

Abstract
Amygdala lesions impair, but do not prevent, acquisition of cerebellum-dependent eyeblink conditioning suggesting that the amygdala modulates cerebellar learning. Two-factor theories of eyeblink conditioning posit that a fast-developing memory within the amygdala facilitates slower-developing memory within the cerebellum. The current study tested this hypothesis by impairing memory consolidation within the amygdala with inhibition of protein synthesis, transcription, and NMDA receptors in rats. Rats given infusions of anisomycin or DRB into the central amygdala (CeA) immediately after each eyeblink conditioning session were severely impaired in contextual and cued fear conditioning, but were completely unimpaired in eyeblink conditioning. Rats given the NMDA antagonist ifenprodil into the CeA before each eyeblink conditioning session also showed impaired fear conditioning, but no deficit in eyeblink conditioning. The results indicate that memory formation within the CeA is not necessary for its modulation of cerebellar learning mechanisms. The CeA may modulate cerebellar learning and retention through an attentional mechanism that develops within the training sessions.
Amygdala lesions impair, but do not prevent, acquisition of cerebellum-dependent eyeblink conditioning in rodents and rabbits (Neufeld and Mintz 2001; Lee and Kim 2004; Blankenship et al. 2005; Burhans and Schreurs 2008; Farley et al. 2016; Pochiro and Lindquist 2016). This effect has generally been interpreted as evidence for a two-factor model of eyeblink conditioning in which the amygdala first forms a fear memory that then facilitates the development of a motor memory in the cerebellum. The fast-developing amygdala-mediated fear memory is therefore necessary for facilitating the slower-developing cerebellar learning mechanisms. The amygdala contribution to eyeblink conditioning is thought to decrease as the eyeblink conditional response (CR) develops, and the cerebellum may even inhibit the amygdala through a feedback mechanism (Magal and Mintz 2014; Pochiro and Lindquist 2016).
Two recent studies cast doubt on the two-factor model by demonstrating a severe deficit in eyeblink conditioning with inactivation of the central amygdala (CeA) after animals had reached asymptotic levels of conditioning (Siegel et al. 2015; Farley et al. 2016). The severe deficit in retention of eyeblink conditioning with CeA inactivation was correlated with a nearly complete suppression of learning-specific ramping activity within the anterior interpositus nucleus, which is necessary for the eyeblink CR (Farley et al. 2016). These findings suggest that the CeA continues to play a crucial role in cerebellum-mediated conditional responses (CRs), even after the motor memory is well consolidated.
Another crucial part of the two-factor model of eyeblink conditioning is that the fast-developing memory within the amygdala is necessary for its modulation of slower-developing cerebellar learning. The recent inactivation studies suggest that memory within the amygdala might be necessary for its role in the retention of cerebellar learning as well (Siegel et al. 2015; Farley et al. 2016). It is also possible however, that memory formation within the amygdala is not necessary for modulation of acquisition or retention of eyeblink conditioning. This alternative nonmnemonic mechanism might be a within-session increase in attention to the CS, a role previously attributed to the CeA in appetitive conditioning (Gallagher et al. 1990).
The current study was designed to determine whether memory consolidation within the amygdala is necessary for its modulation of eyeblink conditioning in rats. We used three different approaches for affecting memory consolidation within the amygdala that are well established in the fear conditioning literature. We inhibited protein synthesis, mRNA transcription, and NMDA receptors during eyeblink conditioning (Schafe and LeDoux 2000; Rodrigues et al. 2001; Wilensky et al. 2006). Anisomycin and DRB were infused into the CeA after each eyeblink conditioning session to inhibit protein synthesis and transcription, respectively. The NMDA receptor antagonist ifenprodil was infused into the CeA before each session. Ifenprodil was used instead of APV because previous studies showed that APV can impair retention of fear conditioning, whereas ifenprodil more selectively impairs acquisition (Maren et al. 1996; Rodrigues et al. 2001). Separate tests were given to assess conditional freezing to verify the effects of the manipulations on amygdala-dependent memory.
Results
Cannula placements
Figure 1 displays the locations of the tips of the infusion cannulae in each rat in coronal sections, separated by the four experimental groups. All of the placements were within the CeA or <1.0 mm away from it. Cannula placements did not differ systematically between groups. Data from seven rats were excluded because of missed cannula placements. The performance of rats in the treatment groups with missed cannula placements did not differ significantly from the controls.
Figure 1.

