

Natsume et al. show that MCM2-depleted cells activate a DNA damage response pathway and generate replication-associated DNA double-strand breaks (DSBs). These cells maintain some DNA synthesis in the absence of MCM2, and this requires the MCM8–9 complex, a paralog of the MCM2–7 replicative helicase.
Keywords: DNA replication, homologous recombination, fork restart, MCM proteins, genome maintenance, conditional degron
Abstract
DNA replication fork progression can be disrupted at difficult to replicate loci in the human genome, which has the potential to challenge chromosome integrity. This replication fork disruption can lead to the dissociation of the replisome and the formation of DNA damage. To model the events stemming from replisome dissociation during DNA replication perturbation, we used a degron-based system for inducible proteolysis of a subunit of the replicative helicase. We show that MCM2-depleted cells activate a DNA damage response pathway and generate replication-associated DNA double-strand breaks (DSBs). Remarkably, these cells maintain some DNA synthesis in the absence of MCM2, and this requires the MCM8–9 complex, a paralog of the MCM2–7 replicative helicase. We show that MCM8–9 functions in a homologous recombination-based pathway downstream from RAD51, which is promoted by DSB induction. This RAD51/MCM8–9 axis is distinct from the recently described RAD52-dependent DNA synthesis pathway that operates in early mitosis at common fragile sites. We propose that stalled replication forks can be restarted in S phase via homologous recombination using MCM8–9 as an alternative replicative helicase.
The replication of genomic DNA is essential for cell proliferation and propagation of genetic information to the next generation. Although DNA replication occurs with remarkable fidelity, many of the genomic alterations found in proliferating cells seem to arise from sites of perturbed DNA replication forks (Durkin and Glover 2007; Aguilera and Gomez-Gonzalez 2008). In human cells, replication forks can travel over many kilobases of template DNA and must overcome any roadblock or template abnormality encountered along the way. It is known that progression of the replication forks can be impeded at certain difficult to replicate genomic regions because of the presence of DNA secondary structures or adducts or because the replication machinery (replisome) collides with a transcribing RNA polymerase complex (Mirkin and Mirkin 2007; Zeman and Cimprich 2014). To avoid underreplication leading to genomic instability, cells must possess mechanisms to deal with the consequences of fork stalling and replisome disruption/disassembly.
In eukaryotic cells, DNA replication is initiated at multiple origins, which then generate bidirectional replication fork movement (Masai et al. 2010). In addition to origins that fire in each S phase, there are many dormant origins that are used only when cells are exposed to replication stress (Blow et al. 2011). The combination of multiple replication forks operating simultaneously and dormant origins creates an efficient fail-safe system to guard against permanent fork arrest because arrested forks can be rescued by a new converging fork (Supplemental Fig. S1A). This fork convergence plays a critical role in the maintenance of genomic integrity, as evidenced by the finding that cells with a reduced number of replication origins are susceptible to replication stress (Woodward et al. 2006; Ge et al. 2007; Ibarra et al. 2008). Interestingly, a recent report showed that human cells experience at least one replicon failure per S phase, where two forks are irreversibly arrested without there being an intervening origin to rescue them (Moreno et al. 2016). This feature of S phase is particularly pertinent to regions of the genome that are origin-poor, meaning that some forks must travel over a considerable distance without breakdown. Therefore, cells must have backup systems to deal with situations in which two or more forks are irreversibly stalled; otherwise, the completion of DNA replication would be compromised.
In prokaryotes, fork restart at nonorigin sites plays a crucial role in the protection against irreversible fork stalling. This is particularly required in these organisms because only a single genomic origin generates a pair of forks that replicate the entire genome before converging at a defined termination region. The main fork restart pathway is dependent on the homologous recombination (HR) machinery and is initiated at a DNA double-strand break (DSB) at the stalled fork (Cox et al. 2000; Michel et al. 2004). For example, replication forks in Escherichia coli can be stalled by inactivation of the replicative DnaB helicase (Michel et al. 1997). This generates a one-ended DSB at the stalled fork that triggers RecBCD- and RuvABC-dependent recombination between sister chromatids (Seigneur et al. 2000). Following RecA-mediated displacement loop (D-loop) formation and the action of the PriA–PriB–DnaT “primosome” complex, DnaB is reloaded for reassembly of the replisome (Seigneur et al. 1998; Heller and Marians 2006). Thus, E. coli has an efficient system for reassembly of the replisome via HR triggered by a one-ended DSB.
In eukaryotes, the form of HR repair used to deal with one-ended DSBs is known as break-induced replication (BIR) and plays an important role in both telomere maintenance and replication fork restart (McEachern and Haber 2006; Llorente et al. 2008; Verma and Greenberg 2016). BIR has been characterized in budding yeast through the analysis of interchromosomal HR induced by a one-ended DSB (Morrow et al. 1997; Bosco and Haber 1998). DNA synthesis during BIR is carried out by DNA polymerase δ (Pol δ), which is coupled to Pif1 helicase-dependent migration of a DNA D-loop structure (Saini et al. 2013; Wilson et al. 2013). The noncatalytic Pol32 subunit of Pol δ is essential for BIR but not bulk DNA replication (Lydeard et al. 2007). In mammalian cells, BIR is poorly characterized, partly because of a lack of defined assays. However, it has been shown that the POLD3 subunit (Pol32 homolog) of Pol δ is also required for BIR and alternative telomere maintenance in human cells (Costantino et al. 2014; Dilley et al. 2016). However, in contrast to yeast, mammalian POLD3 is essential for cell viability (Murga et al. 2016). Importantly, the mechanism of BIR in mammalian cells is still unclear, and it remains to be confirmed that it plays a key role in rescuing irreversibly stalled replication forks.
Replication forks in eukaryotes are driven by the hexameric MCM2–7 helicase, which forms the so-called CMG replicative holohelicase along with CDC45 and the GINS complex (Ilves et al. 2010). MCM2–7 activity is tightly controlled during the cell cycle (Blow and Dutta 2005; Masai et al. 2010). The loading of MCM2–7 at origins is temporally separated from helicase activation, with the former occurring in late M and G1 phases, and the latter occurring only in S phase. Importantly, the loading of additional MCM2–7 is suppressed in S phase, ensuring that DNA replication takes place only once per cell cycle. This implies that, unlike in E. coli, the replisome cannot be reassembled at a stalled fork by the reloading of the MCM2–7 helicase.
Most eukaryotic species, with the exception of yeasts and nematodes, have additional MCM family proteins, known as MCM8 and MCM9 (Liu et al. 2009). We and others reported that MCM8 and MCM9 form a distinct complex that is involved in HR repair (Lutzmann et al. 2012; Nishimura et al. 2012; Park et al. 2013). MCM9 was also shown recently to interact with mismatch repair (MMR) proteins and work with MCM8 as a helicase during MMR (Traver et al. 2015). MCM8 and MCM9 are both required for gametogenesis and tumor suppression in mice (Hartford et al. 2011; Lutzmann et al. 2012), and mutations in the human MCM8 or MCM9 genes are associated with premature onset of menopause (He et al. 2009; Wood-Trageser et al. 2014). Many lines of evidence point to a role for the MCM8–9 complex as a helicase in DNA repair, particularly in HR repair. However, there are conflicting views as to whether MCM8–9 is required for an early process (e.g., DNA end resection) or a later process in HR (Lutzmann et al. 2012; Nishimura et al. 2012; Lee et al. 2015).
In order to define the processes required for rescue of stalled forks in human cells and the possible role of MCM8–9 in these processes, we generated a human cell line in which the MCM2–7 helicase could be inactivated in a controlled manner. For this purpose, we used auxin-inducible degron (AID) technology, whereby a degron-tagged protein can be rapidly degraded by adding the plant hormone auxin (Nishimura et al. 2009; Natsume et al. 2016). This approach was adopted in order to create a situation in which the rescue of stalled forks by fork convergence is not possible (Supplemental Fig. S1B). We demonstrate that, in response to MCM2 degradation, stalled forks are converted to DSBs that are rescued in a RAD51-dependent manner. Crucially, this rescue requires MCM8–9 to promote new DNA synthesis. Although this reaction is superficially similar to the recently described mitotic DNA synthesis (MiDAS) at fragile sites (Minocherhomji et al. 2015; Bhowmick et al. 2016), we show that MCM8–9-dependent DNA synthesis is distinct from MiDAS. We propose that MCM8–9 is required for HR-mediated fork restart and acts as an alternative replicative helicase to promote DNA synthesis.
