChRs) to influence learning and memory, via the release of neurotransmitters and neurotrophic factors. The specific contributions of selective nAChR subunits influencing learning and memory, particularly during development, are poorly understood. Genetic deletion of α2* nAChRs leads to subtle, yet discernible, potentiation of the first 2 d of nicotine self-administration, context specific withdrawal, emotional memory processing (Lotfipour et al. 2013), and baseline deficits in hippocampus-dependent memory (Kleeman et al. 2016). Developmental dysregulation of neural circuits in the absence of α2* nAChRs may be responsible for these modifications. α2 (and β2) mRNA are expressed within midbrain (interpeduncular nucleus) and limbic brain regions (hippocampus, amygdala, and neocortex) (Deneris et al. 1988; Wada et al. 1988, 1989; Ishii et al. 2005; Upton and Lotfipour 2015; Hilscher et al. 2017). During early postnatal rodent development, particularly during the first 2 wk of life, heightened expression patterns of α2* nAChRs are observed within the hippocampus and cortex (Son and Winzer-Serhan 2006). Due to the expression pattern of α2* nAChRs, studies have focused on dissecting the functional roles of these subunits within these brain regions (Nakauchi et al. 2007; Salas et al. 2009).
Within the dorsal through ventral CA1 of the hippocampus, α2* nAChRs are located on oriens lacunosum moleculare (OLM) GABAergic interneurons (Sudweeks and Yakel 2000; Son and Winzer-Serhan 2006; Nakauchi et al. 2007, 2015; Leao et al. 2012; Chen et al. 2016; Kleeman et al. 2016). Studies suggest that α2* nAChRs play an important role in CA1 hippocampal OLM GABAergic interneurons (Ishii et al. 2005; Nakauchi et al. 2007, 2015; Chen et al. 2016; Kleeman et al. 2016). Deletion of Chrna2 induces an absence of nicotine facilitation and suppression of hippocampal long-term potentiation (LTP) (Nakauchi et al. 2007), likely via a dysregulation of OLM GABA neurotransmitter release (Leao et al. 2012). Furthermore, early developmental chronic nicotine exposure during peak mRNA expression periods of α2* nAChRs (i.e., the first 2 wk of rodent life) is known to influence hippocampal synaptic plasticity and learning and memory during adolescence, likely via α2* nAChRs (Nakauchi et al. 2015; Chen et al. 2016; Kleeman et al. 2016). Our current study assessed whether deletion or functional enhancement of α2* nAChRs are important for baseline and acute nicotine's effects on learning and memory processing. We evaluated this using a dorsal CA1 hippocampus-dependent task of preexposure-dependent contextual fear conditioning in adolescent male mice (Fanselow 1990; Rudy et al. 2002; Cushman et al. 2011). Our findings provide evidence for a role of α2* nAChRs in hippocampus-dependent learning and memory and nicotine-facilitated synaptic plasticity in mice.
Results
Adolescent Chrna2L9′S/L9′S generation, and developmental stereotypical behavioral characteristics
We genetically engineered the Chrna2L9′S/WT mice in our laboratory (Fig. 1). Subsequently, Chrna2L9′S/WT mice were backcrossed with C57Bl/6J mice for 3–4 generations. Heterozygous Chrna2L9′S/WT × Chrna2L9′S/WT mice were mated for experiments and offspring had expected Mendelian genotype (1:2:1) and sex ratios (50% M, F). The adolescent male Chrna2L9′S/L9′S mice exhibited good health, no gross anatomical morphological changes, no differences in stereotypical behaviors (Table 1a1; Fig. 2), and no differences in weight gain compared with wild-type littermate control mice (data not shown).
Figure 1.

Genetic design of the hypersensitive Chrna2L9′S/L9′S mouse line. (A) The generation of the hypersensitive Chrna2L9′S/L9′S mouse line has been described in the Materials and Methods. Briefly, a leucine to serine substitution was engineered using gene synthesis. The targeting vector was electroporated into 129S4/SvJae embryonic stem (ES) cells and homologous recombinants were confirmed through DNA sequencing (B) and Southern blot analysis (using the probe denoted by the single red line) (C). Targeted ES cells were microinjected into C57Bl/6J blastocyst embryos and implanted in pseudopregnant female mice, with the DNA sequence of the germline transmitted offspring confirming the AGC (Serine)/CTC (Leucine) heterozygous genomic modification (denoted by *). Genotyping of mice of was through tail biopsy, MyTaq HS Red Mix (Bioline) with PCR primers (A2L9S_1.10, A2L9S_1.11, denoted by arrows flanking the 34 bp FRT Site) designed upstream of and downstream from the deleted PGK-NEO, as described (FRT site denoted by the single arrow).
Table 1.
Statistics table
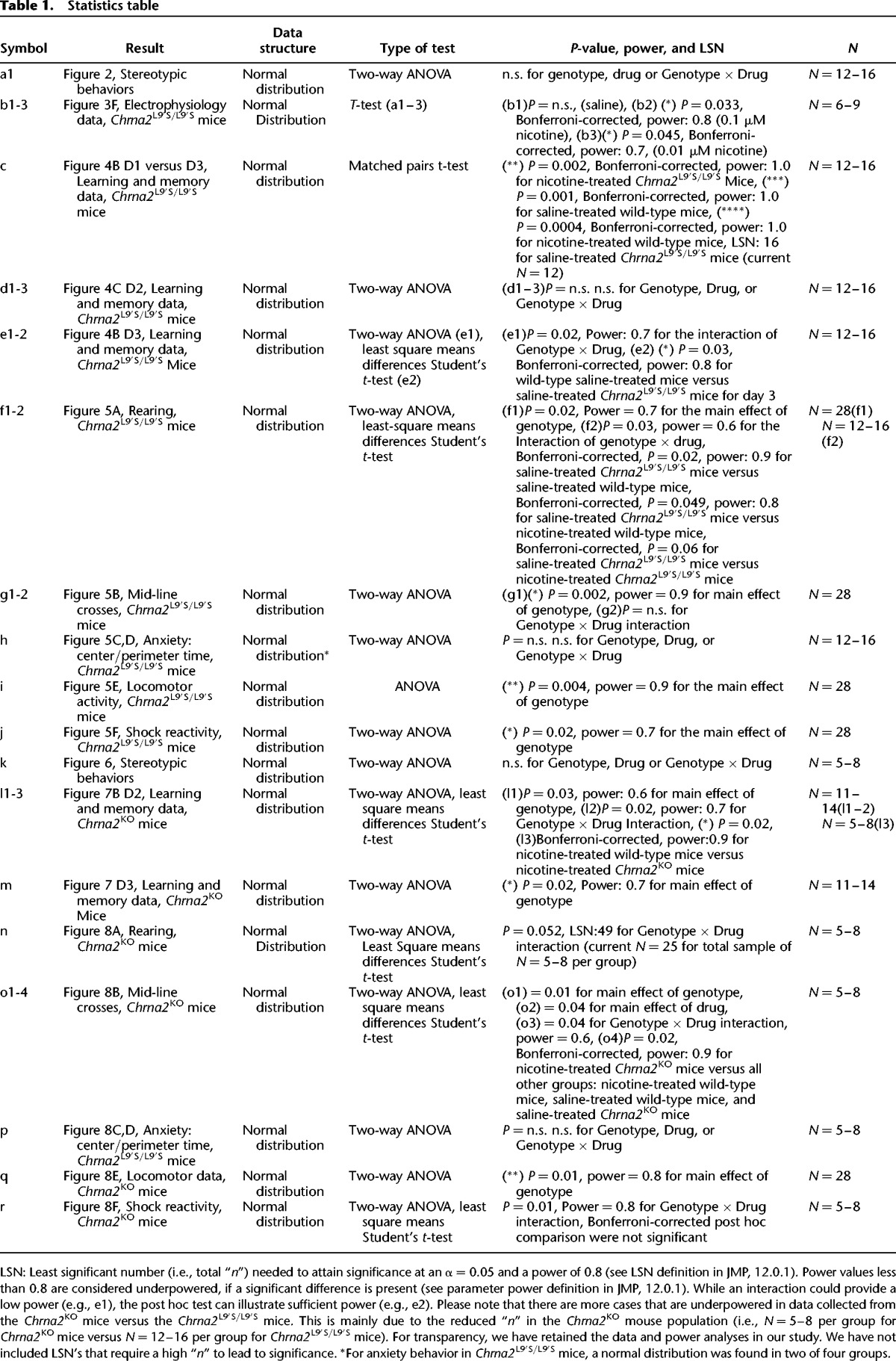
Figure 2.
Stereotypic behaviors were assessed in wild-type and Chrna2L9′S mice on day 1. Results illustrated that stereotypic behaviors do not differ based on drug treatment or genotype. N = 12–16 per group.
Oocyte expression
The use of oocyte expression studies (Fig. 3A) was applied as an initial screen to determine the functionality of a substitution of a serine for a leucine (L9′S) in the α2 nAChR subunit. In the presence of acetylcholine (ACh), fitted Hill equations for α2β2 wild-type nicotinic receptors had an EC50 of 3.7 ± 1.0 µM, which is similar to that previously reported for α4β2 nAChRs, 5.07 ± 2.4 µM (Lipovsek et al. 2008). α2L9′ mutations produced an increase in the apparent affinity for ACh when expressed in Xenopus oocytes together with the β2 subunit (Fig. 3A). Thus, the α2L9′S mutation produced a 100-fold (EC50: 0.041 ± 0.015), the α2L9′A a 10-fold (EC50: 0.37 ± 0.03) and the α2L9′T a fivefold decrease in the ACh EC50 (EC50: 0.91 ± 0.04) when compared with α2WT-containing receptors (EC50: 3.7 ± 1.0) (n = 3 per group). Our results confirmed earlier findings in oocytes that had a serine for a leucine (L9′S) substitution in a nAChR subunit induces a 100-fold leftward shift in the acetylcholine EC50 dose response curves (for review, please see Lester et al. 2003). Based on these data, α2L9′S mutations were chosen for the design and development of the hypersensitive Chrna2L9′S/L9′S mouse line (Fig. 1A), because they produced the greatest increase in ACh apparent affinity (Fig. 3A).
Figure 3.