Drawings of coronal sections depicting the locations of the infusion cannulae tips in all of the rats used in this study. The placements for the vehicle (n = 8), anisomycin (n = 8), DRB (n = 8), and ifenprodil (n = 8) groups are displayed separately. The values to the right of the sections indicate their anterior–posterior stereotaxic coordinates.
Fear conditioning
Anisomycin or DRB was infused bilaterally into the CeA immediately following each of the first five sessions of CS–US paired eyeblink conditioning within Context A. Ifenprodil was infused before each of these paired sessions. Prior to each session, the rats were placed in the conditioning chamber and freezing was recorded for 5 min to assess context conditioning during training to Context A and to assess baseline freezing during test sessions in other contexts (see below). A significant deficit in context conditioning to Context A was found in the drug groups relative to the vehicle group [F(3,28) = 4.69, P = 0.009; Figs. 2A, 3A, 4A]. This result indicates that anisomycin, DRB, or ifenprodil infusions into the CeA impaired the development of contextual freezing during eyeblink conditioning.
Figure 2.
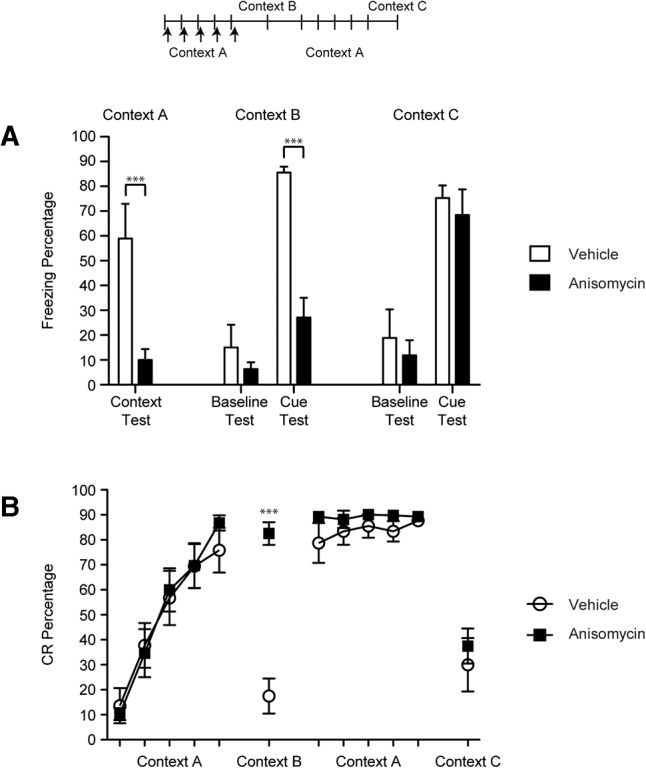
Percentage of freezing (A) and eyeblink CRs (B) for rats given anisomycin or vehicle after each of the first five eyeblink conditioning sessions in Context A (50 trials per session). Tests for cued fear conditioning (five CS-alone trials each) were given after eyeblink conditioning acquisition sessions in Context B and after overtraining of eyeblink conditioning in Context C. Eyeblink CRs and conditional freezing were measured in each testing session. (***) P < 0.01.
Figure 3.
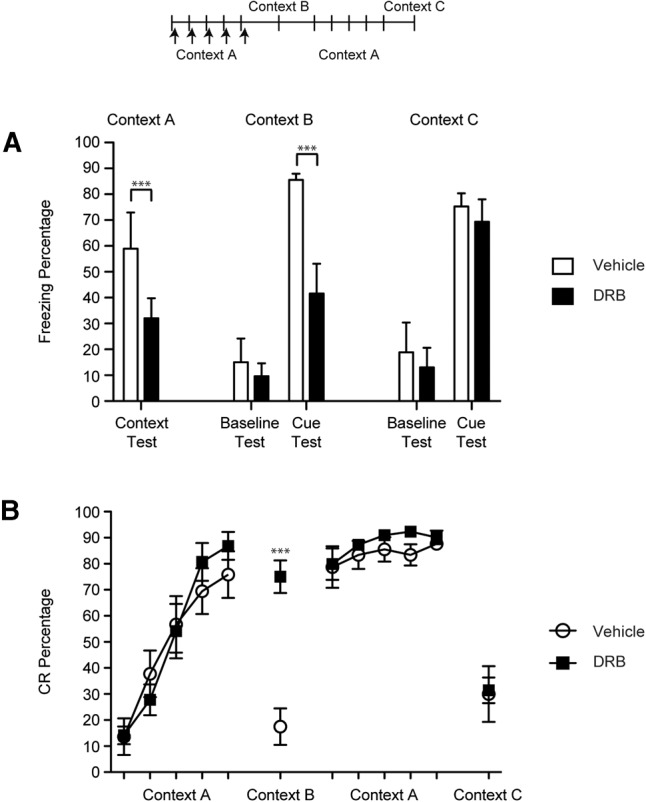
Percentage of freezing (A) and eyeblink CRs (B) for rats given DRB or vehicle after each of the first five eyeblink conditioning sessions in Context A (50 trials per session). Tests for cued fear conditioning (five CS-alone trials each) were given after eyeblink conditioning acquisition sessions in Context B and after overtraining of eyeblink conditioning in Context C. Eyeblink CRs and conditional freezing were measured in each testing session. (***) P < 0.01.