Results
Construction of an MCM2 degron cell line for artificial fork stalling
To characterize how human cells deal with stalled replication forks, we took inspiration from studies in prokaryotes in which the replicative DnaB helicase had been conditionally inactivated using a temperature-sensitive mutation of DnaB (Michel et al. 1997; Seigneur et al. 1998, 2000). In our case, we aimed to induce the rapid degradation of MCM2, a component of the replicative MCM2–7 helicase, by taking advantage of the AID technology (Fig. 1A; Nishimura et al. 2009; Natsume et al. 2016). To achieve this, we tagged both alleles of the MCM2 gene with mini-AID (mAID) using CRISPR–Cas9 in the HCT116 human colorectal cancer line (Fig. 1B; Supplemental S2A,B; Cong et al. 2013; Mali et al. 2013). Subsequently, we introduced an AFB2 gene (derived from Arabidopsis thaliana), which encodes a paralog of TIR1 (Supplemental Fig. S2C; Havens et al. 2012). In the resultant cells, the MCM2 fusion protein (MCM2-mAID) was degraded efficiently within 4 h of the addition of auxin (Fig. 1C), leading to an accumulation of cells in S phase, as expected (Supplemental Fig. S2D). To analyze this more systematically, we synchronized cells in the G1 phase using lovastatin and then treated them with or without auxin before releasing them into S phase (Fig. 1D,E; Javanmoghadam-Kamrani and Keyomarsi 2008). In control cells not exposed to auxin, DNA replication was generally completed within 19 h of release from G1 (Fig. 1E; control). In sharp contrast, a profound defect in DNA replication was observed in cells treated with auxin (Fig. 1E, +auxin). Importantly, this clear S-phase defect could not be seen using siRNA-mediated depletion methods because of the presence of residual MCM2 protein (Supplemental Fig. S2E,F).
Figure 1.
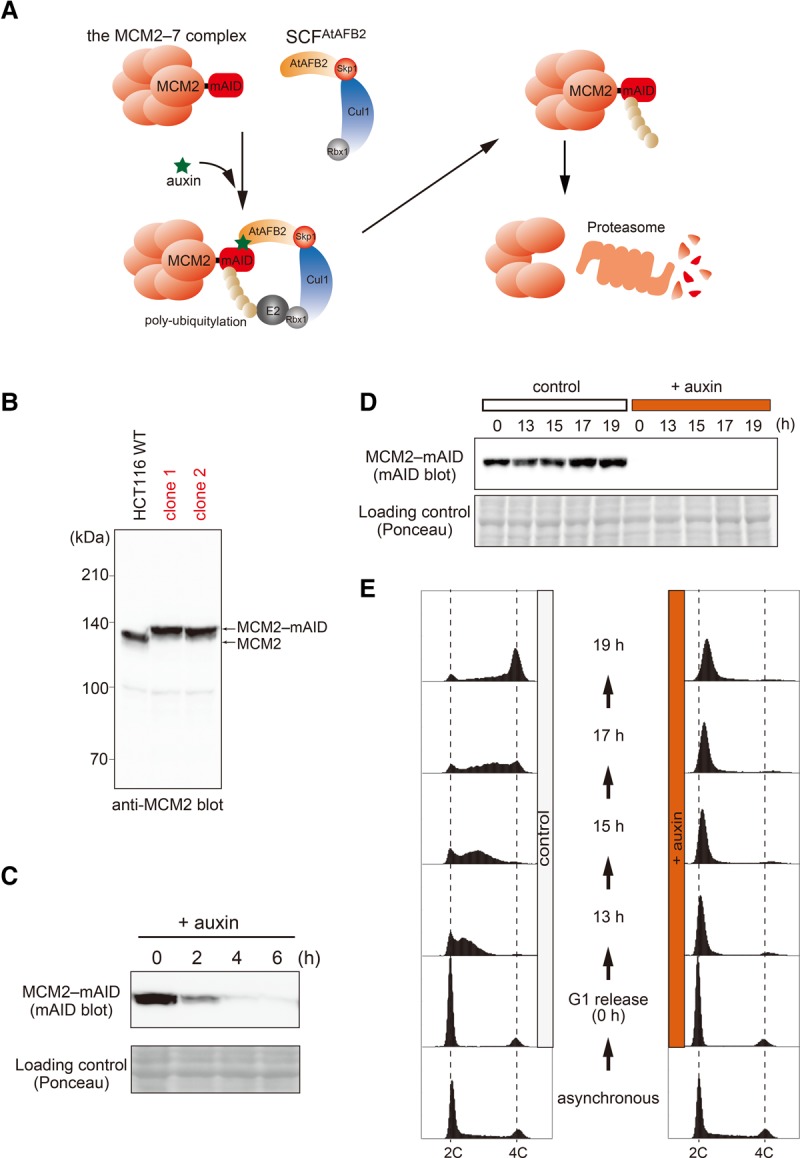
Construction of MCM2-mAID cells for artificial fork stalling. (A) A schematic representation of auxin-mediated proteolysis of MCM2-mAID. An auxin-dependent F-box protein, AFB2 of A. thaliana (AtAFB2), forms an E3 ubiquitin ligase with the endogenous SCF components. In the presence of auxin, MCM2-mAID is targeted by AtAFB2 for polyubiquitylation and subsequent destruction by the proteasome. (B) Evidence that clones 1 and 2 express the MCM2-mAID protein. (C) Time course of proteolysis of MCM2-mAID. Asynchronously growing MCM2-mAID cells were treated with auxin (indole-3-acetic acid [IAA]) before harvesting at the indicated time points. Ponceau staining shows a loading control. (D,E) MCM2-mAID cells were arrested in G1 phase and released into S phase after MCM2-mAID depletion. In control cells, DMSO replaced auxin. MCM2-mAID proteins were detected by immunoblotting using the anti-mAID antibody (D), and DNA content was measured by flow cytometry (E).
Fork stalling by induced proteolysis of MCM2 during S phase
In order to induce fork stalling only after the cells had initiated S phase, we synchronized the MCM2-mAID cells in G1 phase as in Figure 1E but then only added auxin 13 h after release, when most of the cells were in early S phase (Fig. 2A,B). In control cells not exposed to auxin, DNA replication was generally completed by the 19-h time point, and the cells then progressed through to mitosis (Fig. 2B, control). In contrast, the auxin-treated cells failed to carry out bulk DNA replication and eventually lost viability (Fig. 2B, +auxin; data not shown). The MCM2-mAID protein was undetectable by 2 h after auxin addition, suggesting that most replication forks would be inactivated at that point or soon afterward (Fig. 2A, +auxin 15 h). We also confirmed that the chromatin-bound fraction of MCM2-mAID, which is associated with forks and origins, was efficiently degraded (Supplemental Fig. S3A). Intriguingly, we noted that the peak of DNA content drifted toward a 4C DNA content between the 15- and 21-h time points (Fig. 2B, +auxin). More detailed flow cytometric analysis of cells containing a more than 2C DNA content showed that the F1 and F2 populations decreased, while the F3 and F4 populations increased between 15 and 21 h (Fig. 2C). This alternative DNA synthesis that occurs without the MCM2–7 helicase is addressed below.
Figure 2.

MCM2-mAID degradation in synchronized S-phase cells. (A,B) MCM2-mAID cells were arrested in G1 phase and then released into S phase. When most of the released cells were in early S phase (13 h), auxin was added to induce degradation of MCM2-mAID before taking samples at the indicated time points. In control cells, DMSO replaced auxin. MCM2-mAID proteins were detected by immunoblotting using the anti-mAID antibody (A), and DNA content was measured by flow cytometry (B). (C) The auxin-treated cells in early to late S phase were divided into four arbitrary fractions (F1, F2, F3, and F4). The frequencies of each fraction are indicated.
Because AID technology functions at the protein level, we took advantage of the ability to rapidly replenish the MCM2-depleted cells by removal of auxin from the medium. For this, we induced the rapid degradation of MCM2-mAID from 13 to 17 h after release from G1 phase, when the cells were in early S phase, and then removed auxin to allow MCM2-mAID re-expression (Supplemental Fig. S3B,C). Re-expression of MCM2-mAID was detectable 2 h after auxin removal (Supplemental Fig. S3B; 19 h). However, this failed to rescue the defective replication due to MCM2-mAID depletion (Supplemental Fig. S3C; left), consistent with the concept that the replication licensing system in eukaryotes prevents the replicative helicase from being reloaded to chromosomes in S phase (Blow and Dutta 2005). This is in contrast to the system operating in bacteria for reloading of the DnaB helicase (Marians 2000).
DNA DSBs are induced following fork stalling
Stalled replication forks can be converted into DSBs (Petermann et al. 2010). We analyzed whether DSBs were formed after MCM2-mAID depletion. For this, we initially looked at the 53BP1 protein, which forms nuclear foci upon DSB formation (Fig. 3A; Schultz et al. 2000; Anderson et al. 2001; Rappold et al. 2001). We observed that the auxin-treated cells accumulated 53BP1 foci in a manner similar to control cells treated with bleomycin, a known DSB-generating agent (Fig. 3B). To detect DSBs directly in the genomic DNA, we performed pulsed-field gel electrophoresis (PFGE) (Ray Chaudhuri et al. 2012). This revealed that DSBs started to accumulate 4 h after the cells were treated with auxin (Fig. 3C, 17 h). We then analyzed the localization of RAD51 and γH2AX, which form nuclear foci at the sites of damaged DNA (Fig. 4A). We observed a significant enrichment of RAD51 and γH2AX foci in the MCM2-mAID-depleted cells (Fig. 4A, +auxin), indicating that the cells were accumulating DNA damage. Consistent with this, we detected activation of the CHK1 kinase in cells treated with auxin (Supplemental Fig. S4A). Taken together, these results indicate that fork stalling induced by degradation of MCM2-mAID generates DNA DSBs.
Figure 3.
