


Abstract
Contact-dependent growth inhibition (CDI) is an important mechanism of inter-bacterial competition found in many Gram-negative pathogens. CDI+ cells express cell-surface CdiA proteins that bind neighboring bacteria and deliver C-terminal toxin domains (CdiA-CT) to inhibit target-cell growth. CDI+ bacteria also produce CdiI immunity proteins, which specifically neutralize cognate CdiA-CT toxins to prevent self-inhibition. Here, we present the crystal structure of the CdiA-CT/CdiIYkris complex from Yersinia kristensenii ATCC 33638. CdiA-CTYkris adopts the same fold as angiogenin and other RNase A paralogs, but the toxin does not share sequence similarity with these nucleases and lacks the characteristic disulfide bonds of the superfamily. Consistent with the structural homology, CdiA-CTYkris has potent RNase activity in vitro and in vivo. Structure-guided mutagenesis reveals that His175, Arg186, Thr276 and Tyr278 contribute to CdiA-CTYkris activity, suggesting that these residues participate in substrate binding and/or catalysis. CdiIYkris binds directly over the putative active site and likely neutralizes toxicity by blocking access to RNA substrates. Significantly, CdiA-CTYkris is the first non-vertebrate protein found to possess the RNase A superfamily fold, and homologs of this toxin are associated with secretion systems in many Gram-negative and Gram-positive bacteria. These observations suggest that RNase A-like toxins are commonly deployed in inter-bacterial competition.
INTRODUCTION
Bacteria have evolved several mechanisms to sense, communicate and compete with other microbes in the environment. Contact-dependent growth inhibition (CDI) is a common mode of inter-cellular interaction in which Gram-negative bacteria exchange protein toxins upon direct cell-to-cell contact (1–3). CDI toxin delivery is mediated by the CdiB/CdiA family of two-partner secretion proteins. CdiB is an Omp85 β-barrel protein that exports and displays CdiA effectors on the cell surface. CdiA proteins are very large (180–630 kDa), and each effector molecule is predicted to form an elongated filament that extends several hundred angstroms from the cell (3–5). CdiA recognizes specific receptors on neighboring bacteria, then transfers its C-terminal toxin region (CdiA-CT) into the target cell through an incompletely understood translocation pathway (6,7). The CdiA-CT region varies widely in sequence between effector proteins, but is typically demarcated by highly conserved peptide motifs such as VENN in enterobacteria and ELYN in the Burkholderiales (1,8,9). In accordance with CdiA-CT sequence diversity, CdiA effectors carry toxins with distinct activities, many of which are nucleases with unusual specificities (1,6,7,9–11). CDI+ bacteria exchange toxins with sibling cells, and therefore must also produce CdiI immunity proteins to neutralize CdiA-CT toxicity (2,3,12). Because CDI toxin-immunity pairs are polymorphic, immunity proteins do not protect against non-cognate toxins deployed by other bacteria. Thus, CDI allows bacteria to suppress the growth of non-isogenic neighbors, suggesting that these systems play a prominent role in bacterial competition.
The extraordinary diversity of CDI toxin-immunity proteins makes them an intriguing model to explore the evolution of protein-protein interactions. During the course of our studies to characterize CDI toxin families, we have solved the crystal structures of six CdiA-CT/CdiI complexes belonging to five different toxin-immunity sequence types (10,11,13–15). These complexes exhibit unique protein–protein interfaces; and though the toxins are all nucleases, each exhibits a distinct substrate specificity. The CdiA-CTECL toxin from Enterobacter cloacae ATCC 13047 is a member of the novel bacterial toxin Ntox21 family and resembles the C-terminal nuclease domain of colicin E3 (10). Like colicin E3, CdiA-CTECL cleaves 16S ribosomal RNA to block protein synthesis. CdiA-CTEC536 from uropathogenic Escherichia coli 536 contains an α-helical Ntox28 domain with a unique ribonuclease fold (13,16). The CDI toxins deployed by E. coli TA271 and Burkholderia pseudomallei isolates 1026b and E479 are all members of the PD-(D/E)XK phosphodiesterase superfamily, though the domains share no discernable sequence homology (11,14,15). Most PD-(D/E)XK nucleases are specific for DNA, and the CdiA-CTTA271 toxin is a metal-dependent DNase (11,14). However, the B. pseudomallei CdiA-CT1026b and CdiA-CTE479 toxins have no activity on DNA and instead cleave tRNA at distinct positions (11,15). The fact that three apparently unrelated toxin types share the PD-(D/E)XK core fold raises the possibility that other uncharacterized CDI toxins could also be members of the superfamily. Moreover, these observations suggest that many CdiA-CT domains may belong to known protein families, but the relationships are difficult to discern based solely on sequence comparisons and structure predictions.
In this report, we extend our analysis to the CDI toxin and immunity protein from Yersinia kristensenii ATCC 33638, which exhibit no sequence similarity to functionally characterized proteins in the NCBI database. Surprisingly, the crystal structure reveals that the C-terminal domain of CdiA-CTYkris adopts the same fold as RNase A superfamily members, which have hitherto only been identified in vertebrates. In accordance with this structural similarity, we find that CdiA-CTYkris has robust RNase activity in vivo and in vitro, though the toxin's catalytic triad is distinct from the RNase A superfamily. Finally, we identify many other CdiA-CTYkris toxin homologs throughout Gram-negative and Gram-positive bacteria. These homologs are typically found at the C-terminus of anti-bacterial effector proteins secreted through type V, type VI and type VII secretion systems, suggesting that RNase A-like toxins are commonly deployed in inter-bacterial conflict.