Figure 4.
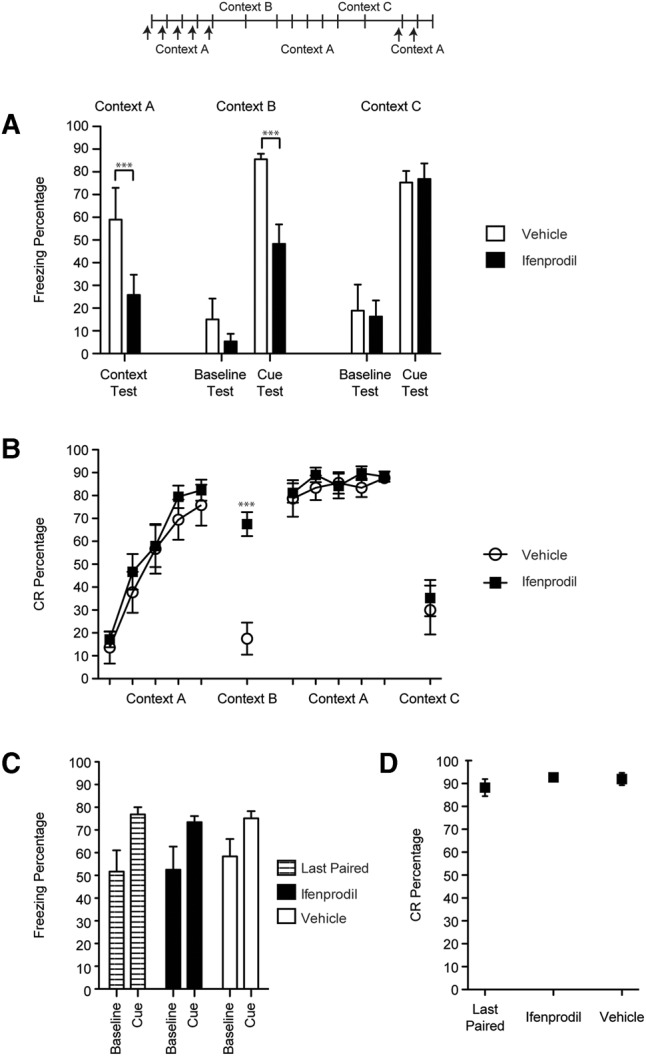
Percentage of freezing (A) and eyeblink CRs (B) for rats given ifenprodil or vehicle before each of the first five eyeblink conditioning sessions in Context A (50 trials per session). Tests for cued fear conditioning (five CS-alone trials each) were given after eyeblink conditioning acquisition sessions in Context B and after overtraining of eyeblink conditioning in Context C. Following the Context C test, the rats were trained for one eyeblink conditioning session without infusions in Context A (Last Paired). They were then given either ifenprodil or vehicle prior to the next two eyeblink conditioning sessions (the order of infusions was counterbalanced). Eyeblink CRs and conditional freezing were measured in each testing session. (***) P < 0.01.
The rats were then tested in Context B with five CS-alone trials. The CS used in the test sessions was the same as used during eyeblink conditioning sessions (i.e., 400 msec). Both the vehicle and drug groups exhibited little pre-CS baseline freezing to the new context and did not differ from each other significantly (Figs. 2A, 3A, 4A). However, after the CS was presented the vehicle group showed significantly more freezing than the drug groups [F(3,28) = 9.04, P = 0.00; Figs. 2A, 3A, 4A].