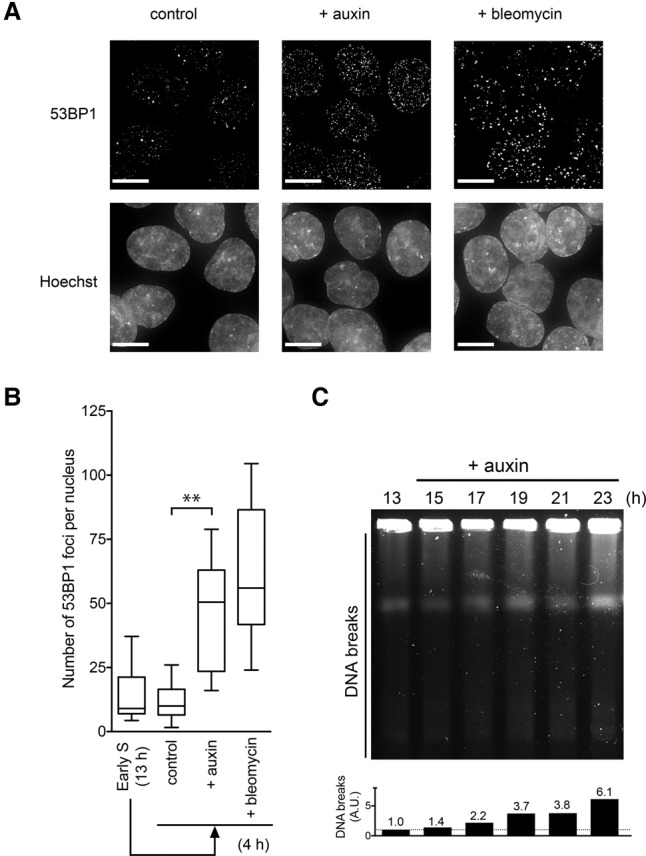
DSBs are generated following fork stalling. (A) 53BP1 focus formation following fork stalling induced by MCM2-mAID proteolysis. The MCM2-mAID cells were synchronized as in Figure 2 and were treated with auxin or bleomycin. At the 17-h time point, the cells were fixed and stained with an anti-53BP1 antibody. Bars, 10 µm. (B) Quantification of the number of 53BP1 foci. (C) PFGE was used to examine accumulation of DNA breaks after fork stalling. The MCM2-mAID cells were synchronized and treated as in Figure 2. At the indicated time points, genomic DNA from 2 × 105 cells was examined by PFGE. While intact genomic DNA remains in wells, DNA fragments generated by DSBs migrate into the gel. The level of DNA breaks (DNA fragments that migrate into the gel) is shown below. The level of fragmented DNA at time 13 h is denoted as 1.0.
Figure 4.
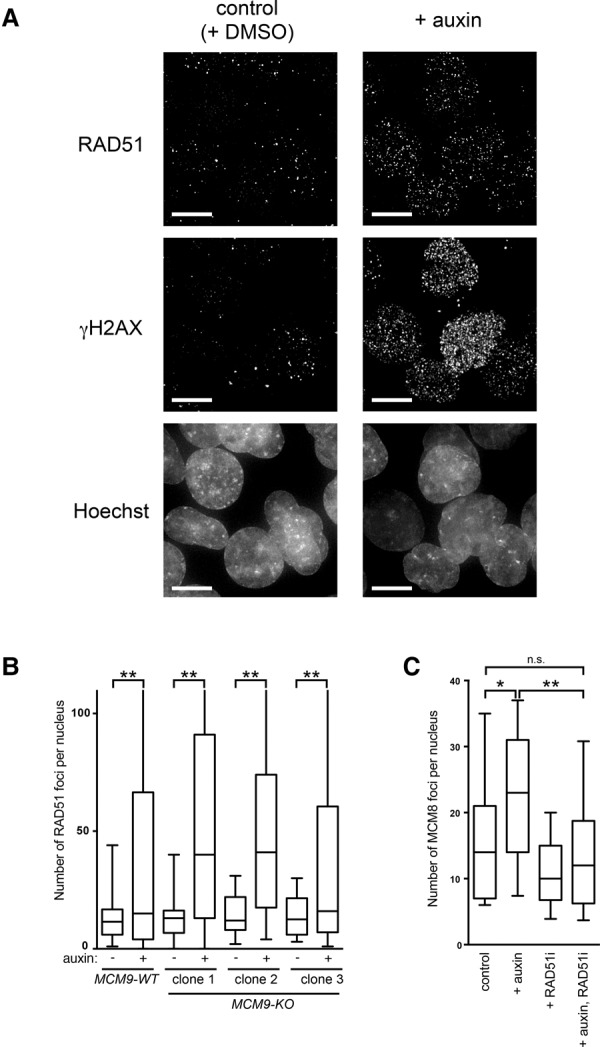
Fork stalling induces a DNA damage response. (A) DNA damage focus formation after MCM2-mAID degradation. The MCM2-mAID cells were synchronized and treated with auxin as in Figure 2. The cells were fixed at the 19-h time point and stained with the anti-γ-H2AX and anti-RAD51 antibodies. Bars, 10 µm. (B) MCM9 is not required for RAD51 focus formation. The MCM2-mAID cells in MCM9 wild-type (MCM9-WT) or knockout (MCM9-KO) backgrounds were treated with auxin for 8 h, and the number of RAD51 foci was quantified following immunostaining. (C) MCM8 focus formation upon MCM2-mAID degradation requires RAD51. The MCM2-mAID cells synchronized in early S phase were treated with auxin, a RAD51 inhibitor (RAD51i), or both together for 4 h. The number of MCM8 foci was quantified following immunostaining.
The presence of γH2AX and 53BP1 nuclear foci after MCM2-mAID depletion is indicative of replication-associated DSBs (Figs. 3A,B, 4A). However, we were intrigued by the colocalization of RAD51 with these DSBs, suggestive of the activation of some form of HR at the stalled forks. We therefore analyzed whether the MCM8–9 complex might be required for RAD51 focus formation. To this end, we disrupted the MCM9 gene in the MCM2-mAID background (Supplemental Fig. S4B–D). The MCM9 knockout (MCM9-KO) cells did not show a significant growth defect under normal growth conditions, in line with our previous observation that MCM9 loss is not detrimental to the growth of chicken DT40 cells (Supplemental Fig. S4E; Nishimura et al. 2012). Moreover, consistent with the observation that MCM8 and MCM9 function together in a complex, the formation of DNA damage-induced MCM8 foci was absent from the MCM9-KO cells (Supplemental Fig. S4F; Lutzmann et al. 2012; Park et al. 2013). We then analyzed RAD51 focus formation in the MCM9-KO cells (Fig. 4B). We observed that three independent clones of MCM9-KO cells formed RAD51 foci similarily to wild-type cells (MCM9-WT), indicating that MCM9 is not essential for RAD51 loading. Conversely, we obtained evidence that RAD51 is required for the loading of MCM8–9. In cells treated with the RAD51 inhibitor RI-1 (RAD51i) (Budke et al. 2012), we observed that MCM8 focus formation was significantly reduced (Fig. 4C).
MCM8–9 promotes DNA synthesis as a backup of DNA replication
As noted in Figure 2B, we observed that DNA synthesis continued to some extent for several hours after degradation of MCM2-mAID. Although this effect might reflect incomplete MCM2-mAID degradation, we considered the possibility that removal of this core component of the replisome might activate an alternative mechanism of DNA synthesis. We therefore investigated whether MCM8–9 might contribute to this MCM2–7-independent DNA synthesis. For this, we synchronized MCM9-WT and MCM9-KO cells in G1 and then released them into S phase as in Figure 2. In early S phase, auxin was added to induce MCM2-mAID depletion, and then time course samples were taken (Fig. 5A; Supplemental Fig. S5A). The MCM9-WT and MCM9-KO cells showed very similar replication profiles in the absence of auxin, indicating that MCM9 is dispensable for normal bulk DNA replication (Fig. 5A, control). In contrast, in the auxin-treated cells in which MCM2-mAID was degraded (Supplemental Fig. S5A, +auxin), the MCM9-KO cells showed a reduced level of DNA synthesis compared with the MCM9-WT cells between the 17- and 21-h time points (Fig. 5A, +auxin). This result was confirmed by analysis of the percentage of MCM9-KO cells present in the late stages of S phase (F3 and F4) (Supplemental Fig. S5B,C) and was also observed in an independent MCM9-KO clone (data not shown). To quantify this DNA synthesis defect in the MCM9-KO cells, we used bromodeoxyuridine (BrdU) to label nascent DNA 4 h after auxin addition. The population of cells labeled with BrdU was reduced in the MCM9-KO cells compared with the MCM9-WT cells (Fig. 5B, dotted circle).
Figure 5.