(A) α2L9′S nicotinic receptor modifications potentiated ACh-induced ion channel conductance by 100-fold (closed circles) versus wild-type α2-containing nicotinic receptors (open circles) in oocyte expression studies, 10-fold against α2L9′A (closed squares), fivefold against α2L9′T (closed diamonds)-containing nAChRs. Based on this data, α2L9′S mutations were chosen for the design of the hypersensitive Chrna2L9′S/L9′S mouse line, because they produced the greatest increase in ACh apparent affinity. (B–F) Lower concentrations of nicotine facilitate LTP induction at CA3–CA1 synapses in Chrna2L9′S/L9′S mice. (B) There was no significant difference between slices from wild-type and Chrna2L9′S/L9′S mice in the stimulus–response relationship. (C,F) LTP was not induced in the absence of nicotine in both wild-type and Chrna2L9′S/L9′S mice. (D–F) Bath application of nicotine (0.01 µM nicotine in C and 0.1 µM in D) facilitated LTP induction in Chrna2L9′S/L9′S, but not wild-type, mice. Administration of nicotine is indicated by the horizontal bar. Traces above each graph (C–E) are representative waveforms recorded before and 50–60 min after weak TBS. Scale bar: 1 mV, 10 msec. (E) Histograms show the percent change (mean ± SEM) in the slope of fEPSPs measured 50–60 min after delivery of weak TBS. Numbers in parentheses indicate the numbers of experiments, (*) P < 0.05, Bonferroni-corrected.
Dorsal/intermediate CA1 hippocampal nicotinic facilitation of LTP
We measured the impact of α2L9′S nAChR expression on synaptic transmission at the Schaffer collateral (SC) pathway by monitoring stimulus–response relationships. We found no significant differences between slices from control and Chrna2L9′S/L9′S mice in the stimulus–response relationships (Fig. 3B), suggesting that the expression of α2L9′S nAChRs had no significant effect on basal synaptic transmission at the SC pathway. We then monitored LTP induction at the SC pathway using weak theta burst stimulation (TBS) protocol. Previous studies demonstrated that endogenous ACh-mediated activation of α2* nAChR-containing OLM cells did not occur during weak TBS at the SC pathway, but bath application of nicotine activated α2*-nAChR-containing OLM cells to induce LTP (Nakauchi et al. 2007). Indeed, in both wild-type and Chrna2L9′S/L9′S mice, LTP was not induced in the absence of nicotine (Fig. 3C,F; wild-type, 102.0 ± 2.8%, N = 6 and Chrna2L9′S/L9′S, 100.7 ± 5.7%, N = 6b1). In wild-type mice, 1 µM nicotine was required for LTP induction (Nakauchi et al. 2007), whereas we found that in Chrna2L9′S/L9′ mice, 0.1 µM nicotine (Fig. 3E,F; wild type, 107.3 ± 3.4%, n = 9 versus Chrna2L9′S/L9′S, 159.3 ± 21.4%, N = 6, P = 0.033, Bonferroni-correctedb2) or 0.01 µM nicotine (Fig. 3D,F; wild type, 99.4 ± 7.8%, N = 7 versus Chrna2L9′S/L9′S, 133.7 ± 9.3%, N = 8, P = 0.045, Bonferroni-correctedb3) was sufficient for LTP induction. These findings suggest that α2L9′S nAChRs were activated by lower concentrations of nicotine to induce LTP. Overall, the findings indicate that α2* nAChRs are capable of participating in nicotinic facilitation of LTP at the SC pathway.
Preexposure-dependent contextual fear conditioning in adolescent mice
We assessed nicotine modulated hippocampus-dependent learning and memory in our Chrna2L9′S/L9′S mouse line compared with their wild-type littermate controls (heterozygous mice were not assessed in these studies) (Fig. 4A–C). The learning and memory task we used was a preexposure-dependent contextual fear conditioning procedure, which is dorsal hippocampus-dependent (Fanselow 1990; Wiltgen et al. 2001; Rudy et al. 2002; Kenney and Gould 2008; Cushman et al. 2011). In the current studies, we hypothesized that Chrna2L9′S/L9′S mice would exhibit modifications in nicotine-facilitated learning and memory. As a measure of conditioning, we examined changes in individual animals freezing from day 1 (preexposure) to day 3 (context test) using matched-pair t-tests (Bonferroni-corrected for multiple comparisons holding α = 0.05b) ((**) P = 0.002, Bonferroni-corrected; (***) P = 0.001, Bonferroni-corrected, (****) P = 0.0004, Bonferroni-corrected, N = 12–16 per group) (Fig. 4c). A significant enhancement of freezing occurred for all genotypes and drug treatments on context test, except for the Chrna2L9′S/L9′S saline-treated mice (Fig. 4B). For day 2, using a two-way ANOVA, we observed no main effects for Genotype (F(1,52) = 0.07, P = n.s.d1), Drug (Nicotine versus Saline) (F(1,52) = 1.68, P = n.s.d2), or Genotype × Drug (F(1,52) = 1.51, P = n.s.d3) interaction (Fig. 4C). For day 3, a two-way ANOVA revealed a Genotype × Drug Treatment (Nicotine versus Saline) interaction (F(1,52) = 6.12, P = 0.02, N = 12–16 per groupe1, Fig. 4B). Bonferroni-corrected post hoc analysis demonstrated saline-treated Chrna2L9′S/L9′S mice had significantly reduced freezing behavior when compared with saline-treated wild-type mice (P = 0.03e2, Fig. 4B). The reduced freezing behavior in Chrna2L9′S/L9′S mice was absent after nicotine treatment. The findings support the conclusion that saline-treated Chrna2L9′S/L9′S mice failed to exhibit enhanced freezing on day 3 when compared with day 1, likely due to a deficit to acquire and encode appropriate contextual representations during preexposure day 1. We refer to this loss of freezing behavior in saline-treated Chrna2L9′S/L9′S mice as a baseline deficit in hippocampus-dependent learning and memory.
Figure 4.

Chrna2L9′S/L9′S mice exhibit deficits in a hippocampus-dependent learning and memory preexposure contextual fear conditioning behavioral task. (A) On day 1 (preexposure, PE), adolescent mice were administered saline or nicotine (0.09 mg/kg/inj, i.p.) 3–5 min prior to being preexposed to a novel environment for 8 min where freezing and automated locomotor activity was recorded. On day 2 (Immediate-Shock, IS), mice received a 2-sec 0.75-mA shock and shock reactivity was assessed. On day 3, (Context Test, CT) animals were administered a second vehicle or nicotine injection 3–5 min prior to being tested for freezing behavior for 8 min. (B) As a measure of conditioning, we examined changes in individual animals freezing from day 1 (preexposure) to day 3 (context test) using matched-pair t-tests (Bonferroni-corrected for multiple comparisons holding α = 0.05a). Bonferroni-corrected post hoc comparisons illustrate that (i) saline-treated adolescent Chrna2L9′S/L9′S mice exhibited deficits in the enhancement freezing behavior post-shock, and (ii) nicotine treatment enhanced freezing behavior on context day in adolescent Chrna2L9′S/L9′S mice. (**) P = 0.002 Bonferroni-corrected; (***) P = 0.001, Bonferroni-corrected, (****) P = 0.0004, Bonferroni-corrected, N = 12–16 per group. For day 3 (CT), post hoc analysis demonstrates saline-treated wild-type mice have significantly greater freezing behavior than Chrna2L9′S/L9′S saline-treated mice. N = 12–16 per group (*) P = 0.03, Bonferroni-corrected. (C) No significant effects were observed for genotype or drug treatment on day 2 (IS) in adolescent Chrna2L9′S/L9′S mice.
Activity measures in Chrna2L9′S/L9′S mice
We tested whether or not changes in learning and memory (as assessed by freezing behavior) were confounded by modifications in other types of behaviors, including locomotor, sensory, and anxiety behavior (Fig. 5). After an acute treatment to saline or nicotine, we measured automated locomotor activity (a robust measure of locomotor activity, Cushman et al. 2011), rearing, mid-line crosses (exploratory behavior), center and perimeter time (anxiety behavior) as animals were preexposed to the conditioning chamber (i.e., a novel environment) on day 1. We also assessed shock reactivity (sensory response) on day 2 across the 2 sec of the 0.75 mA shock and the 2 sec after.
Figure 5.

Locomotor (rearing/vertical, mid-line crosses/vertical, automated vertical/horizontal), anxiety (center/perimeter time) and sensory response (shock reactivity) were assessed in wild-type littermate control and Chrna2L9′S/L9′S mice. Behaviors were assessed in the contextual fear conditioning chamber during the preexposure period on day 1 (PE, Fig. 5A). Shock reactivity was assessed on day 2 (IS, Fig. 5A) using the Med Associated Automated video tracking system. (A) For rearing behavior, Bonferroni-corrected post hoc comparisons illustrate saline-treated Chrna2L9′S/L9′S mice exhibited enhanced vertical movements versus saline and nicotine-treated wild-type mice littermate control mice. N = 12–16 per group, (**) P = 0.02, (*) P = 0.049 B. For mid-line crosses, Chrna2L9′S/L9′S mice exhibited enhanced activity, independent of drug treatment. N = 28 per group, (**) P = 0.002 for mid-line crosses. (C,D) For anxiety, center or perimeter time did not differ in Chrna2L9′S/L9′S of wild-type littermate control mice, independent of drug treatment. N = 12–16 per group. (E) For automated locomotor activity, Chrna2L9′S/L9′S mice exhibited enhanced activity, independent of drug treatment. N = 28 per group, (**) P = 0.004 for automated locomotion. (F) For sensory response (shock reactivity), Chrna2L9′S/L9′S versus wild-type littermate control mice exhibited enhanced shock reactivity on day 2, independent of drug treatment on preexposure day 1. N = 28 per group, (*) P = 0.02.