The rats were then placed back into Context A and trained for five more eyeblink conditioning sessions, this time without infusions to establish robust fear conditioning in the drug groups. A final test session with five CS-alone trials was given in Context C. Similar to the Context B test, all of the groups exhibited low levels of freezing during the pre-CS baseline period (Figs. 2A, 3A, 4A). However, all groups exhibited high levels of freezing during the CS presentations and did not differ from each other (Figs. 2A, 3A, 4A). Thus, anisomycin, DRB, and ifenprodil did not have lasting effects on the development of freezing during eyeblink conditioning, indicating that the deficits in cue and context conditioning were not caused by lesion-like effects.
Eyeblink conditioning
All of the groups exhibited similar rates of acquisition during the first five sessions of eyeblink conditioning (Figs. 2B, 3B, 4B). No significant differences were found between the groups but a main effect of Session [F(4,112) = 114.46, P = 0.000] was found, indicating that significant learning took place across the sessions in all of the groups. During the Context B test the drug groups exhibited a significantly higher percentage of eyeblink CRs than the vehicle group [F(3,28) = 25.30, P = 0.000; Figs. 2B, 3B, 4B]. The rats then received five sessions of eyeblink conditioning in Context A and no significant differences were observed between the groups during these overtraining sessions. Finally, the rats were given a test session in Context C and all of the groups showed significantly fewer eyeblink CRs than the prior session, but no significant group differences were found [F(3,28) = 0.25, P = 0.860]. There were no group differences in CR latency or amplitude. Taken together, these data indicate that the drug treatments had no effect on CRs formation. However, the drug infusions appear to have made eyeblink conditioning insensitive to the context change.
Additional testing with ifenprodil
Additional test sessions were given for the ifenprodil group and three of the rats in the vehicle group to verify that ifenprodil was producing effects on conditional freezing by impairing learning and not by disruption of synaptic transmission within the CeA. Following the Context C test, the rats were trained for one eyeblink conditioning session without infusions in Context A (Fig. 4C,D, Last Paired). They were then given either ifenprodil or vehicle prior to the next two sessions (the order of infusions was counterbalanced). Ifenprodil did not affect either conditional freezing (baseline or cued, Fig. 4C) or eyeblink conditioning (Fig. 4D). The results from these additional testing sessions indicate that ifenprodil did not impair the development of conditional freezing during the initial sessions of eyeblink conditioning by blocking synaptic transmission within the CeA.
Discussion
All three treatments that impaired memory consolidation of fear conditioning had no effect on eyeblink conditioning. Thus, inhibition of translation, transcription, or blockade of GluN2B receptors all impaired consolidation of contextual and cued fear conditioning, but had no effect on acquisition or retention of eyeblink CRs.
The findings of this study are not consistent with two-factor theories of eyeblink conditioning (Neufeld and Mintz 2001; Lee and Kim 2004; Farley et al. 2016; Pochiro and Lindquist 2016). These theories posit that a memory for the emotional significance of the CS is first formed within the amygdala and facilitates the subsequent development of a memory necessary for generating a learned motor response within the cerebellum. All three manipulations demonstrate that if consolidation mechanisms within the amygdala are inhibited, the freezing CR is diminished, whereas the eyeblink CR is completely unaffected. And yet, it is clear from several previous studies that the amygdala modulates acquisition and retention of eyeblink conditioning by gating input to the CS pathway (Taub and Mintz 2010; Farley et al. 2016; Pochiro and Lindquist 2016). This CS gating mechanism must therefore develop rapidly within training sessions.
We further hypothesize that amygdala-dependent CS facilitation is associative and not due to increased arousal or sensitization. We are hypothesizing a mechanism that requires the CS–US association because many behavioral studies have shown that acquisition of eyeblink conditioning is impaired, not facilitated, following unpaired presentations of the CS and US or just the US (Gormezano et al. 1983). Thus, nonassociative and contextual conditioning processes established by unpaired presentations of the US or US-alone trials are not mechanisms that would facilitate eyeblink conditioning. The proposed CS–US associative mechanism, mediated by the CeA, is thought to boost attention to the CS, as previously demonstrated for appetitive conditioning (Gallagher et al. 1990).