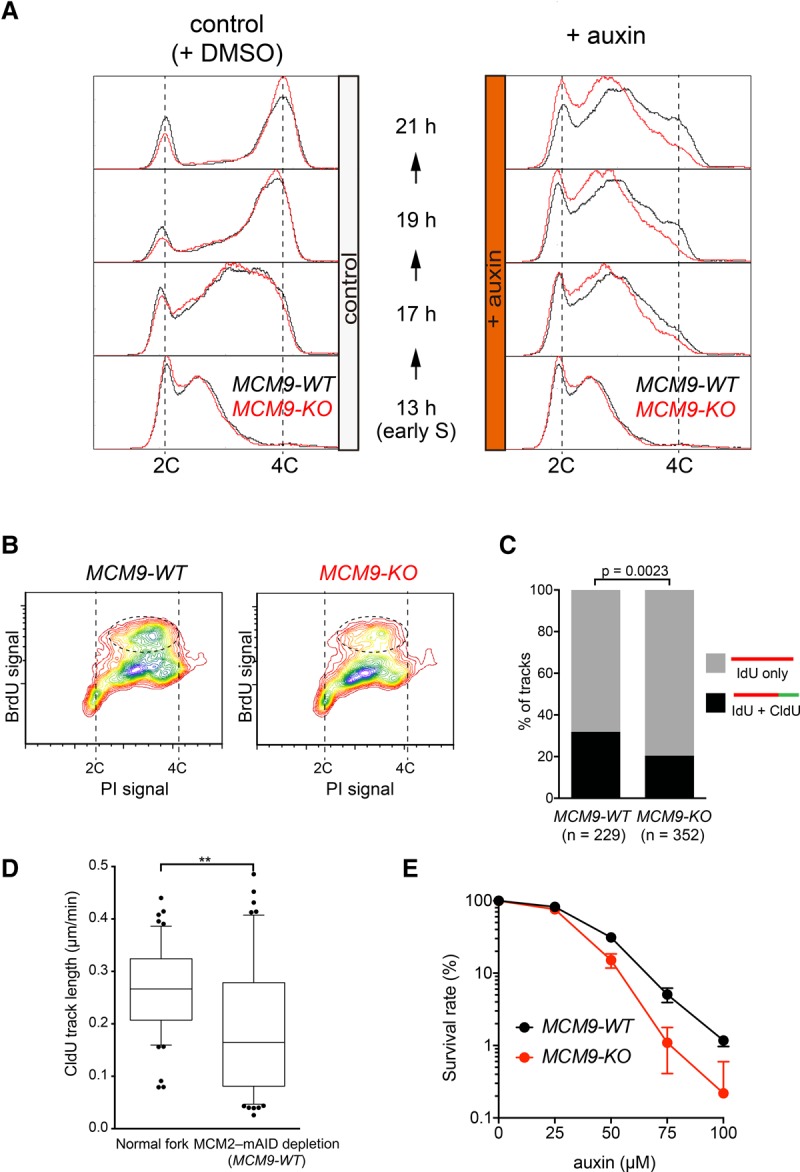
MCM8–9 promotes DNA synthesis after fork stalling. (A) MCM9-WT and MCM9-KO derivatives of the MCM2-mAID cells (shown with black and red lines, respectively) were synchronized and treated as in Figure 2. DNA content was then measured using flow cytometry after either DMSO (left) or auxin (right) treatment. (B) Incorporation of BrdU after fork stalling was examined using flow cytometry in the MCM9-WT and MCM9-KO backgrounds. (C) DNA synthesis after fork stalling was examined using isolated DNA fibers. The MCM9-WT and MCM9-KO derivatives of the MCM2-mAID cells were synchronized and treated as in Figure 2. At the 21-h time point, the cells were labeled with iododeoxyuridine (IdU) followed by chlorodeoxyuridine (CldU) labeling. The percentage of DNA fibers labeled with both IdU and CldU was compared with those that contained IdU only. n = 229 IdU; n = 352 CldU. (D) CldU tract length was quantified in the wild-type (normal fork) and MCM2-mAID cells with MCM9-WT. (E) Cells with a reduced amount of MCM2–7 are more reliant on MCM8–9 for their survival. MCM9-WT and MCM9-KO derivatives of the MCM2-mAID cells (shown in black and red lines, respectively) were allowed to form colonies in the presence of various doses of auxin. The colony number was counted after crystal violet staining. The survival rate in the absence of auxin was defined as 100%.
To directly quantify ongoing DNA synthesis, we exposed cells sequentially to two thymidine analogs, iododeoxyuridine (IdU) and chlorodeoxyuridine (CldU), and then performed immunodetection of the incorporated IdU and CldU on stretched DNA fibers (Fig. 5C). This revealed that the proportion of DNA fibers labeled with both IdU and CIdU was reduced in the MCM9-KO cells (Fisher's exact test, P = 0.0023). However, we still observed that ∼20% of the DNA fibers incorporated both IdU and CldU in the MCM9-KO cells. This suggests either that there is an MCM8–9-independent mechanism of DNA synthesis in the absence of MCM2-mAID or that degradation of MCM2-mAID was incomplete. We then analyzed the fork speed by measuring the length of CldU tracts (Fig. 5D). The fork speed after MCM2-mAID depletion was significantly slower than that of normal forks. This result supports the notion that MCM2-mAID depletion was efficient and that alternative DNA synthesis with MCM9 is qualitatively different from normal DNA synthesis. To define the location of DNA synthesis in the nucleus, we degraded MCM2-mAID using 4 h of auxin treatment and then labeled the MCM9-WT and MCM9-KO cells with ethynyldeoxyuridine (EdU) for 30 min. We observed that 17% of the large γH2AX foci colocalized with EdU foci (Supplemental Fig. S6A,B). However, colocalization of EdU and γH2AX foci was significantly reduced in the MCM9-KO cells (Supplemental Fig. S6B). Taken together, these results are consistent with the hypothesis that MCM8–9 contributes significantly to DNA synthesis after forks have been stalled due to a lack of the MCM2–7 complex.
It has been shown that a reduction in MCM2–7 expression causes spontaneous fork stalling by an extension in the size of replicons (Moreno et al. 2016). If, as we propose, MCM8–9 functions as a backup replicative helicase, the MCM2–7-depleted cells should have an increased reliance on MCM8–9. To test this hypothesis, we treated the MCM9-WT and MCM9-KO cells with various doses of auxin to reduce, but not eliminate, the cellular level of MCM2-mAID. We observed that the MCM9-WT cells were more resistant to a reduction in the level of MCM2-mAID than the MCM9-KO cells (Fig. 5E), supporting the hypothesis that MCM8–9 functions as a backup replicative helicase.
MCM8–9 promotes DNA synthesis during DSB-induced HR
Considering that MCM8–9 plays a role in HR repair (Lutzmann et al. 2012; Nishimura et al. 2012; Park et al. 2013), we investigated whether DNA synthesis occurring during DSB-induced HR requires MCM8–9. For this, we used an established HR reporter system in chicken DT40 cells, in which a copy of an SCneo substrate is stably introduced at the ovalbumin locus (Nishimura et al. 2012). These cells can repair the neomycin (Neo) resistance gene by HR upon expression of I-SceI, an endonuclease that generates a DSB in S2neo (Fig. 6A). If repaired by HR, two outcomes are possible, depending on whether short-tract gene conversion (STGC) or long-tract gene conversion (LTGC) is used (Johnson et al. 1999). To generate the STGC product, only 300 base pairs (bp) of DNA synthesis is required. In contrast, >3.3 kb of DNA synthesis is required to generate the LTGC product. A similar large product can be generated by sister chromatid exchange (SCE), but this is a rare event (Johnson and Jasin 2000).
Figure 6.
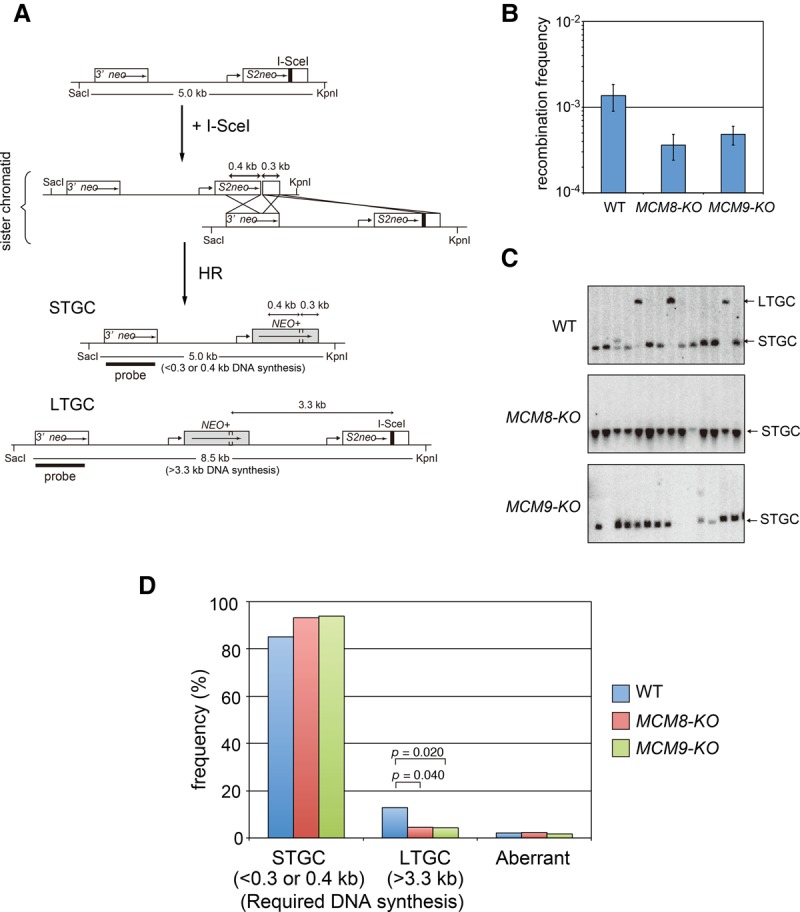
MCM8–9 promotes DNA synthesis in DSB-induced HR. (A) Schematic illustration showing the generation of STGC and LTGC from the SCneo substrate integrated at the ovalbumin locus in the chicken DT40 genome. The expressed I-SceI endonuclease cleaves the I-SceI site in S2neo and induces HR repair between sister chromatids. STGC is generated by copying 0.3–0.4 kb from 3′ neo, while LTGC is produced by a extensive DNA synthesis (>3.3 kb) from 3′ neo. (B) After I-SceI expression in wild-type, MCM8-KO, and MCM9-KO DT40 cells, the recombination frequency was calculated by counting G418-resistant colonies. Error bars indicate standard deviation. n = 3. (C) Southern blotting using a probe containing 3′ neo. Arrows indicate the positions of the STGC and LTGC products. (D) Frequency of each gene conversion event. Clones that were unable to be classified into either STGC or LTGC are shown as “aberrant.”