For rearing (exploratory vertical movement), two-way ANOVA revealed a main effect for Genotype (F(1,52) = 5.75, P = 0.02, N = 28 per groupf1) and an interaction for Genotype × Drug Treatment (F(1,52) = 4.91, P = 0.03, N = 12–16 per groupf2). Bonferroni-corrected t-test post hoc analysis revealed that saline-treated Chrna2L9′S/L9′S mice had significantly greater rearing behavior than wild-type saline (P = 0.02) and nicotine-treated mice (P = 0.049), but not nicotine-treated Chrna2L9′S/L9′S mice (P = 0.06) (Fig. 5A). For mid-line crossing behavior (exploratory horizontal movement), two-way ANOVA revealed a main effect for Genotype (F(1,52) = 10.89, P = 0.002, N = 28 per groupg1) and no interaction for Genotype × Drug Treatment (F(1,52) = 3.33, P = n.s., N = 12–16 per groupg2, Fig. 5B). Significant main effect analysis for Genotype revealed that Chrna2L9′S/L9′S mice have increased mid-line crosses when compared with wild-type mice ((**) P = 0.002). For anxiety behavior, we quantified center and perimeter time on day 1, within the contextual fear conditioning chamber. An increase in center time was regarded as reduced anxiety. For center (and perimeter) time anxiety measures, a two-way ANOVA demonstrated no main effect for Genotype (F(1,52) = 2.92, P = n.s.), Drug (Nicotine versus Saline) (F(1,52) = 0.18, P = n.s.), or Genotype × Drug (F(1,52) = 1.54, P = n.s.h) interaction (Fig. 5C,D). The findings highlighted that anxiety measures were not influenced by genotype or acute drug treatment. Independent of genotype or drug treatment, mice spent significantly greater time on the perimeter versus center of the open field chamber (524.5 sec in perimeter versus 75.5 sec in center, P = 0.0001).
For automated locomotor activity, a two-way ANOVA demonstrated a main effect for Genotype (F(1,52) = 9.37, P = 0.004, N = 28 per groupi), but no other main effect or interaction. Significant main effect analysis for Genotype demonstrated Chrna2L9′S/L9′S versus wild-type mice had greater automated locomotor activity on day 1, independent of drug treatment (Fig. 5E). Thus, for both automated locomotor activity and mid-line crosses, hyperactivity was observed in Chrna2L9′S/L9′S mice (versus wild-type littermate controls), independent of drug treatment. Rearing behavior only resolved hyperactivity in saline-treated Chrna2L9′S/L9′S mice. Hyperactivity may provide additional evidence of impaired hippocampal function in the Chrna2L9′S/L9′S mice, as one effect of hippocampal lesions is hyperactivity (Anagnostaras et al. 1999; Godsil et al. 2005). On the other hand, it is possible that the hyperactivity observed on day 1 in Chrna2L9′S/L9′S mice confounds learning and memory deficits on day 3. If this was the case, we predicted that there would be a significant correlation between activity on day 1 and freezing behavior on day 3, with greater activity associated with reduced freezing behavior. Given our learning and memory findings, we predicted that the relationships should be dependent on both genotype and drug treatment, since nicotine treatment rescued the baseline deficit selectively in Chrna2L9′S/L9′S mice. When we separated by genotype and drug treatment, significant correlations were not observed for any groups: saline-treated Chrna2WT mice: R2 = 0.004, P = n.s.; nicotine-treated Chrna2WT mice: R2 = 0.003, P = n.s.; saline-treated Chrna2L9′S/L9′S mice: R2 = 0.07, P = n.s.; nicotine-treated Chrna2L9′S/L9′S mice: R2 = 0.09, P = n.s., n = 12–16 per group. The results did not illustrate selective correlations in saline or nicotine-treated Chrna2L9′S/L9′S mice. Thus, we do not believe that hyperactivity on day 1 confounded interpretations of our learning and memory results on day 3.
For changes in sensory response, we assessed the impacts of shock reactivity for all groups of mice. For shock reactivity, a two-way ANOVA revealed a significant effect for Genotype (F(1,52) = 5.81, P = 0.02, N = 28 per groupj) and no other main effect or interaction. Significant main effect analysis for Genotype demonstrated that Chrna2L9′S/L9′S mice have greater shock reactivity versus wild-type control mice, independent of drug exposure (Fig. 5F). Increased shock reactivity may indicate increased pain sensitivity (i.e., sensory response). In Chrna2L9′S/L9′S mice, however, this would be expected to produce increased freezing, rather than the decrease that was observed in saline-treated Chrna2L9′S/L9′S mice, arguing that altered sensory response cannot explain the results. Thus, our results suggest that our learning and memory observations are not confounded by modifications in other types of behaviors, including locomotor, sensory, or anxiety behaviors.
Adolescent Chrna2KO stereotypical behavioral characteristics
We have previously described the genetic design, absence of gross anatomical morphological changes and behavioral characterization of adult Chrna2KO mice (Lotfipour et al. 2013). Adolescent Chrna2KO mice had no differences in weight gain or stereotypical behaviors observed in a novel environment compared with wild-type littermate control mice (Fig. 6k). Thus, no growth or development modifications were observed in adolescent Chrna2KO mice.
Figure 6.
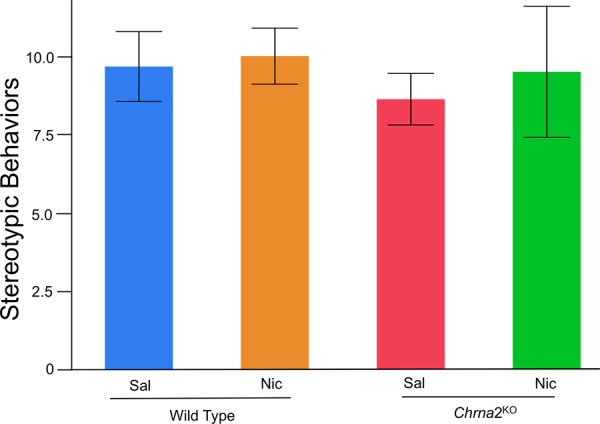
Stereotypic behaviors were assessed in wild-type and Chrna2KO mice on day 1. Results illustrated that stereotypic behaviors do not differ based on drug treatment or genotype. N = 5–8 per group.
Preexposure-dependent contextual fear conditioning in adolescent Chrna2KO mice
Using Chrna2KO mice, which exhibit an absence of nicotine-induced facilitation and depression of LTP during adolescence (Nakauchi et al. 2007), we predicted that we would observe an absence of nicotine-facilitated learning and memory in these mutant mice (Fig. 7). As a measure of conditioning, we examined changes in individual animals freezing from day 1 (preexposure) to day 3 (context test) using Bonferroni-corrected matched-pair t-tests. Data demonstrated a significant enhancement of freezing behavior in all groups ((*) P = 0.048, Bonferroni-corrected, (**) P = 0.01, Bonferroni-corrected, (***) P = 0.004, Bonferroni-corrected, N = 5–8 per group, Fig. 7A). For day 2, using a two-way ANOVA, a main effect was observed for Genotype (F(1,21) = 5.20, P = 0.03l1) as well as a Genotype × Drug Treatment (Nicotine versus Saline) interaction (F(1,21) = 6.88, P = 0.02, N = 5–8 per groupl2). Bonferroni-corrected t-test post hoc analysis revealed that wild-type nicotine-treated mice had significantly greater freezing behavior than nicotine-treated Chrna2KO mice (P = 0.02l3, Fig. 7B). The findings suggest that nicotine-treated adolescent Chrna2KO mice had a deficit in learning and memory on day 2, when compared with nicotine-treated Chrna2WT mice. For day 3, a two-way ANOVA revealed a main effect for Genotype (F(1,21) = 6.56, P = 0.02m) but no Genotype × Drug Treatment (Nicotine versus Saline) interaction. Significant main effect analysis for Genotype revealed that wild-type mice had greater freezing behavior than Chrna2KO mice, independent of drug treatment (Fig. 7). Overall, the findings highlighted that Chrna2KO mice exhibited deficits in contextual learning during the preexposure that were not rescued by acute nicotine exposure.
Figure 7.

Deficits in Chrna2KO adolescent mice were observed on the preexposure-dependent contextual fear conditioning paradigm. As a measure of conditioning, we examined changes in individual animals freezing from day 1 (preexposure) to day 3 (context test) using matched-pair t-tests. (A) day 1 to day 3 analysis illustrated that all mice condition (i.e., have significant enhancement of freezing behavior on day 3 versus day 1), independent of genotype or drug treatment. (*) P = 0.048, Bonferroni-corrected, (**) P = 0.01, Bonferroni-corrected, (***) P = 0.004, Bonferroni-corrected, N = 5–8 per group. On contextual testing day (day 3, CT), Chrna2KO mice had reduced freezing behavior, independent of drug treatment. N = 5–8 per group, (*) P = 0.02. (B) For day 2 (IS), post hoc analysis revealed that wild-type nicotine-treated mice had significantly greater freezing behavior than nicotine-treated Chrna2KO mice. Bonferroni-corrected, N = 11–14 per group, (*) P = 0.02.
Activity measures in Chrna2KO mice
We tested whether or not changes in learning and memory (as assessed by freezing behavior) were confounded by modifications in other types of behaviors, including locomotor, sensory, or anxiety-related behaviors (Fig. 8). After an acute treatment to saline or nicotine, we measured rearing, mid-line crosses, center time, and automated locomotor activity as animals were preexposed to the conditioning apparatus on day 1. We also assessed shock reactivity on day 2 (Fig. 8F). For rearing (exploratory vertical movement) behavior, two-way ANOVA failed to reach a significant main effect or interaction for Genotype × Drug Treatment (F(1,21) = 4.23, P = 0.052, N = 5–8 per groupn, Fig. 8A). For mid-line crossing behavior (exploratory horizontal movement), two-way ANOVA revealed a main effect for Genotype (F(1,21) = 7.34, P = 0.01o1), a main effect for Drug Treatment (F(1,21) = 4.55, P = 0.04o2) and an interactive effect for Genotype × Drug Treatment (F(1,21) = 4.97, P = 0.04, N = 5–8 per groupo3, Fig. 8B). Bonferroni-corrected t-test post hoc analysis revealed nicotine-treated Chrna2KO had significantly greater mid-line crosses than all other groups (P = 0.02o4). For anxiety behavior, we assessed center (Fig. 8C) and perimeter time (Fig. 8D) within the contextual fear conditioning chamber. For anxiety measures, a two-way ANOVA demonstrated no main effect for Genotype (F(1,21) = 0.35, P = n.s.), Drug (Nicotine versus Saline) (F(1,21) = 0.97, P = n.s.), or Genotype × Drug (F(1,52) = 0.74, P = n.s.p) interaction (Fig. 8C,D). The findings highlighted that anxiety, as measured by an increase in center time, was not influenced by genotype or acute drug treatment. Independent of genotype or drug treatment, mice spent significantly greater time on the perimeter versus center of the open field chamber (522.1 sec in perimeter versus 77.9 sec in center, P = 0.0001).