Although there have been several reports showing that lesions or inactivation of the CeA impair eyeblink conditioning in rats (Neufeld and Mintz 2001; Lee and Kim 2004; Blankenship et al. 2005; Farley et al. 2016; Pochiro and Lindquist 2016), rabbits (Burhans and Schreurs 2008), and mice (Siegel et al. 2015), there have been reports demonstrating intact eyeblink conditioning in the absence of the CeA (Weisz et al. 1992; Hesslow 1994; Hesslow and Ivarsson 1996; Hesslow et al. 1999; Bengtsson et al. 2007; Svensson et al. 2010). Weisz et al. (1992) found that electrolytic lesions of the CeA impaired eyeblink conditioning in rabbits with a relatively low intensity auditory CS (65 dB), but not with a high intensity CS (85 dB). Moreover, Hesslow and colleagues have numerous reports showing eyeblink conditioning in cats and ferrets decerebrated just rostral to the red nucleus, thereby severing communication between the CeA and cerebellum. There are procedural differences between the studies that found deficits and the studies that did not find deficits with CeA disruption that might shed light on the parameters that promote CeA modulation. The rabbits in the Weisz et al. (1992) study were unimpaired with an 85-dB tone CS, which is similar to the physical intensity of the auditory CSs used in the studies demonstrating deficits with CeA inactivation or lesions, but was presented through earphones, raising the possibility that it had a relatively higher intensity at the tympanic membrane than auditory CSs presented through speakers. In the decerebration studies, the ferrets were trained with a relatively intense 300-msec electrical stimulation CS (1.0–1.8 mA) delivered through needle electrodes in the forelimb. These observations are consistent with our attentional hypothesis, assuming that weaker stimuli require more attention than intense stimuli during eyeblink conditioning. However, the role of stimulus intensity in CeA modulation of cerebellar learning has not been examined systematically.
A surprising and potentially important finding was the difference in eyeblink CR percentage between the vehicle and drug groups during the cue test sessions in Context B. The vehicle group showed a substantial decrease in CRs when the context switched, whereas the drug groups showed very little decrement. The context-dependence of eyeblink conditioning has been demonstrated in rabbits (Penick and Solomon 1991), but the magnitude of the CR decrement in the current study was greater. The substantial loss of CRs in the vehicle group might be related to a strong context-dependence of the CR in rats relative to rabbits. It is also possible, however, that the change in the sensory qualities of the conditioning chamber plus omission of the US during the Context B test may have combined to be a multidimensional context shift. That is, the chamber and the CS–US contingency both changed, resulting in a more substantial decrement than just changing the chamber. Perhaps most significant is the absence of the CR decrement in the drug groups. Thus, treatments that impaired the consolidation of fear conditioning also made eyeblink conditioning context-independent. This finding suggests that the context representation within the amygdala or in hippocampus–amygdala projections might be necessary for the context-dependence of eyeblink conditioning by serving as an occasion setter for the CS–US representation within the cerebellum. In the absence of the context representation, the CS–US association within the cerebellum becomes context-independent.
Our finding that memory consolidation within the CeA is crucial for retention of fear conditioning is consistent with several previous studies (Goosens and Maren 2001, 2003; Wilensky et al. 2006; Ciocchi et al. 2010). Wilensky et al. (2006) found that inactivation of the CeA severely impaired fear conditioning and protein synthesis inhibition blocked consolidation of fear conditioning. Blockade of NMDA receptors in the CeA also impaired consolidation of cued and contextual fear conditioning (Goosens and Maren 2003). The cellular mechanisms underlying CeA plasticity have not been identified, but might involve synaptic plasticity in projections from the BLA, sensory thalamus, and cortical areas (Orsini and Maren 2012; Keifer et al. 2015). There may also be plasticity at synapses within the CeA between the lateral and medial subdivisions (Ciocchi et al. 2010; Keifer et al. 2015). In the case of context conditioning, the hippocampus plays a crucial role and there could be plasticity in projections from the hippocampus or subiculum to the CeA or in projections between the hippocampus and upstream systems of the CeA, such as the BLA, that send context-related input to the CeA (Orsini and Maren 2012). In the current study, suppression of protein synthesis, transcription, or blockade of NMDA receptors impaired fear memory consolidation, presumably by disrupting the crucial mechanisms underlying CeA plasticity.
The findings of this study indicate that long-term memory formation within the amygdala is not necessary for its facilitation of eyeblink conditioning. Our alternative hypothesis to the traditional two-factor theory is that short-term associative processing within the amygdala during eyeblink conditioning sessions facilitates pontine neuronal responses to the CS (Taub and Mintz 2010; Pochiro and Lindquist 2016) and thereby facilitates cerebellar learning and CR expression (Farley et al. 2016). Amygdala inactivation is therefore functionally equivalent to reducing the intensity of the CS within the cerebellum, which impairs acquisition and expression of the eyeblink CR (Scavio and Gormezano 1974). We further hypothesize that amygdala-dependent CS facilitation is associative (CS–US) and not due to increased arousal or sensitization.