To analyze the putative role of MCM8–9 in DSB-induced HR, we compared wild-type DT40 cells with MCM8-KO or MCM9-KO. Initially, we looked at HR efficiency by quantifying Neo-resistant clones following I-SceI expression. This revealed that the MCM8-KO and MCM9-KO cells showed comparably reduced HR efficiencies, as reported previously (Fig. 6B; Lutzmann et al. 2012; Nishimura et al. 2012). Next, we analyzed the Neo-resistant clones that did arise in the MCM8-KO and MCM9-KO cells to define the outcome of the HR reactions. LTGC products can be distinguished from STGC by the size of a SacI–KpnI restriction fragment (Fig. 6A). In the wild-type background, 17% of clones were generated by LTGC, while this was reduced to 5% in the MCM8-KO and MCM9-KO cells (Fig. 6C,D). Conversely, STGC frequencies were elevated in the MCM8-KO and MCM9-KO cells. Taken together, we conclude that the MCM8–9 complex promotes DNA synthesis during DSB-induced HR.
MCM8–9-dependent DNA synthesis is distinct from RAD52-dependent MiDAS
Recently, it was shown that DNA synthesis could occur at common fragile sites in the human genome in the early stages of mitosis if cells are exposed to replication stress (Minocherhomji et al. 2015). This MiDAS requires RAD52 and is mechanistically distinct from conventional DNA replication (Bhowmick et al. 2016). Therefore, we investigated whether MCM8–9 might function in MiDAS. To this end, we disrupted the MCM8 gene in U2OS cells, in which MiDAS is known to occur very efficiently (Supplemental Fig. S7A–C; Minocherhomji et al. 2015). We then treated MCM8-WT and MCM8-KO U2OS cells with a low dose of the DNA polymerase inhibitor aphidicolin to induce replication stress and with the CDK1 inhibitor RO-3306 to induce a late G2-phase arrest. The cells were then released into mitosis in the presence of EdU to define sites of new DNA synthesis occurring in mitosis. Unexpectedly, we found that two independent clones of MCM8-KO cells showed an increase in EdU incorporation compared with MCM8-WT cells, showing that MiDAS is enhanced in MCM8-KO cells (Fig. 7A,B). Consistent with there being no role for MCM8–9 in MiDAS, the frequency of 53BP1 nuclear bodies in the following G1 phase (a marker of failed MiDAS) was unchanged in the MCM8-KO cells (Supplemental Fig. S7D,E; Minocherhomji et al. 2015). Interestingly, a similar increase in EdU incorporation has been observed previously in RAD51-depleted cells (Bhowmick et al. 2016). Therefore, we analyzed the epistatic relationship between RAD51 and MCM8–9. For this, we depleted RAD51 using siRNA in MCM8-WT and MCM8-KO cells and then quantified MiDAS (Fig. 7C; Supplemental Fig. S7F). We observed that RAD51 depletion enhanced the level of MiDAS in the MCM8-WT cells but not the MCM8-KO cells, consistent with RAD51 and MCM8–9 operating in the same pathway (Fig. 7C). We conclude that the RAD51/MCM8–9 axis promotes a backup form of DNA synthesis that is distinct from MiDAS.
Figure 7.
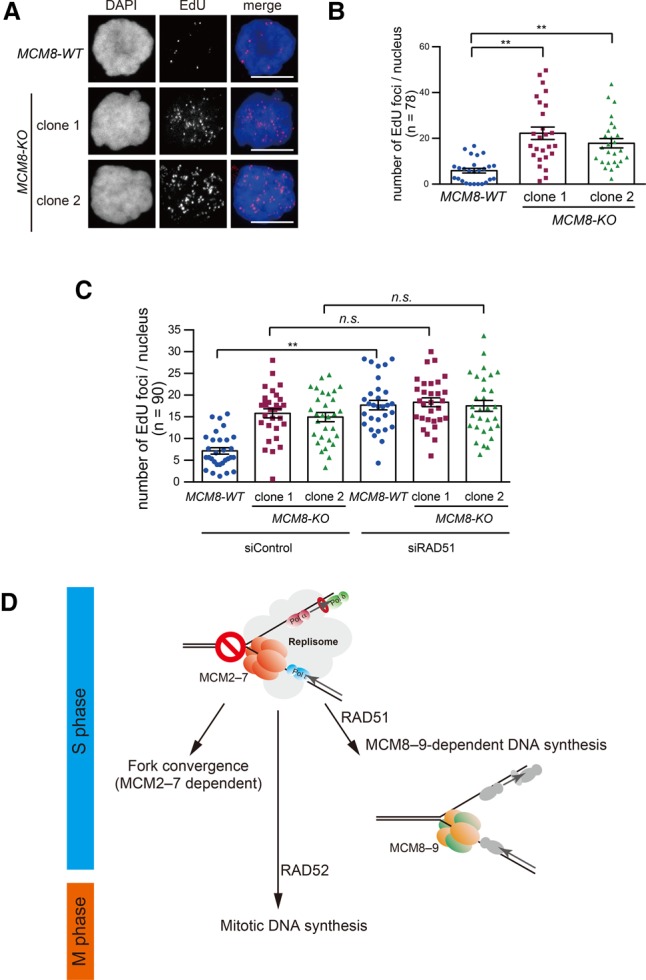
The RAD51–MCM8–9 axis is not involved in MiDAS. (A) MCM8-WT and MCM8-KO U2OS cells were treated with a low dose of aphidicolin and RO-3306 before being released into mitosis in the presence of EdU. Sites of EdU incorporation (MiDAS) were visualized using the Click-iT reaction. Bars, 10 µm. (B) Quantification of the number of EdU foci from A. (C) Quantification of MiDAS in cells with or without RAD51 depletion. (D) Schematic diagram outlining the multiple pathways that contribute to DNA synthesis and ensure the completion of genomic DNA duplication before chromosome segregation (see the text for details).
Discussion
Inactivating MCM2–7 as a novel strategy for stalling replication forks
Replication stress is frequently induced in cells by exposure to aphidicolin or hydroxyurea. These agents can lead to DSB formation, albeit after a long period of drug exposure (Petermann et al. 2010; Toledo et al. 2013). However, it is impossible to study DSB-induced DNA synthesis using these inhibitors, as they inhibit polymerase function. An alternative approach is to use DNA-damaging agents, but these cause the stalling of only a subset of replication forks, making it difficult to avoid fork convergence (Supplemental Fig. S1). Therefore, an efficient experimental system is required to permit the analysis of protective mechanisms invoked in dealing with fork stalling.
In prokaryotes, the response to fork stalling has been studied either by using the replication terminator Ter or through inactivation of the DnaB replicative helicase (Horiuchi and Fujimura 1995; Seigneur et al. 2000). Similarly, protein-mediated endogenous barriers have been used for arresting replication forks in yeasts (Ahn et al. 2005; Calzada et al. 2005; Lambert et al. 2005). Recently, the E. coli Tus/Ter system was transplanted into yeast and mouse cells to serve as a heterologous replication barrier (Larsen et al. 2014; Willis et al. 2014). These studies revealed detailed molecular events following fork pausing, in which recombination proteins often played a prominent part. Importantly, Willis et al. (2014) showed that the HR induced following Tus-induced fork pausing was regulated differently from that induced by a DSB.