Figure 8.

Locomotor (rearing/vertical, mid-line crosses/vertical, automated vertical/horizontal), anxiety (center/perimeter time), and sensory response (shock reactivity) were assessed in wild-type littermate control and Chrna2KO mice. Behaviors were assessed in the contextual fear conditioning chamber during the preexposure period on day 1 (PE, Fig. 8A). Shock reactivity was assessed on day 2 (IS, Fig. 8A) using the Med Associated Automated video tracking system. (A) For rearing behavior, no main effects or interactions were observed, although there was a trend for an interaction. N = 5–8 per group. (B) For mid-line crosses, Chrna2KO versus wild-type littermate control mice exhibit enhanced activity on day 1, independent of drug treatment, N = 11–14 per group, (**) P = 0.01. (C–E) For anxiety, center (C) or perimeter time (D) did not differ in Chrna2KO versus wild-type littermate control mice, independent of drug treatment. N = 11–14 per group. (E) For automated locomotor activity, Chrna2KO versus wild-type littermate control mice exhibit enhanced automated activity on day 1, independent of drug treatment, N = 11–14 per group, (**) P = 0.01. (F) For sensory response (shock reactivity), Bonferroni-corrected t-test post hoc analysis demonstrated that Chrna2KO versus wild-type mice did not exhibit any differences in shock reactivity on day 2 with or without drug treatment, N = 5–8 per group.
For automated locomotor activity, two-way ANOVA demonstrated a main effect for Genotype (F(1,21) = 9.09, P = 0.01q), but no other main effect or interaction. Greater activity was observed on day 1 in Chrna2KO versus wild-type mice, independent of drug treatment (Fig. 8E). To determine whether hyperactivity confounded our learning and memory results, we assessed simple linear correlations between activity on day 1 and freezing behavior on day 3 of context test. When we separated by genotype and drug treatment, significant correlations were not observed for any groups: saline-treated Chrna2WT mice: R2 = 0.11, P = n.s.; nicotine-treated Chrna2WT mice: R2 = 0.36, P = n.s.; saline-treated Chrna2KO mice: R2 = 0.02, P = n.s.; nicotine-treated Chrna2KO mice: R2 = 0.002, P = n.s., n = 5–8 per group. Additionally, when we collapsed by drug treatment, again we observed no significant correlation between activity on day 1 and freezing behavior on day 3 of context test: Chrna2WT mice: R2 = 0.12, P = n.s.; Chrna2KO mice: R2 = 0.02, P = n.s.; n = 11–14. The results did not illustrate selective correlations in Chrna2KO mice. Thus, we do not believe that hyperactivity on day 1 confounded interpretations of our learning and memory results on day 3.
For shock reactivity across the 2 sec of the 0.75-mA shock and the 2 sec after, two-way ANOVA revealed a significant interaction for Genotype × Drug Treatment (F(1,21) = 9.05, P = 0.01, N = 5–8 per groupr) with no main effects observed. Bonferroni-corrected t-test post hoc analysis failed to reach statistical significance for any of the comparisons (Fig. 8F). The findings provide convergent evidence for impaired hippocampal function, similar to the pattern seen in Chrna2L9′S/L9′S mice. The findings strongly suggest that our learning and memory observations are not confounded by modifications in other types of behaviors.
Discussion
The current study tested the hypothesis that genetic mutations of α2* nAChRs in Chrna2L9′S/L9′S, but not Chrna2KO mice, would produce modifications in nicotine-induced hippocampus-dependent learning and memory. When we substituted a serine for a leucine (L9′S) in the α2 subunit (encoded by the Chrna2 gene) to make a hypersensitive nAChR, we confirmed the mutation potentiates acetylcholine-induced ion channel flux in oocytes. Subsequently, in adolescent Chrna2L9′S/L9′S mice, we observed a potentiation of nicotine-induced facilitation of synaptic plasticity in hippocampal electrophysiological studies at subthreshold doses of nicotine (0.01 and 0.1 µM). Using a preexposure-dependent contextual fear paradigm (Wiltgen et al. 2001; Rudy et al. 2002; Stote and Fanselow 2004; Cushman et al. 2011), we observed an impairment in freezing behavior (i.e., baseline deficit) in both adolescent Chrna2L9′S/L9′S and Chrna2KO mice. A subthreshold dose of nicotine (0.09 mg/kg per injection), was able to rescue the baseline deficit in Chrna2L9′S/L9′S, but not Chrna2KO mice, which have potentiated and impaired nicotine-facilitated CA1 hippocampal synaptic plasticity, respectively. The findings support the hypothesis that α2* nAChRs influence hippocampus-dependent learning and memory via alterations in nicotine-facilitated CA1 hippocampal synaptic plasticity in adolescent mice (Fig. 9). These effects were not influenced by locomotor (hyperactivity), sensory (pain response), or anxiety-related measures (center time); which suggests that our learning and memory observations are not confounded by modifications in other types of behaviors.
Figure 9.

Electrophysiological and behavioral phenotypes of wild-type, Chrna2KO and Chrna2L9′S/L9′S mice. The findings indicate that α2* nAChRs are capable of participating in nicotinic facilitation of long-term potentiation in the hippocampal CA1 and influence the effects of nicotine on hippocampus-dependent learning and memory.
The α2 nAChR subunits are expressed on GABAergic OLM interneurons in the stratum oriens (SO) of the CA1 of the mouse (Nakauchi et al. 2007; Leao et al. 2012). These OLM cells are anatomically positioned such that they can regulate neurotransmitter release and local circuit activity. Activation of these neurons demonstrates that they can regulate long-term potentiation via GABA release (Leao et al. 2012). Interestingly, 50% or more of the OLMα2 neurons are somatostatin (SOM) positive neurons within the SO of the CA1 (Jia et al. 2010; Leao et al. 2012), highlighting that the Chrna2-positive neurons may represent a distinct molecular genetic target within the hippocampus (Leao et al. 2012). Optogenetic studies using blue light activation of OLMα2 cells expressing channelrhodopsin in Chrna2-Cre mice, confirm that these GABAergic interneurons influence long-term potentiation within the CA1 hippocampus through GABAergic release (Leao et al. 2012). Based on these findings, our current results provide additional evidence for the hypothesis that α2 nAChR subunit assemblies within the OLMα2 CA1 hippocampal brain region have functional consequences in synaptic plasticity, which may underlie hippocampus-dependent learning and memory in mice (Kleeman et al. 2016).
Previous studies have demonstrated that deletion of the α2 nAChR subunit results in enhancements in the acquisition of intravenous nicotine self-administration (in the first 2 d after transitioning from food reinforcement), context dependent nicotine withdrawal, sex-dependent nicotine-facilitated fear conditioning (Lotfipour et al. 2013), and hippocampus-dependent memory impairments (Kleeman et al. 2016). Using a dorsal hippocampus-dependent behavioral task, known as the preexposure-dependent contextual fear conditioning, we extended these results and demonstrated that adolescent male hypersensitive α2 nAChR subunit mutant mice displayed baseline deficits in hippocampus-dependent learning and memory. Acute low-dose nicotine exposure (0.09 mg/kg, i.p; equivalent to blood plasma levels reached by an average cigarette (∼10 ng/mL) (Portugal et al. 2012)) rescued learning and memory deficits during adolescence in the hypersensitive α2 nAChR mice. The result confirms that modifications in the α2 nAChR subunit induced functional changes in hippocampus-dependent learning and memory during adolescence. Based on the in vivo electrophysiology studies in the Chrna2L9′S/L9′S mutant mouse line, we did not predict that baseline behavior would be different, as we found no significant differences between slices from control and Chrna2L9′S/L9′S mice in the stimulus–response relationships (Fig. 3B). When we performed the behavioral tasks, however, we observed a hippocampal deficit in saline-treated Chrna2L9′S/L9′S versus wild-type mice (i.e., reduced freezing behavior), which was rescued by an acute exposure to nicotine. We propose the mechanisms mediating the nicotine-induced rescue of learning and memory are through enhanced potentiation of CA1 hippocampus-synaptic plasticity, via direct actions on GABAergic OLMα2 neurons. Alternatively, the increase in freezing may be due to excessive plasticity in response to nicotine. Future studies are needed to assess the mechanisms mediating the baseline deficit in hippocampus-dependent learning in Chrna2L9′S/L9′S and Chrna2KO mice. We speculate this may be mediated via alterations in cholinergic tone, induced by enhanced or absent activity within α2-containing GABAergic interneurons. The alteration in activity may influence medial septum and/or CA1 hippocampal OLMα2 neurons that regulate fimbria fornix septal-cholinergic input, thereby disrupting learning and memory (Ishii et al. 2005; Lovett-Barron et al. 2014). The hypothesis is derived from studies illustrating that dendritic inhibition of hippocampal neurons influence fear memory (Lovett-Barron et al. 2014).
The interpretation of our learning and memory results is based on assessing freezing behavior on days 2 and 3 using the preexposure-dependent contextual fear conditioning paradigm. A failure to exhibit enhanced freezing on days 2 and 3 is likely due to the inability to acquire appropriate contextual representations during preexposure day 1. In our results, greater variability in freezing behavior was observed on day 2 after immediate-shock versus day 3 of contextual testing in Chrna2L9′S/L9′S and Chrna2KO mice. Previous studies using alcohol demonstrated that day 2 freezing behavior does not always predict day 3 freezing data (Cushman et al. 2011). While alcohol was able to reduce freezing on day 2 after an immediate-shock, it had no effect on day 3. The findings highlight that alcohol may impair the mechanisms mediating pattern completion in the presence of an immediate-shock; this deficit is not present during pattern completion of the shock–context pairing on day 3. Pattern completion refers to the ability of the hippocampus to use a subset of features experienced previously to recall or activate a conjunctive representation (Rudy et al. 2002, 2004; Matus-Amat et al. 2004; Nakashiba et al. 2012). In our current analysis, we did not observe a matched-pair correlation between days 2 and 3 freezing behavior in Chrna2L9′S/L9′S and Chrna2KO mice. The data confirmed that freezing behavior on day 2 did not predict freezing on day 3. The prior results with alcohol are in contrast with our current study, which illustrate that behavioral deficits are more prevalent on day 3 of contextual testing. This is likely due to a difference in methods, i.e., alcohol versus nicotine treatment.