Materials and Methods
Subjects
The subjects were 39 male Long-Evans rats (250–300 g). The rats were housed in the animal colony in Spence Laboratories of Psychology at the University of Iowa (Iowa City, IA). All rats were maintained on a 12 h light–dark cycle and given ad libitum access to food and water. The rats were randomly assigned to one of four groups: Vehicle (n = 8), Anisomycin (n = 8), DRB (n = 8), or Ifenprodil (n = 8). The data from 7 of the 39 rats could not be used due to misplaced cannulae. All procedures were approved by the University of Iowa's Institutional Animal Care and Use Committee.
Surgery
One week before training, rats were removed from their home cages and anesthetized with isoflurane. After the onset of anesthesia, the rats were fitted with differential electromyography (EMG) electrodes (stainless steel) implanted into the upper left orbicularis oculi muscle. The ground electrode was a silver wire attached to a stainless steel skull screw. The EMG electrode leads terminated in gold pins in a plastic connector. A bipolar stimulating electrode (Plastics One) for delivering the shock US was implanted subdermally, caudal to the left eye. A 27-gauge guide cannula was implanted bilaterally dorsal to the central amygdala. A 32-gauge stylet was inserted into the guide cannula and extended 0.5 mm from the end of the guide. The stereotaxic coordinates taken from bregma for the cannula were 1.5 mm posterior, 3.6 mm lateral, and 5.5 mm ventral from skull surface. The plastic connector housing the EMG electrode leads, bipolar stimulating electrode, the guide cannula, and skull screws were secured to the skull with Osteobond copolymer bone cement. The rats were given analgesics for 48 h after surgery.
Infusion procedure
For each infusion the stylet was removed from the guide cannula and replaced with a 32-gauge infusion cannula that extended 1.0 mm beyond the guide cannula. The infusion cannula was connected to polyethylene tubing (PE 10), which was connected to a 10 µL gas tight syringe (Hamilton). The syringe was placed in an infusion pump (Harvard Apparatus), and 0.3 µL of anisomycin (ANI; 125 µg/µL), 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB; 20 ng/µL), ifenprodil (3.3 µg/µL), or vehicle (0.9% saline) was infused over 3 min at a rate of 6.0 µL/h. Infusions of ANI and DRB were made immediately following the sessions, whereas ifenprodil was infused before each session. The vehicle group included five rats that received infusions after sessions and three rats that received infusions before sessions to match handling and infusion exposure with the drug groups. No significant differences or even trends were found between vehicle group rats that received infusions before or after sessions. After the infusion, the infusion cannula was left for 3 min in order to allow diffusion of the drug. The infusion cannula was then removed and replaced with the 32 gauge stylet.
Apparatus
The conditioning apparatus consisted of two small-animal sound-attenuating chambers (BRS/LVE). Within each sound-attenuating chamber was a small-animal conditioning chamber (BRS/LVE). One wall of the conditioning chamber was fitted with two speakers. An exhaust fan on one of the walls provided a 65-dB masking noise. The tone CS used in training was a 2000-Hz pure tone (85 dB). The electrode leads from the rat's headstage were connected to peripheral equipment by lightweight cables that allowed the rat to move freely during conditioning. A desktop computer was connected to the peripheral equipment. Computer software controlled the delivery of stimuli and the recording of eyelid EMG activity (JSA Designs). One circuit was used to deliver the shock stimulus through a stimulus isolator (model 365A; World Precision Instruments). Another circuit amplified differentially (×2000), filtered (500–5000 Hz), and integrated the EMG activity.
Digital network cameras were secured outside the conditioning chambers. Two small LED lights were placed outside the conditioning chamber (not visible to the rats) to indicate the occurrence of the CS for the video recordings.
Conditioning procedure
Rats recovered from surgery for 1 wk prior to the initiation of testing. All rats completed 12 consecutive daily sessions of training. The first five sessions were paired eyeblink conditioning in which five blocks of nine paired CS–US presentations and 1 CS-alone probe trial were presented. The CS was 400 msec (2 kHz,85 dB) and the US was a 25 msec periorbital stimulation (2.5 mA). Rats received infusions either immediately before (ifenprodil) or following (anisomycin or DRB) each of these sessions. These sessions were conducted in Context A, which included no house light, cleaned with ETOH prior to each rat, large grid floor, and clear plastic walls. Following the fifth eyeblink conditioning session, the rats were switched to Context B and presented with five CS-alone trials. Context B included a house light, cleaned with vinegar prior to each rat, medium grid floor, and black and white bars on the wall. The rats were then given five more eyeblink conditioning sessions in Context A. The rats were then changed to Context C and received five CS-alone trials. Context C had a house light on, cleaned with a mint odor prior to each animal, small grid floor, and checkered pattern on the walls.