Fork stalling caused by the inactivation of the replicative MCM2–7 helicase has been achieved using the heat-inducible degron system in budding yeast (Labib et al. 2001). Analogous studies using the same experimental system have not been possible in human cells until now because of the requirement for such a drastic temperature shift to induce protein degradation. We have now overcome this problem and achieved inactivation of MCM2–7 at replication forks for the first time in human cells by using AID technology (Nishimura et al. 2009; Natsume et al. 2016). Although simultaneous stalling of all forks is not expected to occur regularly under physiological conditions (and therefore some caution must be applied to interpretation using the MCM2-mAID cells), we suggest that this system could have broad applicability in the study of fork stalling in the future. Consistent with a previous report in yeast (Labib et al. 2001), we showed that inactivation of MCM2–7 in human cells causes fork stalling. Interestingly, this revealed the presence of residual DNA synthesis that occurred via an MCM8–9-dependent process. This new DNA synthesis was apparently not robust enough for the cells to complete S phase, indicating that MCM8–9-dependent DNA synthesis is either incapable of rescuing all stalled forks or lacks the processivity to cover the full genome. Alternatively, efficient fork restart might not be possible due to a limitation in the cellular level of HR repair factors in cases where multiple forks collapse simultaneously. MCM8–9-dependent synthesis might be designed to operate less efficiently than conventional S-phase replication and might normally be called into action at only a very small number of irreversibly stalled replication forks. Indeed, the MCM8–9 system might represent a double-edged sword for the maintenance of genome integrity. In support of this idea, the expression level of MCM8–9 is >100 times lower than that of MCM2–7 (Beck et al. 2011), and we note that overexpression of MCM8–9 is detrimental to proliferation (our unpublished data).
The MCM8–9 complex promotes HR-mediated DNA synthesis
The MCM8–9 complex has been implicated previously in HR, although there is a lack of consensus concerning the process involved (Nishimura et al. 2012; Park et al. 2013; Lee et al. 2015). We demonstrated that the formation of MCM8 foci is RAD51-dependent (Fig. 4B,C) and that MCM8–9 is required for both DNA synthesis after fork stalling induced by MCM2-mAID degradation (Fig. 5) and DSB-induced HR (Fig. 6). These results are consistent with previous observations showing that loss of the MCM8 homolog in mice and flies affects meiotic recombination only at a stage after the loading of the meiosis-specific RAD51 homolog DMC1 (Blanton et al. 2005; Lutzmann et al. 2012). Considering that the CMG holohelicase and the MCM8–9 complex each possess helicase activity (Ilves et al. 2010; Traver et al. 2015), we propose that MCM8–9 promotes DNA synthesis following RAD51-dependent DNA strand invasion between sister chromatids by acting as an alternative replicative helicase (Fig. 7D). In the future, it will be interesting to test whether DNA synthesis driven by MCM8–9 is semiconservative, like in normal DNA replication, or conservative in nature. This is because recent reports indicated that DNA synthesis occurring during BIR in yeast and DNA synthesis occurring during both the alternative lengthening of telomeres process and MiDAS in human cells are conservative events (Saini et al. 2013; Bhowmick et al. 2016; Roumelioti et al. 2016). It should be noted that, even though we propose that MCM8–9 promotes DNA synthesis in HR, it is possible that MCM8–9 is also involved in the resection of DSB ends (Lee et al. 2015). Both the human HCT116 and the chicken DT40 cells that we studied are recombination-proficient, and therefore any defect in resection might be masked by the availability of redundant resection systems dependent on BLM, DNA2, or EXO1 (Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008).
HR involved in interstrand cross-link (ICL) repair
We reported previously that the MCM8–9 complex is involved in HR-mediated ICL repair (Nishimura et al. 2012). Studies using Xenopus egg extracts showed that ICL repair in a replication-competent plasmid proceeds only after the convergence of replication forks on either side of the ICL (Raschle et al. 2008; Zhang et al. 2015). DSBs generated by ICL unhooking are repaired using HR. Indeed, MCM8–9 has been detected at sites of ICLs in the Xenopus system but was found not to be essential for ICL repair (Park et al. 2013). On the other hand, cells deficient in MCM8 or MCM9 are hypersensitive to ICL-inducing anti-cancer agents but only marginally sensitive to ionizing radiation, which generates two-ended DSBs (Nishimura et al. 2012; Park et al. 2013). We hypothesize that most ICLs are repaired through a two-ended DSB intermediately following fork convergence at the ICL. However, a small number of ICLs are converted into one-ended DSBs, the repair of which requires extensive DNA synthesis using MCM8–9. If true, this model might be particularly relevant to the repair of one-ended DSBs induced by ICLs that arise at chromosome fragile sites, where a single fork might have to travel over a long distance.
Noncanonical DNA syntheses as a backup of DNA replication
We showed that the RAD51/MCM8–9 axis operates separately from MiDAS. We therefore propose that cells have at least three systems to deal with instances of fork stalling (Fig. 7D): fork convergence, MCM8–9-dependent DNA synthesis, and MiDAS. We hypothesize that MCM8–9-dependent DNA synthesis and MiDAS are mechanistically related by being functionally analogous to yeast BIR but use a different set of HR factors.
Archaeal replication was carried out by an MCM homohexameric helicase. It is likely that the archaeal MCM helicase can carry out both origin-dependent and HR-mediated DNA replication (Hawkins et al. 2013). We speculate that, in eukaryotes, the MCM2–7 and MCM8–9 helicases evolved from a single ancestral MCM in order to catalyze origin-dependent DNA replication and HR-mediated DNA synthesis, respectively. This division of labor might have occurred in response to the establishment of the replication licensing system in eukaryotes (Blow and Dutta 2005). Evolutional loss of the MCM8 and MCM9 genes in yeast might be due to the fact that the number and distribution of origins on chromosomes evolved for optimal fork convergence so that MCM8–9-dependent DNA synthesis was dispensable in the ancestor of yeast (Liu et al. 2009; Newman et al. 2013). Further analysis of the fate of stalled forks and the role of MCM8–9 in their repair will hopefully reveal the relationship between conventional DNA replication and HR-mediated DNA synthesis in human cells.
Materials and methods
Cell lines
Genetically engineered HCT116 cell lines used in this study were as follows: HCT116 MCM2-mAID (clone 1: #269; clone 2: #270), HCT116 MCM2-mAID AtAFB2 (#310), HCT116 MCM2-mAID AtAFB2 MCM8–mCherry2 (#353), and HCT116 MCM2-mAID AtAFB2 MCM8–mCherry2 MCM9-KO (clone 1: #395; clone 2: #396; clone 3: #398). Genetically engineered U2OS cell lines used in this study were as follows: U2OS MCM8-KO (clone 1: #513; clone 2: #514).
Cell culture, transfection, and cloning
Human cell culture was undertaken as described previously (Natsume et al. 2016). To induce the degradation of MCM2-mAID, 500 µM indole-3-acetic acid (IAA; a natural auxin; Nacalai Tesque) was added to the culture medium unless otherwise noted. The RAD51 inhibitor RI-1 (Abcam) and bleomycin (Nippon Kayaku) were used at concentrations of 100 µM and 10 µg/mL, respectively. Transfection was performed using FuGENE HD (Promega). Transfected cells were selected with 1 µg/mL puromycin, 700 µg/mL G418, or 100 µg/mL HygroGold (InvivoGen). The detailed procedure for generation of mutant cells was described previously (Natsume et al. 2016).
Cell synchronization
HCT116 cells were synchronized in the G1 phase as described previously (Javanmoghadam-Kamrani and Keyomarsi 2008). Briefly, asynchronously growing HCT116 cells were treated with 20 µM lovastatin (LKT Laboratories) for 24 h to arrest them in G1 phase. Following that, the cells were washed twice with fresh medium and then grown in medium containing 2 mM mevalonic acid (Sigma-Aldrich).
RNAi
For depletion of the MCM2 and MCM5 proteins, HCT116 cells were transfected with 50 nM Silencer Select siRNAs (Thermo Fisher Scientific) using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific) following the manufacturer's instructions. The transfected cells were harvested after 72 h. RAD51 depletion in U2OS cells was performed as described previously (Bhowmick et al. 2016). We used the following siRNAs: siCONT (negative control; 4390843), MCM2-1 (s8586), MCM2-2 (s8587), MCM2-3 (s8588), MCM5-1 (s8595), and Mcm5-1i and Mcm5-2i (Ge et al. 2007).
Plasmid construction
We used pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene, 42230) for the construction of CRISPR plasmids following a published protocol (Ran et al. 2013). To construct a donor plasmid for the expression of A. thaliana AFB2 (AtAFB2) from the AAVS1 locus, pMK232 was modified (Natsume et al. 2016). The donor plasmid for tagging MCM2 with mAID was constructed by using PCR-amplified homology arms (1 kb each) and pMK286. The donor plasmid for tagging MCM8 with mCherry2 was constructed by using PCR-amplified homology arms (850 bp each) and pMK281. The donor plasmid for generating MCM8-KO in a U2OS background was constructed by using PCR-amplified homology arms (900 bp each) and a puromycin-resistant gene from pMK194.
Genomic PCR
To prepare genomic DNA, cells were lysed in buffer (100 mM Tris-HCl at pH 8.0, 200 mM NaCl, 5 mM EDTA, 1% SDS, and 0.6 mg/mL proteinase K) for 1 h at 55°C. After isopropanol precipitation, DNA pellets were washed with 70% ethanol and resuspended in TE containing 50 µg/mL RNase A overnight at 37°C. Genomic PCR was performed using Tks Gflex DNA polymerase (Takara Bio) according to the manufacturer's instructions (30 cycles of the following protocol: 10 sec at 98°C, 15 sec at 55°C, and 0.5 min at 68°C per kilobase).