For studies performed in the absence of a preexposure day 1, zero to minimal freezing behavior (i.e., immediate-shock deficit) is regularly observed post-shock, illustrating a deficit in contextual encoding. In the presence of preexposure day 1, a rescue of the immediate-shock deficit is observed, associated with a significant enhancement of freezing behavior post-shock. The findings highlight that preexposure to a context develops a conjunctive representation of that environment which can be recalled on immediate-shock day to enhance freezing behavior (Rudy et al. 2002). In the current analysis, enhanced freezing behavior (>15%) was observed on day 2 in Chrna2L9′S/L9′S and Chrna2KO mice and their wild-type littermate control mice. The only exception was minimal freezing behavior on day 2 of nicotine-treated Chrna2KO mice. For all but one group, the data suggest that the immediate-shock deficit has been rescued in these mice given preexposure day 1. Therefore, the formation of a conjunctive representation of the context was observed in all groups, except for nicotine-treated Chrna2KO mice. This would suggest that the ability of a subset of features to activate a conjunctive representation in the presence of a shock (i.e., pattern completion) is normal in Chrna2KO and Chrna2L9′S/L9′S mice, with the exception of nicotine-treated Chrna2KO mice (Rudy et al. 2002). However, when memory of the shock–context pairing was tested on day 3, a deficit in freezing behavior was observed in saline-treated Chrna2L9′S/L9′S and all Chrna2KO mice. Thus, we have reason to believe that shock–context memory is altered by α2* nAChRs (Rudy et al. 2002, 2004; Matus-Amat et al. 2004). Prior results have demonstrated that Chrna2KO mice exhibit deficits in hippocampus-dependent memory (Kleeman et al. 2016). Thus, in combination with current results, day 3 data appear to best represent further evidence that α2* nAChRs are important in hippocampus-dependent learning and memory. We speculate that the deficits observed on day 3 versus day 2 are mediated through mechanisms that relate to more permanent memory traces (Frankland et al. 2001).
Please note, in supplementary studies, we have assessed the impacts of preexposure on the contextual fear conditioning procedure in adolescent wild-type and Chrna2KO mice (S Lotfipour et al., in prep.). The methods used for the no preexposure control group were similar to those described in our article, except animals were not given the 10-min preexposure on day 1. Based on previous studies (Cushman et al. 2011), we predicted and observed that animals in the no preexposure control group would fail to exhibit freezing behavior post-shock (day 2) and show minimal increase (<10%) on context test (day 3). The findings help validate our preexposure model used in our current studies. We have not included these data in the current article as they are integrated in a supplemental study.
We believe our effects demonstrate direct hippocampus-dependent learning memory modifications rather than indirect effects (e.g., locomotor hyperactivity, sensory, or anxiety-related behaviors). When we measured exploratory behavior in the adolescent hypersensitive Chrna2L9′S/L9′S and Chrna2KO mice, our results demonstrated that the mutant mice have heightened activity. If hyperactivity on day 1 was confounding learning and memory deficits on day 3, we predicted that nicotine-treated Chrna2L9′S/L9′S or Chrna2KO mice independent of nicotine treatment, would illustrate significant association between hyperactivity and freezing behavior. When Chrna2L9′S/L9′S, Chrna2KO or their wild-type littermate control mice were separated by treatments and/or genotype, activity on day 1 did not predict freezing behavior on day 3. Given that the results did not illustrate selective correlations in nicotine-treated Chrna2L9′S/L9′S or Chrna2KO mice independent of nicotine treatment, we conclude that hyperactivity on day 1 does not confound interpretations in our learning and memory results on day 3. Indeed, the hyperactivity that is induced by dorsal hippocampal lesions has been argued to result from the same learning impairment that disrupts contextual learning: impaired ability to encode the multisensory features of the environment into a stable contextual representation, i.e., the “common-deficit hypothesis” (Godsil et al. 2005). Further exploration is needed on whether similar mechanisms as proposed by Godsil and colleagues may be influencing our results. Prior results have demonstrated that Chrna2KO mice exhibit deficits in hippocampus-dependent memory (Kleeman et al. 2016). Thus, our findings fit well with recent data in the Chrna2KO mice as assessed via an alternative behavioral task, i.e., the object location but not the hippocampus-independent novel object recognition task (Kleeman et al. 2016). If locomotor modifications confound learning and memory deficits, we would predict that both behaviors would be altered in Chrna2KO mice, which was not observed. Thus, we conclude that the hippocampus-dependent learning and memory observations in the current studies were not confounded by modifications in locomotor hyperactivity.
Our data also suggest that our learning and memory results are not influenced by changes in sensory response (i.e., pain sensitivity as determined by shock reactivity) or anxiety-related behavior (center time). The preexposure-dependent contextual fear procedure was specifically designed to minimize any potential analgesic effects of memory modulating drugs by separating drug administration from shock presentation. For sensory response, we observed increased shock reactivity in Chrna2L9′S/L9′S mice, independent of drug exposure. If increased shock reactivity is a measure of enhanced sensory response, we would predict increased freezing behavior during contextual testing in saline-treated Chrna2L9′S/L9′S mice; rather, we observed a decrease in freezing behavior. No significant effects were observed for shock reactivity in Chrna2KO mutant mice. Thus, our findings argue that altered sensory response cannot explain changes in freezing behavior. Previous studies in adult Chrna2KO mice have indicated differences in nicotine-induced hyperalgesia in the hotplate test, but not when assessing baseline or nicotine-induced analgesia to the tail withdrawal or the formalin test (Lotfipour et al. 2013). The findings suggest that nicotine-induced pain response to selective behaviors may be influenced in Chrna2KO and Chrna2L9'S/L9′S mice. However, given that (i) our nicotine exposure did not occur during day 2 immediate-shock presentation, (ii) adolescent Chrna2KO mice did not exhibit significant differences in shock reactivity, and (iii) Chrna2L9'S/L9′S mice had increased shock reactivity with reduced (instead of increased) freezing behavior in saline-treated mice, we do not believe that pain sensitivity is a plausible explanation of our results. In regards to anxiety-related behaviors, our current studies did not observe differences in center time in Chrna2L9'S/L9′S or Chrna2KO mice. Given that ventral (versus dorsal) hippocampal circuits are more involved in mediating anxiety-related behaviors and we did not observe differences in center time, we are confident that our results are not confounded by changes in anxiety-related circuits. Therefore, indirect effects, including sensory response to shock or anxiety, do not appear to account for our findings. Rather they implicate learning and memory processes. Our findings are consistent with previous studies, which found that hippocampal nAChR antagonists disrupt nicotine-facilitated learning and memory processing, as tested by contextual fear conditioning (Davis et al. 2007). Furthermore, genetic studies have demonstrated an absence of nicotine-facilitated contextual fear conditioning in β2−/− (but not α7−/−, β3−/−, or β4−/−) mutant mice (Wehner et al. 2004; Davis and Gould 2007; Semenova et al. 2012). Such findings suggest that nAChRs assembled from select subunits regulate nicotine's effects on learning and memory processing.
Our results are not without limitations. For example, authors have argued that nAChR expression and/or function in oocytes may not be identical to native tissues (Buller and White 1990; Sivilotti et al. 1997). In particular, differences in nAChR subunit composition and/or molar concentrations of individual subunits are known to influence “channel open times, ion conductance, desensitization rates” as well as the “sensitivity to agonists and antagonists” (Boulter et al. 1987; Deneris et al. 1988; Wada et al. 1988; Papke et al. 1989; Luetje and Patrick 1991; Paradiso et al. 2001; Grady et al. 2007; Lipovsek et al. 2008; McIntosh et al. 2009; Jin and Steinbach 2011, 2015). Neurons in animal and human studies contain a diversity of neuronal α/β nAChR subunits at different levels, which would differently alter the subunit composition and stoichiometry. For example, interneurons of the stratum oriens (where OLM neurons reside) contain more than α2 and β2 nAChR subtypes (Sudweeks and Yakel 2000; Jia et al. 2009). Thus, applicability of our studies in oocytes to function in neurons in vivo needs to be interpreted cautiously. Our studies in oocytes were applied only as an initial screen to confirm the functionality of the serine for a leucine (L9′S) substitution in the α2 nAChR subunit. Future studies could use, for example, single-cell electrophysiology and neurotransmitter release assays or in vivo imaging coupled with behavioral techniques to better confirm the biophysical and pharmacological properties of the hypersensitive nAChR in Chrna2L9'S/L9′S mice (Sudweeks and Yakel 2000; Grady et al. 2007; Jia et al. 2009; Lovett-Barron et al. 2014). Another potential limitation authors have reported regarding oocytes studies is the possibility of low level endogenous expression of peripheral α1 nAChR subunits (Buller and White 1990). Such a finding will not have an impact in our studies, given that neuronal α2 or β2 nAChR subunits when expressed, alone, in oocytes do not form functional nAChRs (Boulter et al. 1987; Deneris et al. 1988; Wada et al. 1988). Prior work has used similar oocyte expression methodologies assessing knock-in mutations for different nAChRs to validate behavioral outcomes in mice and humans (Lester et al. 2003; Klaassen et al. 2006; Lipovsek et al. 2008). Thus, the oocyte expression studies have been helpful in identifying function in vivo, with our current studies providing supportive evidence for their validation.