The rats with pretraining infusions of ifenprodil or vehicle during training received three additional test sessions in Context A to examine if ifenprodil impaired fear expression. Following the Context C test, the same rats were trained for one eyeblink conditioning session without infusions in Context A. They were then given either ifenprodil or vehicle prior to the next two sessions. Thus, rats from the original vehicle and ifenprodil groups were given two test sessions, one with vehicle and one with ifenprodil, with the order of infusions counterbalanced.
Eyeblink CRs were defined as eyelid EMG activity that exceeded a threshold of 0.4 units (amplified and integrated units) above the baseline mean during the CS period, but after 80 msec. EMG responses that exceeded the threshold during the first 80 msec of the CS period were defined as startle responses. On CS-alone probe trials, the duration for scoring CRs was extended beyond the CS to the end of the trial period (0.5 sec). URs were defined as responses that crossed the threshold after the onset of the US. CR onset latency and amplitude were calculated from paired and CS-alone trials.
Freezing measures
Eyeblink conditioning and fear conditioning use very different parameters, which could result in different patterns of conditional freezing. Our eyeblink conditioning procedure uses a shorter CS (400 msec versus 10–30 sec), US (25 msec versus 0.5–2.0 sec), inter-stimulus interval (400 msec versus 10–30 sec) and inter-trial interval (30 sec versus 1–10 min) than the standard fear conditioning procedures. Eyeblink conditioning also involves more training trials because it is acquired much more slowly than conditional fear (300–500 trials versus 1–5 trials). Despite these parametric differences, the conditional freezing in the current control group was very strong during the testing sessions.
All training sessions were videotaped with digital network cameras. Two small LED lights were placed outside the conditioning chamber (not visible to the rats) to indicate the occurrence of the CS for the video recordings. Video files were imported into MATLAB to perform automated frame-by-frame coding of movement (Ng and Freeman 2014). A threshold was set to ensure that movements produced by respiration were not counted. The absence of movement throughout an entire 1 sec time interval was counted as freezing. Before each session the rat was placed in the conditioning chamber for 5 min prior to the initiation of the first trial. The percentage of time freezing (1 sec intervals) during this pre-CS period was used as an index of contextual fear conditioning to Context A in the first phase of training. The pre-CS period was also used to assess baseline freezing in Contexts B and C prior to the cue tests. The percentage of time freezing was calculated during each CS (400 msec + 600 msec after the CS) and for an additional 10 sec after each CS during the cue tests in Contexts B and C.
Histology
After training, the rats were deeply anesthetized with an injection of sodium pentobarbital (150 mg/kg) and transcardially perfused with physiological saline followed by 10% neutral buffered formalin. After perfusion, the brains were cryo-protected in a 30% sucrose in formalin solution, and subsequently sectioned at 50 µm with a sliding microtome. Sections were then stained with thionin. The location of the cannula placements was verified using a light microscope (Leica DMLS) and a stereotaxic brain atlas.
Acknowledgments
This work was supported by NINDS grant NS088567 to J.H.F.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.045310.117.