Flow cytometry
Cells were collected and fixed in 70% ethanol. Fixed cells were washed once with PBS and then resuspended in PBS containing 1% BSA, 50 µg/mL RNase A, and 40 µg/mL propidium iodide. After incubation for 30 min at 37°C, the cells were filtered though a nylon mesh filter (42-µm pore size). DNA content was measured using an Accuri C6 flow cytometer (BD Biosciences) and analyzed by FCS Express 4 software (De Novo Software). For the analysis of DNA synthesis, cells were pulse-labeled with 30 µM BrdU before fixation with 90% ethanol. After washing with PBS, the fixed cells were treated with 2 M HCl and 0.5% Triton X-100 for 30 min for denaturing of genomic DNA. The cells were gently resuspended in 0.1 M Na2B4O7 (pH 8.5) and incubated for 30 min. After washing once with the antibody solution (1% BSA, 0.2% Tween 20 in PBS), cells were treated with the anti-BrdU antibody (BD Biosciences, B44) diluted in the antibody solution for 30 min. After washing once, the cells were treated with the FITC-conjugated anti-mouse antibody (Jackson Laboratory) diluted in the antibody solution for 30 min. Finally, the cells were treated with the antibody solution containing 50 µg/mL RNase A and 40 µg/mL propidium iodide before flow cytometer analysis.
Immunofluorescence staining
HCT116 cells were cultured in a glass-bottomed dish (MatTek) before fixation with 3.7% formaldehyde/PBS for 15 min. After washing twice with PBS, the cells were permeabilized with 0.5% Triton X-100/PBS for 20 min followed by a blocking treatment with 3% skim milk/PBS for 1 h. After washing twice with PBS, primary antibodies diluted in 1% BSA/PBS were applied before incubation for 1 h at room temperature. After washing three times with 0.05% Tween 20/PBS (PBS-T), secondary antibodies diluted in 1% BSA/PBS were applied before incubation for 1 h at room temperature. The cells were washed twice with PBS-T and once with PBS before DNA staining with 5 µg/mL Hoechst 33342 in PBS for 30 min. The coverslips were overlaid with VectaShield mounting medium (Vector Laboratories). Incorporated EdU was visualized using Click-iT Plus Alexa fluor 647 imaging kit (Thermo Fisher Scientific) before primary antibody treatment following the manufacturer's instruction.
Protein detection
To prepare whole-cell extracts, cells were lysed in RIPA buffer (25 mM Tris-HCl at pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) containing a protease inhibitor cocktail (Complete EDTA-free, Roche). Protein concentration was then measured using a Bradford assay kit (Bio-Rad). Tris-SDS (2×) sample buffer (125 mM Tris-HCl at pH 6.8, 4% SDS, 20% glycerol, 0.01% bromophenol blue, 10% 2-mercaptoethanol) was then added, and the samples were incubated for 5 min at 95°C. Preparation of chromatin-bound proteins was performed as described previously (Nishitani et al. 2014). Proteins were separated using SDS-PAGE, transferred to a nitrocellulose membrane (Protran Premium NC 0.45, GE Healthcare Life Sciences), and then incubated with antibodies after blocking with 5% skim milk/TBS-T for 30 min at room temperature. Detection was performed using ECL Prime detection reagent (GE Healthcare Life Sciences) with a ChemiDoc touch imaging system (Bio-Rad).
Antibodies
Antibodies used for immunoblotting and immunofluorescence were as follows: anti-MCM8 and anti-MCM9 antibodies (raised in rabbits; in-house antibodies), anti-MCM2 antibody (Santa Cruz Biotechnology, sc-9839), anti-MCM5 antibody (Santa Cruz Biotechnology, sc-22780), anti-mAID antibody (MBL, M214-3), anti-RFP antibody (MBL, M204-3), anti-RAD51 antibody (BioAcademia, 70001), anti-γH2AX antibody (Millipore, 05-636), anti-53BP1 antibody (Santa Cruz Biotechnology, sc-22760), anti-Histone H3 (Abcam, ab1791; and Active Motif, 39763), anti-α-tubulin (Sigma, 00020911), and anti-phospho-CHK1 (Ser345) antibody (Cell Signaling, 2341). For detection of MCM8-mCherry2, RFP-Booster ATTO 594 (Chromotek, rba594) was used.
Microscopy
Cells were visualized using a DeltaVision microscope equipped with deconvolution software, an incubation chamber, and a CO2 supply (GE Healthcare Life Sciences). For live-cell imaging, HCT116 cells were cultured in a glass-bottomed dish (MatTek) at 37°C with 5% CO2. To visualize nuclei in live cells, 10 µg/mL Hoechst 33342 was added to the medium before observation. DNA damage foci were analyzed using the Volocity software (PerkinElmer).
PFGE
PFGE was performed using a published protocol (Zellweger et al. 2015). To prepare cell plugs, 2 × 105 cells were embedded in 1% 2-hydroxyethylagarose (Sigma-Aldrich) using the 50-well plug mold (Bio-Rad). To lyse cells, the cell plugs were incubated in the plug lysis buffer (100 mM EDTA at pH 8.0, 1% sarkosyl, 0.2% sodium deoxycholate, 1 mg/mL proteinase K) overnight at 37°C. The cell plugs were washed once with the plug wash buffer (20 mM Tris-HCl at pH 8.0, 50 mM EDTA) before insertion into the wells of a 0.9% agarose gel. PFGE was performed using a CHEF Mapper XA PFGE system (Bio-Rad) with 0.5× TBE for 21 h at 14°C as follows: block 1: 9 h, 120° pulse angle, 5.5 V/cm, 30 sec to 18 sec switch time; block 2: 6 h, 117° pulse angle, 4.5 V/cm, 18 sec to 9 sec switch time; and block 3: 6 h, 112° pulse angle, 4.0 V/cm, 9 sec to 5 sec switch time. The gel was stained with GelGed (Biotium), and images were acquired using a ChemiDoc touch imaging system (Bio-Rad) and analyzed using Image Lab software (Bio-Rad).
DNA fiber assays
DNA fiber assays were performed following a published protocol with minor modifications (Schwab and Niedzwiedz 2011). Cells were pulse-labeled with 25 µM IdU for 30 min followed by a second labeling with 250 µM CldU for 30 min. The labeled cells were collected in ice-cold medium. Two microliters of cell suspension was spotted onto the Aminosilane-coated glass slide (Matsunami). Seven microliters of the fiber lysis solution (200 mM Tris-HCl at pH 7.5, 50 mM EDTA, 0.5% SDS) was applied onto the cells, followed by a gentle stirring. After 5 min, the glass slide was tilted at an angle of 15°, allowing DNA fibers to spread. After air-drying, the slide was treated with methanol/acetic acid (3:1) for 10 min to fix DNA fibers and then with 2.5 M HCl for 80 min to denature DNA. After washing three times with PBS, the slide was treated with PBS containing 5% BSA for 30 min for blocking. The slide was treated with antibodies (mouse B44 [BD Biosciences] and rat BU1/75 [Abcam] for IdU and CldU, respectively) for 1 h at room temperature. After washing three times with PBS, the slide was treated with Alexa fluor 594-conjugated anti-mouse IgG antibodies (Thermo Fisher Scientific, A-11032) and Alexa fluor 488 anti-rat IgG antibodies (Thermo Fisher Scientific, A-11006) for 1 h at room temperature. After three washes with PBS, the slide was sealed with a coverslip using VectaShield mounting medium (Vector Laboratories). Images were captured using an Olympus BX51 microscope with a DP72 CCD camera.
I-SceI-induced gene conversion assay
The I-SceI-induced gene conversion assay was performed as described previously (Yamamoto et al. 2005).
Detection of MiDAS
The assay was performed as described previously with minor modifications (Minocherhomji et al. 2015). Briefly, the assay used Click-iT chemistry according to the manufacturer's instructions but with a 1× final concentration of the Click-iT EdU buffer additive (Click-iT EdU Alexa fluor 594 imaging kit, Thermo Fisher Scientific). Asynchronously growing cells were treated with low-dose APH (0.4 µM) and RO-3306 (9 µM) (Sigma) for 16 h. Cells synchronized in late G2 were released into early prophase by vigorous washing (three to four times for up to 5 min each with 1× PBS prewarmed to 37°C). Subsequently, cells in early prophase were maintained for 30 min at 37°C in a humidified atmosphere containing 5% CO2 in prewarmed fresh medium supplemented with10 µM EdU (Thermo Fisher Scientific). Loosely attached mitotic cells were then shaken off and seeded on polylysine-coated slides and kept for 10 min at room temperature before simultaneous fixation and permeabilization using PTEMF buffer and subsequent EdU detection using Click-iT chemistry.
Supplementary Material
Acknowldgments
We thank Akemi Mizuguchi and Kaoru Iwai for experimental support. U2OS cells were a gift from Dr. Daiju Kitagawa. T.N. is supported by Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) grants (25891026, 15K18482, and 17K15068). M.T.K. is supported by JSPS KAKENHI grants (25131722 and 16K15095); a Japan Science and Technology Agency PRESTO (Precursory Research for Embryonic Science and Technology) program (JPMJPR13A5); and a research grant from the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the SGH Foundation, and the Sumitomo Foundation. I.D.H. is supported by grants from the Danish National Research Foundation (DNRF115), the European Research Council, and the Nordea Foundation. S.M. and R.B. were supported by post-doctoral fellowships from the Danish Medical Research Council.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.297663.117.