In addition to limitations on oocyte studies, other concerns should be highlighted. In particular, heightened expression of α2* nAChRs appear during the first 2 wk of rodent life, while our behavioral and electrophysiological studies took place during adolescence (i.e., 4 wk of life). Therefore, learning and memory modifications induced by developmental nicotine exposure could be potentiated if nicotine was delivered during this heightened period of expression during the first weeks of early postnatal development. Furthermore, it is possible that Chrna2L9′S/L9′S mice have modified α2* nAChRs expression patterns within the brain, particularly since prior results demonstrate that knock-in mutations within the TM2 may decrease receptor expression, but not mRNA levels (O'Neill et al. 2013). Therefore, future studies should aim to determine whether the Chrna2L9′S/L9′S mice exhibit differences in nAChR binding within the brain. Given the selectivity of α2* nAChRs within the stratum oriens GABAergic OLM interneurons, it is possible that such effects may be negligible in the dorsal hippocampal CA1. Prior studies have demonstrated that maternal nicotine exposure does reduce the number of α2* cells and α2 nAChR subunit mRNA expression by 10%–15 % in the dorsal hippocampal CA1, i.e., one brain region where the current electrophysiological slice recording took place in the current study (Chen et al. 2016). Therefore, it is possible that similar effects would be observed within the Chrna2L9′S/L9′S mice, with likely more significant consequences observed within the ventral (versus dorsal) hippocampus (Chen et al. 2016). Given our anxiety results, we do not believe that ventral hippocampus nAChRs would confound interpretation of our current findings. Indeed, we found no significant differences between slices from control and Chrna2L9′S/L9′S mice in the stimulus–response relationships in our electrophysiological studies within the dorsal/intermediate CA1 hippocampus (Fig. 3B), suggesting that the modified expression of α2L9′S/L9′S nAChRs had no significant effect on basal synaptic transmission at the SC pathway.
Another limitation of our current studies is that we have not assessed sex-dependent effects. Given prior findings that emotional memory processing is modified in a sex-dependent manner in adult Chrna2KO mice (Lotfipour et al. 2013), future studies should assess sex-dependent effects in hippocampal-dependent learning and memory in adolescent Chrna2KO and Chrna2L9′S/L9′S mice. In addition, future studies should assess nicotine dose–response relationships and age-dependent effects using the Chrna2KO and Chrna2L9′S/L9′S mice. Our current results used the 0.09 mg/kg/injection nicotine dose based on adult data illustrating enhancement in wild-type mice (Kenney and Gould 2008; Kutlu et al. 2016). Therefore, we were initially surprised that 0.09 mg/kg/injection had no effects in wild-type mice. Subsequent studies, however, confirmed our findings in adolescent C57Bl/6J mice using the 0.09 mg/kg/injection nicotine dose (Kutlu et al. 2016). Kutlu et al. (2016), have performed a nicotine dose response during adolescence and adulthood using the preexposure-dependent contextual fear conditioning paradigm. They have demonstrated that adolescent C57BL/6J wild-type mice are less sensitive to low-dose nicotine exposure, at the 0.09 mg/kg dose, which we have replicated with our findings in adolescent wild-type mice.
Our findings illustrated that while low-dose nicotine exposure had no effect on adolescent wild-type mice, as reported (Kutlu et al. 2016), nicotine exposure was able to rescue the baseline deficit in Chrna2L9′S/L9′S, but not Chrna2KO mice. The findings support the rationale that the hypersensitive mutation potentiated synaptic plasticity in addition to hippocampus-dependent learning and memory at nicotine doses that did not influence wild-type mice. Given that nicotine pretreatment was not able to rescue the baseline deficit in hippocampal-dependent learning and memory in Chrna2KO mice, we propose that this was mediated by the known absence of nicotine-facilitated synaptic plasticity (Nakauchi et al. 2007). Thus, nicotine-facilitated synaptic plasticity plays an important role in how nicotine can rescue baseline deficits in learning and memory. Whether these results remain at higher doses of nicotine needs to be tested in future studies. Because the Chrna2KO mice were backcrossed for >12 generations (>99.98% of the percentage of the C57BL/6J host strain) while the Chrna2L9′S/L9′S mice were backcrossed for significantly fewer generations (N = 3–4, 75%–87.5% of C57BL/6J host strain), the reader is cautioned to the interpretations made when comparing and contrasting the effects of L9′S with those of the null mutation (% host strain attained from Fox and Witham 1997).
Materials and Methods
All animal procedures were conducted in accordance with the National Institute of Health Guide for the care and use of laboratory animals and with protocols approved by the Institutional Animal Care and Use Committee at the University of California, Los Angeles and Irvine. Mice used in this study were placed on a 12-h dark–light cycle with ad lib access to food and water. Mice were group housed and tested during the light phase of the light cycle.
Animals
Construction and characterization of the Chrna2 null mutant mouse line (backcrossed to C57BL/6J, N17-18 generations, RRID:MGI_3790875) were described earlier (Lotfipour et al. 2013). Chrna2L9′S/WT mice (backcrossed to C57BL/6J, N3-4 generations) were engineered similar to (Taranda et al. 2009), with the exception that (i) an FRT flanked positive selection marker phosphoglycerate kinase gene promoter-neomycin resistance (PGK-NEO) cassette was inserted within an AgeI restriction enzyme genomic site, upstream of the fifth exon of the Chrna2 gene (Fig. 1A) and (ii) a gene synthesized (GENWIZ) Aat II/NdeI segment of DNA with a modified AGC codon for Serine corresponding to the 9′ amino acid within the transmembrane region 2 (TM2) (Revah et al. 1991; Labarca et al. 1995; Lester et al. 2003; Tapper et al. 2004; Plazas et al. 2005; Drenan et al. 2008) was inserted within the 5th exon of the Chrna2 gene (Fig. 1; Klaassen et al. 2006). The targeting vector was electroporated into SvJ129 embryonic stem (ES) cells and homologous recombinants were confirmed through DNA sequencing (Fig. 1B) and Southern blot (Fig. 1C) using a XmnI/NheI 1007 base pair fragment upstream of exon 1. Genomic DNA from electroporated ES cells were cut with BstZ17I/SwaI restriction enzymes with a targeted Southern blot fragment of 9500 bp (Fig. 1C). Targeting efficiency of homologous recombination within ES cells was calculated at 43% (31 of 72 ES cells had the targeted fragment). Targeted ES cells were microinjected into C57Bl/6J blastocyst embryos at the UCLA Transgenic Core and implanted into pseudopregnant female mice. Germline transmitted agouti colored offspring were DNA sequenced at the UCLA genomic core to confirm that they were carriers of the AGC/CTC heterozygous genomic modification within exon 5 (Fig. 1B). The introduction of the AGC site at the correct location within the fifth exon introduced a SacI restriction enzyme site, which acted as a quick screening strategy for confirmation of the genetic mutation (not shown). After confirming the presence of the mutation and the correct orientation of the FRT flanked PGK-NEO cassette, the mice were mated with congenic FLPeR mice (#009086, The Jackson Laboratory) to delete the positive selection marker, leaving a 34-bp FRT finger print in the intronic genomic region. PCR primers (A2L9S_1.10) GGCTAGTCAGAACAGCTGCATT and (A2L9S_1.11) GAACAAGGAAGAAATCGGCAGG (Eurofins MWG Operon LLC) were designed upstream of and downstream from the deleted PGK-NEO cassette (Fig. 1A) to provide PCR-amplified fragments for wild-type and mutant carriers (not shown).
Oocyte expression studies
The use of oocyte expression studies (Fig. 3A) was applied as an initial screen to determine the functionality of a substitution of a serine for a leucine (L9′S) in the α2 nAChR subunit. Nicotinic receptor subunits were subcloned into a pSGEM vector, which contains Xenopus 5′-UTR and 3′-UTR segments that markedly increase expression of cloned cDNAs (Liman et al. 1992; Lipovsek et al. 2008). Plasmid constructs, where the L9′ amino acid (located in TM2) was replaced either with serine, threonine, or alanine, were linearized with Nhe I to generate templates for in vitro transcription. cRNA prepared from wild-type mouse α2 plasmid, as well as the mutants L9′S, L9′A, and L9′T mutants, where coinjected in oocytes along with the wild-type mouse β2 cRNA at a 1:1 equimolar ratio (Fig. 3A). Electrophysiological recordings and concentration–response curves to ACh were performed under two-electrode voltage clamp, as described in Lipovsek et al. (2008).
Hippocampal synaptic plasticity studies
Slice preparation and extracellular field recordings within the dorsal/intermediate CA1 hippocampal slice were performed using the methods described by Nakauchi et al. (2007), Figure 3B–F. Briefly, slices of 375–400 µm were attained from 15 wild-type and Chrna2L9′S/L9′S adolescent mice (4 wk of age). Animals included in the study were: (i) C57BL/6J wild-type male mice (n = 5), (ii) Chrna2L9′S/L9′S male (n = 5) homozygous S9′S mice, (iii) Chrna2L9′S/L9′S wild-type male (n = 3) and female (n = 2) mice, with the reported “n” representing the numbers of experiments in our study. Slices from these animals were maintained in artificial cerebral spinal fluid at 30°C, containing (mM): NaCl, 124; KCl, 5; NaH2PO4, 1.25; MgSO4, 2; CaCl2, 2.5; NaHCO3, 22; and glucose, 10, and oxygenated with 95% O2 and 5% CO2. A stimulating electrode activated the glutamatergic stratum radiatum schaffer collateral projections arriving from the CA3 hippocampus. Field excitatory postsynaptic potentials (fEPSPs) were recorded within the stratum radiatum using a glass electrode filled with 2 M NaCl (3–8 MΩ). We monitored LTP induction at the SC pathway using a subthreshold stimulation protocol (weak θ burst stimulation (TBS): two θ bursts of four pulses at 100 Hz), which induces LTP in the presence, but not absence, of the activation of α2*-nAChR-expressing O/A interneurons (Nakauchi et al. 2007). To evaluate the magnitudes of LTP, the mean value for the slopes of fEPSPs recorded 50–60 min after weak TBS was calculated and expressed as a ratio of the mean value of the initial baseline slope of fEPSPs. For the hippocampal synaptic plasticity studies, we used two subthreshold doses of nicotine (0.01 and 0.1 µM) to assess nicotinic facilitation of LTP. The rationale for the use of the subthreshold doses was based on the observation that our systemic nicotine treatments, at the 0.09 mg/kg/injection dose, were subthreshold to influence learning and memory in wild-type mice, but not in the Chrna2L9′S/L9′S mice. The majority of brain tissue slices from individual animals (n = 9) were exposed to all vehicle and drug conditions, with a few individual animal brain slices exposed to drug exposures alone (n = 2), or additional vehicle (n = 3) and/or drug exposures (n = 3).