References
- Bengtsson F, Jirenhed DA, Svensson P, Hesslow G. 2007. Extinction of conditioned blink responses by cerebello-olivary pathway stimulation. Neuroreport 18: 1479–1482. [DOI] [PubMed] [Google Scholar]
- Blankenship MR, Huckfeldt R, Steinmetz JJ, Steinmetz JE. 2005. The effects of amygdala lesions on hippocampal activity and classical eyeblink conditioning in rats. Brain Res 1035: 120–130. [DOI] [PubMed] [Google Scholar]
- Burhans LB, Schreurs BG. 2008. Inactivation of the central nucleus of the amygdala abolishes conditioning-specific reflex modification of the rabbit (Oryctolagus cuniculus) nictitating membrane response and delays classical conditioning. Behav Neurosci 122: 75–88. [DOI] [PubMed] [Google Scholar]
- Ciocchi S, Herry C, Grenier F, Wolff SB, Letzkus JJ, Vlachos I, Ehrlich I, Sprengel R, Deisseroth K, Stadler MB, et al. 2010. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468: 277–282. [DOI] [PubMed] [Google Scholar]
- Farley SJ, Radley JJ, Freeman JH. 2016. Amygdala modulation of cerebellar learning. J Neurosci 36: 2190–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M, Graham PW, Holland PC. 1990. The amygdala central nucleus and appetitive Pavlovian conditioning: lesions impair one class of conditioned behavior. J Neurosci 10: 1906–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, Maren S. 2001. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem 8: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosens KA, Maren S. 2003. Pretraining NMDA receptor blockade in the basolateral complex, but not the central nucleus, of the amygdala prevents savings of conditional fear. Behav Neurosci 117: 738–750. [DOI] [PubMed] [Google Scholar]
- Gormezano I, Kehoe EJ, Marshall BS. 1983. Twenty years of classical conditioning with the rabbit. Prog Psychobiol Physiol Psychol 10: 197–275. [Google Scholar]
- Hesslow G. 1994. Inhibition of classically conditioned eyeblink responses by stimulation of the cerebellar cortex in the decerebrate cat. J Physiol 476: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslow G, Ivarsson M. 1996. Inhibition of the inferior olive during conditioned responses in the decerebrate ferret. Exp Brain Res 110: 36–46. [DOI] [PubMed] [Google Scholar]
- Hesslow G, Svensson P, Ivarsson M. 1999. Learned movements elicited by direct stimulation of cerebellar mossy fiber afferents. Neuron 24: 179–185. [DOI] [PubMed] [Google Scholar]
- Keifer OP Jr, Hurt RC, Ressler KJ, Marvar PJ. 2015. The physiology of fear: reconceptualizing the role of the central amygdala in fear learning. Physiology (Bethesda) 30: 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Kim JJ. 2004. Differential effects of cerebellar, amygdalar, and hippocampal lesions on classical eyeblink conditioning in rats. J Neurosci 24: 3242–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magal A, Mintz M. 2014. Inhibition of the amygdala central nucleus by stimulation of cerebellar output in rats: a putative mechanism for extinction of the conditioned fear response. Eur J Neurosci 40: 3548–3555. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Stote DL, Fanselow MS. 1996. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav Neurosci 110: 1365–1374. [DOI] [PubMed] [Google Scholar]
- Neufeld M, Mintz M. 2001. Involvement of the amygdala in classical conditioning of eyeblink response in the rat. Brain Res 889: 112–117. [DOI] [PubMed] [Google Scholar]
- Ng KH, Freeman JH. 2014. Amygdala inactivation impairs eyeblink conditioning in developing rats. Dev Psychobiol 56: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Maren S. 2012. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev 36: 1773–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penick S, Solomon PR. 1991. Hippocampus, context, and conditioning. Behav Neurosci 105: 611–617. [DOI] [PubMed] [Google Scholar]
- Pochiro JM, Lindquist DH. 2016. Central amygdala lesions inhibit pontine nuclei acoustic reactivity and retard delay eyeblink conditioning acquisition in adult rats. Learn Behav 44: 191–201. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, Schafe GE, LeDoux JE. 2001. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci 21: 6889–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavio MJ, Gormezano I. 1974. CS intensity effects on rabbit nictitating membrane conditioning, extinction and generalization. Pavlov J Biol Sci 9: 25–34. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. 2000. Memory consolidation of auditory Pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20: RC96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ, Taylor W, Gray R, Kalmbach B, Zemelman BV, Desai NS, Johnston D, Chitwood RA. 2015. Trace eyeblink conditioning in mice is dependent upon the dorsal medial prefrontal cortex, cerebellum, and amygdala: behavioral characterization and functional circuitry(1,2,3). eNeuro 2 10.1523/ENEURO.0051-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson P, Jirenhed DA, Bengtsson F, Hesslow G. 2010. Effect of conditioned stimulus parameters on timing of conditioned Purkinje cell responses. J Neurophysiol 103: 1329–1336. [DOI] [PubMed] [Google Scholar]
- Taub AH, Mintz M. 2010. Amygdala conditioning modulates sensory input to the cerebellum. Neurobiol Learn Mem 94: 521–529. [DOI] [PubMed] [Google Scholar]
- Weisz DJ, Harden DG, Xiang Z. 1992. Effects of amygdala lesions on reflex facilitation and conditioned response acquisition during nictitating membrane response conditioning in rabbit. Behav Neurosci 106: 262–273. [DOI] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE. 2006. Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci 26: 12387–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]