References
- Aguilera A, Gomez-Gonzalez B. 2008. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet 9: 204–217. [DOI] [PubMed] [Google Scholar]
- Ahn JS, Osman F, Whitby MC. 2005. Replication fork blockage by RTS1 at an ectopic site promotes recombination in fission yeast. EMBO J 24: 2011–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L, Henderson C, Adachi Y. 2001. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 21: 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. 2011. The quantitative proteome of a human cell line. Mol Syst Biol 7: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, Hickson ID. 2016. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol Cell 64: 1117–1126. [DOI] [PubMed] [Google Scholar]
- Blanton HL, Radford SJ, McMahan S, Kearney HM, Ibrahim JG, Sekelsky J. 2005. REC, Drosophila MCM8, drives formation of meiotic crossovers. PLoS Genet 1: e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow JJ, Dutta A. 2005. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 6: 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow JJ, Ge XQ, Jackson DA. 2011. How dormant origins promote complete genome replication. Trends Biochem Sci 36: 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco G, Haber JE. 1998. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150: 1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. 2012. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 40: 7347–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. 2005. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev 19: 1905–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. 2014. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. 2000. The importance of repairing stalled replication forks. Nature 404: 37–41. [DOI] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA. 2016. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539: 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, Glover TW. 2007. Chromosome fragile sites. Annu Rev Genet 41: 169–192. [DOI] [PubMed] [Google Scholar]
- Ge XQ, Jackson DA, Blow JJ. 2007. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev 21: 3331–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP. 2008. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22: 2767–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartford SA, Luo Y, Southard TL, Min IM, Lis JT, Schimenti JC. 2011. Minichromosome maintenance helicase paralog MCM9 is dispensible for DNA replication but functions in germ-line stem cells and tumor suppression. Proc Natl Acad Sci 108: 17702–17707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens KA, Guseman JM, Jang SS, Pierre-Jerome E, Bolten N, Klavins E, Nemhauser JL. 2012. A synthetic approach reveals extensive tunability of auxin signaling. Plant Physiol 160: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins M, Malla S, Blythe MJ, Nieduszynski CA, Allers T. 2013. Accelerated growth in the absence of DNA replication origins. Nature 503: 544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Kraft P, Chen C, Buring JE, Pare G, Hankinson SE, Chanock SJ, Ridker PM, Hunter DJ, Chasman DI. 2009. Genome-wide association studies identify loci associated with age at menarche and age at natural menopause. Nat Genet 41: 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Marians KJ. 2006. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7: 932–943. [DOI] [PubMed] [Google Scholar]
- Horiuchi T, Fujimura Y. 1995. Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J Bacteriol 177: 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, Schwob E, Mendez J. 2008. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci 105: 8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves I, Petojevic T, Pesavento JJ, Botchan MR. 2010. Activation of the MCM2–7 helicase by association with Cdc45 and GINS proteins. Mol Cell 37: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanmoghadam-Kamrani S, Keyomarsi K. 2008. Synchronization of the cell cycle using lovastatin. Cell Cycle 7: 2434–2440. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. 2000. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RD, Liu N, Jasin M. 1999. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 401: 397–399. [DOI] [PubMed] [Google Scholar]
- Labib K, Kearsey SE, Diffley JF. 2001. MCM2–7 proteins are essential components of prereplicative complexes that accumulate cooperatively in the nucleus during G1-phase and are required to establish, but not maintain, the S-phase checkpoint. Mol Biol Cell 12: 3658–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. 2005. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121: 689–702. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Sass E, Suski C, Mankouri HW, Hickson ID. 2014. The Escherichia coli Tus–Ter replication fork barrier causes site-specific DNA replication perturbation in yeast. Nat Commun 5: 3574. [DOI] [PubMed] [Google Scholar]
- Lee KY, Im JS, Shibata E, Park J, Handa N, Kowalczykowski SC, Dutta A. 2015. MCM8–9 complex promotes resection of double-strand break ends by MRE11–RAD50–NBS1 complex. Nat Commun 6: 7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Richards TA, Aves SJ. 2009. Ancient diversification of eukaryotic MCM DNA replication proteins. BMC Evol Biol 9: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente B, Smith CE, Symington LS. 2008. Break-induced replication: what is it and what is it for? Cell Cycle 7: 859–864. [DOI] [PubMed] [Google Scholar]
- Lutzmann M, Grey C, Traver S, Ganier O, Maya-Mendoza A, Ranisavljevic N, Bernex F, Nishiyama A, Montel N, Gavois E, et al. 2012. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol Cell 47: 523–534. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, Haber JE. 2007. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448: 820–823. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ. 2000. PriA-directed replication fork restart in Escherichia coli. Trends Biochem Sci 25: 185–189. [DOI] [PubMed] [Google Scholar]
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. 2010. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem 79: 89–130. [DOI] [PubMed] [Google Scholar]
- McEachern MJ, Haber JE. 2006. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75: 111–135. [DOI] [PubMed] [Google Scholar]
- Michel B, Ehrlich SD, Uzest M. 1997. DNA double-strand breaks caused by replication arrest. EMBO J 16: 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Grompone G, Flores MJ, Bidnenko V. 2004. Multiple pathways process stalled replication forks. Proc Natl Acad Sci 101: 12783–12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. 2015. Replication stress activates DNA repair synthesis in mitosis. Nature 528: 286–290. [DOI] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. 2007. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71: 13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno A, Carrington JT, Albergante L, Al Mamun M, Haagensen EJ, Komseli ES, Gorgoulis VG, Newman TJ, Blow JJ. 2016. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc Natl Acad Sci 113: E5757–E5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P. 1997. ‘Break copy’ duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M, Lecona E, Kamileri I, Diaz M, Lugli N, Sotiriou SK, Anton ME, Mendez J, Halazonetis TD, Fernandez-Capetillo O. 2016. POLD3 is haploinsufficient for DNA replication in mice. Mol Cell 63: 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, Kanemaki MT. 2016. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep 15: 210–218. [DOI] [PubMed] [Google Scholar]
- Newman TJ, Mamun MA, Nieduszynski CA, Blow JJ. 2013. Replisome stall events have shaped the distribution of replication origins in the genomes of yeasts. Nucleic Acids Res 41: 9705–9718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. 2009. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6: 917–922. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Ishiai M, Horikawa K, Fukagawa T, Takata M, Takisawa H, Kanemaki MT. 2012. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol Cell 47: 511–522. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Morino M, Murakami Y, Maeda T, Shiomi Y. 2014. Chromatin fractionation analysis of licensing factors in mammalian cells. Methods Mol Biol 1170: 517–527. [DOI] [PubMed] [Google Scholar]
- Park J, Long DT, Lee KY, Abbas T, Shibata E, Negishi M, Luo Y, Schimenti JC, Gambus A, Walter JC, et al. 2013. The MCM8–MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Mol Cell Biol 33: 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. 2010. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37: 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J. 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol 153: 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. 2008. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134: 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. 2012. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19: 417–423. [DOI] [PubMed] [Google Scholar]
- Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, Gagos S. 2016. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep 17: 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, Malkova A. 2013. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502: 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. 2000. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 151: 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab RA, Niedzwiedz W. 2011. Visualization of DNA replication in the vertebrate model system DT40 using the DNA fiber technique. J Vis Exp 10.3791/3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Bidnenko V, Ehrlich SD, Michel B. 1998. RuvAB acts at arrested replication forks. Cell 95: 419–430. [DOI] [PubMed] [Google Scholar]
- Seigneur M, Ehrlich SD, Michel B. 2000. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol Microbiol 38: 565–574. [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. 2013. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103. [DOI] [PubMed] [Google Scholar]
- Traver S, Coulombe P, Peiffer I, Hutchins JR, Kitzmann M, Latreille D, Mechali M. 2015. MCM9 is required for mammalian DNA mismatch repair. Mol Cell 59: 831–839. [DOI] [PubMed] [Google Scholar]
- Verma P, Greenberg RA. 2016. Noncanonical views of homology-directed DNA repair. Genes Dev 30: 1138–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. 2014. BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510: 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, et al. 2013. Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature 502: 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-Trageser MA, Gurbuz F, Yatsenko SA, Jeffries EP, Kotan LD, Surti U, Ketterer DM, Matic J, Chipkin J, Jiang H, et al. 2014. MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am J Hum Genet 95: 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. 2006. Excess Mcm2–7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 173: 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Hirano S, Ishiai M, Morishima K, Kitao H, Namikoshi K, Kimura M, Matsushita N, Arakawa H, Buerstedde JM, et al. 2005. Fanconi anemia protein FANCD2 promotes immunoglobulin gene conversion and DNA repair through a mechanism related to homologous recombination. Mol Cell Biol 25: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. 2015. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC. 2015. DNA interstrand cross-link repair requires replication-fork convergence. Nat Struct Mol Biol 22: 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.