Nicotine studies in adolescent mice using the preexposure-dependent contextual fear conditioning procedure
Male adolescent (4 wk of age) Chrna2KO, Chrna2L9′S/L9′S or wild-type littermate control mice of each respective line were used in the current studies. Importantly, Chrna2WT mice used for the Chrna2L9′S/L9′S studies were littermates of the Chrna2L9′S/L9′S mice. Similarly, Chrna2WT mice used for the Chrna2KO studies were littermates of the Chrna2KO mice. Three days before day 1 of preexposure (Fig. 4A, PE), animals were handled and habituated to their transport cage for 5 min for 2 d, similar to Kenney and Gould (2008) and Kutlu et al. (2016). On the third day, animals were briefly weighed, exposed to their transport cage, and experiments began on the next day. On day 1 of preexposure, mice were transferred to the behavioral testing suite and allowed to acclimate to the new environment for at least 1 h prior to experimentation. Subsequently, on day 1 of preexposure, using random assignment, mice were placed into transport cages, administered either a pretraining injection of vehicle or nicotine (0.09 mg/kg, i.p., base) 3–5 min prior to being preexposed to a novel conditioning chamber for 10 min. Chamber dimensions were four identical 30 cm × 24 cm × 21 cm (Med Associates, Inc.) boxes fitted with grid floors and a digital video camera (Fig. 4A, PE). On day 2 (Immediate-Shock, IS, Fig. 4A), mice were placed in the same chamber and within 10 sec of entry given a single electric shock (0.75 mA, 2 sec) and assessed for shock reactivity and freezing behavior post-shock (30 sec). On day 3 (Context Test, CT, Fig. 4A), mice were administered a pretesting injection of vehicle or nicotine (0.09 mg/kg, i.p.), and 3–5 min later assessed for freezing behavior in the previously shocked environment for 8 min. The nicotine dose and dual injection approach was based on the study of Kenney and Gould (2008) and the time period for PE and CT was based on the study of Cushman et al. (2011). The subthreshold dose of nicotine was previously shown to enhance learning and memory in adult wild-type mice (Kenney and Gould 2008), therefore this dose was chosen for the current studies. The time periods for these two phases are based on the fact that different processes are engaged. During the preexposure period the animals are forming a new hippocampus-dependent contextual representation of the conditioning chamber whereas on day 3 they are retrieving this representation and expressing fear based on the strength of the context–shock association. Ten minutes for the preexposure was based on prior piloting to determine the necessary length of time for the contextual representation to be formed. 8 min for the context test is based on the most commonly used length during test day (Kim and Fanselow 1992; Bissiere et al. 2011). For consistency in our analyses between day 1 and 3, only the first 8 min of the 10 min PE period is reported in our results, even though the findings remained consistent if the 10-min period was reported instead. Freezing and shock reactivity were monitored through near infrared lighting and a digital video camera recording at 30 frames per second placed in front of the chambers, and scored through a Med Associates fear conditioning software package (Med Associates Video Freeze system). This system calculates the average motion in the video as activity units, which are determined based on the amount of gray scale pixel change in the image for each frame normalized by background noise when no mouse is present. Freezing was defined as activity below 19 activity units for 1 sec (30 frames). Shock reactivity on day 2 was used as a measure of sensory pain response in the presence of a shock, as determined by the activity units measured during the 2 sec of the 0.75-mA shock and the following 2 sec. Animals which have greater shock reactivity, likely have enhanced pain sensitivity to the shock (King et al. 1996). Freezing varies positively with shock intensity and pain sensitivity, so increased pain sensitivity should increase freezing (Fanselow and Bolles 1979).
To illustrate conditioning (i.e., a significant increase in freezing on context test after shock training), freezing data were graphed together as PE (day 1) and CT (dy 3). The data are visualized using the same time scale on PE (day 1) and CT (day 3) of 8 min. We separated PE/CT and IS (day 2 data) results primarily because of the different time scales used in the analysis: 8 min for PE and CT versus 30 sec for IS (Figs. 4, 7) in order to reduce confusion about the different time scales used. The results remained consistent if data were illustrated by individual day of experimentation instead.
Growth, exploratory, stereotypical, and anxiety behaviors
Animal weight was measured on the day before preexposure day 1 and on day 2 of immediate-shock. Manually scored exploratory (vertical-rearing and horizontal-midline crosses), anxiety (center and perimeter time), and stereotypical mouse behavior were quantified in the conditioning chamber, by a blind observer during the 10-min preexposure period on day 1. Manually scored exploratory behavior was assessed as a secondary measure of automated locomotor activity, as previous work had demonstrated that rearing and crosses did not individually resolve automated locomotor activity (Cushman et al. 2011). Stereotypical behaviors included natural mouse behavior observed in a novel environment, including grooming, scratching, chewing, head nodding, paw tremors, head shakes, jumping and backing behavior (Lotfipour et al. 2013). Center time was quantified by manually recording the seconds spent in the center versus parameter of the apparatus for individual animals. An increase in center time is regarded as reduced anxiety. A mouse was defined as “in center” when its entire body was a tail length away from the nearest wall. Weight gain was used, in part, to calculate the injection volume. Weight gain and stereotypical behaviors were used to determine any growth or development modifications in adolescent Chrna2L9′S/L9′S and Chrna2KO mice. We assessed automated locomotor activity, rearing, mid-line crosses, center time, and shock reactivity, to determine genotype and drug-induced changes in exploratory activity, anxiety and pain sensitivity (as a measure of sensory response).
Statistics
Descriptive data were analyzed using the JMP Pro statistical software (SAS Institute Inc., version 12.0.1) and OriginPro 8.1 (OriginLab). Power analyses were determined using JMP Pro and G*Power 3.1. Electrophysiology data were assessed with t-test analysis and Bonferroni-corrected for multiple comparisons for each of the three doses tested. As our primary interests were within dose assessments for the electrophysiology data, across dose comparisons and multiple comparisons correction were not made. Data from wild-type mice from the C57Bl/6J and Chrna2L9′S/L9′S line or the male and female data did not differ, thus results were pooled for analysis. Each data point collected was assessed as a separate experiment and not as a within measure analysis during t-test analysis. For behavioral data, day 1 versus day 3 freezing data as well as days 2 and 3 freezing data were assessed using a matched-pair within subject analysis, followed by Bonferroni-correction for all four comparisons. The association between day 1 activity and day 3 freezing data were assessed as a simple linear correlation using bivariate analysis. For day 1 stereotypical and activity data as well as days 2 and 3 freezing data, significant effects were assessed using analysis of variance (ANOVA). When there were more than two groups compared, Bonferroni-correction for multiple comparisons was applied. Outliers were determined and excluded based on Box-and-Whisker plot outlier analysis, identifying data outside the interquartile range (Tukey 1977). For the Chrna2L9′S/L9′S mouse line, a total of four mice were identified as outliers for freezing data (the primary outcome variable) and excluded from all analyses. For the Chrna2KO mouse line one mouse was identified as an outlier for freezing data and excluded from all analyses.
Acknowledgments
We thank Meisheng Jiang, Rong Qiao, and the UCLA Transgenic Core for their assistance on this project. We thank Ms. Anjelica Mia Cardenas for her assistance on the manuscript. This study was funded through two Brain and Behavior (formally NARSAD) Research Foundation Young Investigator Awards (21517, 17717) (S.L.), University of California Office of the President Tobacco-Related Disease Research Program Postdoctoral Fellowship (20FT-0072) and Project Grant (22RT-0103A) (S.L.), NIMH Grant MH62122 (M.S.F.), and NIDA Grant DA026458 (K.S.).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.045369.117.
References
- Anagnostaras SG, Maren S, Fanselow MS. 1999. Temporally graded retrograde amnesia of contextual fear after hippocampal damage in rats: within-subjects examination. J Neurosci 19: 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissiere S, Zelikowsky M, Ponnusamy R, Jacobs NS, Blair HT, Fanselow MS. 2011. Electrical synapses control hippocampal contributions to fear learning and memory. Science 331: 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter J, Connolly J, Deneris E, Goldman D, Heinemann S, Patrick J. 1987. Functional expression of two neuronal nicotinic acetylcholine receptors from cDNA clones identifies a gene family. Proc Natl Acad Sci 84: 7763–7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller AL, White MM. 1990. Functional acetylcholine receptors expressed in Xenopus oocytes after injection of Torpedo β, γ, and Δ subunit RNAs are a consequence of endogenous oocyte gene expression. Mol Pharmacol 37: 423–428. [PubMed] [Google Scholar]
- Chen K, Nakauchi S, Su H, Tanimoto S, Sumikawa K. 2016. Early postnatal nicotine exposure disrupts the α2* nicotinic acetylcholine receptor-mediated control of oriens-lacunosum moleculare cells during adolescence in rats. Neuropharmacology 101: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman JD, Moore MD, Jacobs NS, Olsen RW, Fanselow MS. 2011. Behavioral pharmacogenetic analysis on the role of the α4 GABA(A) receptor subunit in the ethanol-mediated impairment of hippocampus-dependent contextual learning. Alcohol Clin Exp Res 35: 1948–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Gould TJ. 2007. β2 subunit-containing nicotinic receptors mediate the enhancing effect of nicotine on trace cued fear conditioning in C57BL/6 mice. Psychopharmacology (Berl) 190: 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, Kenney JW, Gould TJ. 2007. Hippocampal α4β2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. J Neurosci 27: 10870–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneris ES, Connolly J, Boulter J, Wada E, Wada K, Swanson LW, Patrick J, Heinemann S. 1988. Primary structure and expression of β 2: a novel subunit of neuronal nicotinic acetylcholine receptors. Neuron 1: 45–54. [DOI] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, et al. 2008. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity α 6 nicotinic acetylcholine receptors. Neuron 60: 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS. 1990. Factors governing one trial contextual conditioning. Anim Learn Behav 18: 264–270. [Google Scholar]
- Fanselow MS, Bolles RC. 1979. Naloxone and shock-elicited freezing in the rat. J Comp Physiol Psychol 93: 736–744. [DOI] [PubMed] [Google Scholar]
- Fox RR, Witham BA. 1997. Handbook on Genetically Standardized JAX Mice. 5:60. [Google Scholar]
- Frankland PW, O'Brien C, Ohno M, Kirkwood A, Silva AJ. 2001. α-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 411: 309–313. [DOI] [PubMed] [Google Scholar]
- Godsil BP, Stefanacci L, Fanselow MS. 2005. Bright light suppresses hyperactivity induced by excitotoxic dorsal hippocampus lesions in the rat. Behav Neurosci 119: 1339–1352. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ. 2007. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol 74: 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilscher MM, Leao RN, Edwards SJ, Leao KE, Kullander K. 2017. Chrna2-Martinotti cells synchronize layer 5 type A pyramidal cells via rebound excitation. PLoS Biol 15: e2001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K, Wong JK, Sumikawa K. 2005. Comparison of α2 nicotinic acetylcholine receptor subunit mRNA expression in the central nervous system of rats and mice. J Comp Neurol 493: 241–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Yamazaki Y, Nakauchi S, Sumikawa K. 2009. α2 nicotine receptors function as a molecular switch to continuously excite a subset of interneurons in rat hippocampal circuits. Eur J Neurosci 29: 1588–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Yamazaki Y, Nakauchi S, Ito K, Sumikawa K. 2010. Nicotine facilitates long-term potentiation induction in oriens-lacunosum moleculare cells via Ca2+ entry through non-α7 nicotinic acetylcholine receptors. Eur J Neurosci 31: 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Steinbach JH. 2011. A portable site: a binding element for 17β-estradiol can be placed on any subunit of a nicotinic α4β2 receptor. J Neurosci 31: 5045–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Steinbach JH. 2015. Potentiation of neuronal nicotinic receptors by 17β-estradiol: roles of the carboxy-terminal and the amino-terminal extracellular domains. PLoS One 10: e0144631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. 2008. Nicotine enhances context learning but not context-shock associative learning. Behav Neurosci 122: 1158–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. 1992. Modality-specific retrograde amnesia of fear. Science 256: 675–677. [DOI] [PubMed] [Google Scholar]
- King TE, Joynes RL, Meagher MW, Grau JW. 1996. Impact of shock on pain reactivity: II. Evidence for enhanced pain. J Exp Psychol Anim Behav Process 22: 265–278. [DOI] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. 2006. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci 103: 19152–19157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeman E, Nakauchi S, Su H, Dang R, Wood MA, Sumikawa K. 2016. Impaired function of α2-containing nicotinic acetylcholine receptors on oriens-lacunosum moleculare cells causes hippocampus-dependent memory impairments. Neurobiol Learn Mem 136: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu MG, Braak DC, Tumolo JM, Gould TJ. 2016. Adolescent mice are less sensitive to the effects of acute nicotine on context preexposure than adults. Brain Res 1642: 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. 1995. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature 376: 514–516. [DOI] [PubMed] [Google Scholar]
- Leao RN, Mikulovic S, Leao KE, Munguba H, Gezelius H, Enjin A, Patra K, Eriksson A, Loew LM, Tort AB, et al. 2012. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat Neurosci 15: 1524–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester HA, Fonck C, Tapper AR, McKinney S, Damaj MI, Balogh S, Owens J, Wehner JM, Collins AC, Labarca C. 2003. Hypersensitive knockin mouse strains identify receptors and pathways for nicotine action. Curr Opin Drug Discov Devel 6: 633–639. [PubMed] [Google Scholar]
- Liman ER, Tytgat J, Hess P. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9: 861–871. [DOI] [PubMed] [Google Scholar]
- Lipovsek M, Plazas P, Savino J, Klaassen A, Boulter J, Elgoyhen AB, Katz E. 2008. Properties of mutated murine α4β2 nicotinic receptors linked to partial epilepsy. Neurosci Lett 434: 165–169. [DOI] [PubMed] [Google Scholar]
- Lotfipour S, Byun JS, Leach P, Fowler CD, Murphy NP, Kenny PJ, Gould TJ, Boulter J. 2013. Targeted deletion of the mouse α2 nicotinic acetylcholine receptor subunit gene (Chrna2) potentiates nicotine-modulated behaviors. J Neurosci 33: 7728–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, Turi GF, Hen R, Zemelman BV, Losonczy A. 2014. Dendritic inhibition in the hippocampus supports fear learning. Science 343: 857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetje CW, Patrick J. 1991. Both α- and β-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci 11: 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus-Amat P, Higgins EA, Barrientos RM, Rudy JW. 2004. The role of the dorsal hippocampus in the acquisition and retrieval of context memory representations. J Neurosci 24: 2431–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. 2009. α9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol 78: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, Rodriguez Barrera V, Chittajallu R, Iwamoto KS, McBain CJ, et al. 2012. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 149: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakauchi S, Brennan RJ, Boulter J, Sumikawa K. 2007. Nicotine gates long-term potentiation in the hippocampal CA1 region via the activation of α2* nicotinic ACh receptors. Eur J Neurosci 25: 2666–2681. [DOI] [PubMed] [Google Scholar]
- Nakauchi S, Malvaez M, Su H, Kleeman E, Dang R, Wood MA, Sumikawa K. 2015. Early postnatal nicotine exposure causes hippocampus-dependent memory impairments in adolescent mice: association with altered nicotinic cholinergic modulation of LTP, but not impaired LTP. Neurobiol Learn Mem 118: 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill HC, Laverty DC, Patzlaff NE, Cohen BN, Fonck C, McKinney S, McIntosh JM, Lindstrom JM, Lester HA, Grady SR, et al. 2013. Mice expressing the ADNFLE valine 287 leucine mutation of the β2 nicotinic acetylcholine receptor subunit display increased sensitivity to acute nicotine administration and altered presynaptic nicotinic receptor function. Pharmacol Biochem Behav 103: 603–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Boulter J, Patrick J, Heinemann S. 1989. Single-channel currents of rat neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. Neuron 3: 589–596. [DOI] [PubMed] [Google Scholar]
- Paradiso K, Zhang J, Steinbach JH. 2001. The C terminus of the human nicotinic α4β2 receptor forms a binding site required for potentiation by an estrogenic steroid. J Neurosci 21: 6561–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plazas PV, De Rosa MJ, Gomez-Casati ME, Verbitsky M, Weisstaub N, Katz E, Bouzat C, Elgoyhen AB. 2005. Key roles of hydrophobic rings of TM2 in gating of the α9α10 nicotinic cholinergic receptor. Br J Pharmacol 145: 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal GS, Wilkinson DS, Kenney JW, Sullivan C, Gould TJ. 2012. Strain-dependent effects of acute, chronic, and withdrawal from chronic nicotine on fear conditioning. Behav Genet 42: 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. 1991. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 353: 846–849. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Barrientos RM, O'Reilly RC. 2002. Hippocampal formation supports conditioning to memory of a context. Behav Neurosci 116: 530–538. [DOI] [PubMed] [Google Scholar]
- Rudy JW, Huff NC, Matus-Amat P. 2004. Understanding contextual fear conditioning: insights from a two-process model. Neurosci Biobehav Rev 28: 675–685. [DOI] [PubMed] [Google Scholar]
- Salas R, Sturm R, Boulter J, De Biasi M. 2009. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci 29: 3014–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova S, Contet C, Roberts AJ, Markou A. 2012. Mice lacking the β4 subunit of the nicotinic acetylcholine receptor show memory deficits, altered anxiety- and depression-like behavior, and diminished nicotine-induced analgesia. Nicotine Tob Res 14: 1346–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivilotti LG, McNeil DK, Lewis TM, Nassar MA, Schoepfer R, Colquhoun D. 1997. Recombinant nicotinic receptors, expressed in Xenopus oocytes, do not resemble native rat sympathetic ganglion receptors in single-channel behaviour. J Physiol 500: 123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JH, Winzer-Serhan UH. 2006. Postnatal expression of α2 nicotinic acetylcholine receptor subunit mRNA in developing cortex and hippocampus. J Chem Neuroanat 32: 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stote DL, Fanselow MS. 2004. NMDA receptor modulation of incidental learning in Pavlovian context conditioning. Behav Neurosci 118: 253–257. [DOI] [PubMed] [Google Scholar]
- Sudweeks SN, Yakel JL. 2000. Functional and molecular characterization of neuronal nicotinic ACh receptors in rat CA1 hippocampal neurons. J Physiol 527(Pt 3): 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranda J, Maison SF, Ballestero JA, Katz E, Savino J, Vetter DE, Boulter J, Liberman MC, Fuchs PA, Elgoyhen AB. 2009. A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol 7: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. 2004. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science 306: 1029–1032. [DOI] [PubMed] [Google Scholar]
- Tukey JW. 1977. Exploratory data analysis. Addison-Wesley, Boston. [Google Scholar]
- Upton M, Lotfipour S. 2015. α2-Null mutant mice have altered levels of neuronal activity in restricted midbrain and limbic brain regions during nicotine withdrawal as demonstrated by cfos expression. Biochem Pharmacol 97: 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K, Ballivet M, Boulter J, Connolly J, Wada E, Deneris ES, Swanson LW, Heinemann S, Patrick J. 1988. Functional expression of a new pharmacological subtype of brain nicotinic acetylcholine receptor. Science 240: 330–334. [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. 1989. Distribution of α 2, α 3, α 4, and β 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 284: 314–335. [DOI] [PubMed] [Google Scholar]
- Wehner JM, Keller JJ, Keller AB, Picciotto MR, Paylor R, Booker TK, Beaudet A, Heinemann SF, Balogh SA. 2004. Role of neuronal nicotinic receptors in the effects of nicotine and ethanol on contextual fear conditioning. Neuroscience 129: 11–24. [DOI] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Behne NS, Fanselow MS. 2001. Sex differences, context preexposure, and the immediate shock deficit in Pavlovian context conditioning with mice. Behav Neurosci 115: 26–32. [DOI] [PubMed] [Google Scholar]