MATERIALS AND METHODS
Plasmid constructions and site-directed mutagenesis
CdiAYkris from Y. kristensenii ATCC 33638 is encoded by ordered locus ykris0001_13530; and CdiIYkris is encoded by ykris0001_13520, though the initiating Met codon of the immunity protein is misidentified in the genome annotation. Coding sequences for CdiA-CTYkris (corresponding to residues Val3116–Pro3396) and CdiIYkris were synthesized by Genscript (Piscataway, NJ, USA) and provided in plasmid pUC57. The cdiA-CT/cdiIYkris coding sequences were amplified with primers CH3751 (5΄-TTT CCA TGG TGG AGA ATA ACT CCA TGA GTG) and CH3752 (5΄-TTT CTC GAG ACT AGT GTT CAT TTG TTT ATA TAT ATG TTT CAA C) and ligated to pET21S using NcoI/XhoI restriction sites to generate plasmid pCH12245. The NcoI/SpeI fragment from pCH12245 was subcloned to generate plasmid pCH12560, which appends an ssrA(DAS) peptide tag onto CdiIYkris for controlled proteolysis mediated activation of CdiA-CTYkrisin vivo (12,17). The cdiIYkris immunity gene was amplified with primers CH3765 (5΄-CCG GTA CCA TGA TTA TTA ACG AAA AAT A) and CH3752 and ligated to plasmids pTrc99A and pET21P through NcoI/SpeI restriction sites to generate plasmids pCH12558 and pCH12559, respectively. Site-directed mutagenesis of putative active-site residues was performed by mega-primer PCR. Plasmid pCH12245 was amplified with CH3752 in conjunction with primers CH4057 (Arg186Ala; 5΄-GCG GAG CTA TTA AAT GCA GTG TCA ACG GGT AAT G), CH3861 (His175Ala; 5΄-CCT AAT CGA TAG GGC TGT TGG AAA AAC AGA A), CH3862 (Thr276Ala; 5΄-GGG TAT AAA ATC ATT GCT GGA TAT CCA ACA CCA) and CH3863 (Tyr278Ala; 5΄-GGG TAT AAA ATC ATT ACT GGA GCT CCA ACA CCA). The resulting products were used as megaprimers to amplify cdiA-CT/cdiIYkris with forward primer CH3751. The mutated fragments were ligated to protein over-production and ssrA(DAS)-tag vectors as described above. The Asn191Ala, Lys193Ala and Ser196Ala mutations were generated through PCR amplification of expression plasmids using primers: Asn191Ala forward (5΄-TTA AAT AGA GTG TCA ACG GGT GCG GTT AAA TCA GCG TCC TCA TTT) and reverse (5΄-AAA TGA GGA CGC TGA TTT AAC CGC ACC CGT TGA CAC TCT ATT TAA), Lys193Ala forward (5΄-GAG TGT CAA CGG GTA ATG TTG CGT CAG CGT CCT CAT TTA CAG) and reverse (5΄-CTG TAA ATG AGG ACG CTG ACG CAA CAT TAC CCG TTG ACA CTC), and Ser196Ala forward (5΄-CGG GTA ATG TTA AAT CAG CGG CTT CAT TTA CAG ATA GGA CTA C) and reverse (5΄-GTA GTC CTA TCT GTA AAT GAA GCC GCT GAT TTA ACA TTA CCC G).
Plasmid-borne chimeric CDI systems were constructed by allelic exchange of the counter- selectable pheS* marker from plasmid pCH10163 (11). cdiA-CT/cdiIYkris sequences were amplified with primers CH3749 (5΄-CAG GTA GGA ACT CGG TTG AGA ATA ATT CCA TGA GTG GAG ATC AAG) and CH3750 (5΄-GGT CTG GTG TCT AAC CTT TGG GTT AGT TCA TTT GTT TAT ATA TAT GTT TCA). The cdiA-CT/cdiIYkris sequences were fused to fragments amplified from regions upstream and downstream of the cdiAEC93 gene. The upstream homology fragment was amplified using primers CH4100 (5΄-GAA GCG ATG AAA GCA GCC AGG) and CH4101 (5΄-CTC AAC CGA GTT CCT ACC TGC CTG), and the downstream fragment with primers CH4102 (5΄-AAC CCA AAG GTT AGA CAC CAG ACC) and CH4103 (5΄-AAG TAG GCA TTC TCG ACC CTG). The three products were then fused to each other using overlap-extension PCR with primers CH4100/CH4103. The final DNA product (100 ng) was electroporated together with plasmid pCH10163 (300 ng) into E. coli DY378 cells (18). Recombinant plasmid clones were selected on yeast extract glucose-agar supplemented with 33 μg/ml chloramphenicol and 10 mM d/l-p-chlorophenylalanine.
Expression and purification for crystallography
Sub-cloning of the cdiA-CT/cdiIYkris coding sequence into pMCSG expression plasmids failed, presumably due to unbalanced production of toxin and immunity proteins. Therefore, an optimized ribosome-binding site was introduced upstream of cdiIYkris through PCR amplification of the pUC57 construct with primers (5΄-GTT TAA CTT TAA GAA GGA GAT ATA CAT ATG ATT ATT AAC GAA AAA TAT CCT TAT TTG TCA TAT CTA CTA A) and (5΄-CTC CTT CTT AAA GTT AAA CAC CAT TCT ATG GTG TTG GAT ATC CAG TAA TGA TTT TAT ACC). The product was treated with T4 DNA polymerase and transformed into E. coli BL21-Gold(DE3). After verification by DNA sequencing, the modified cdiA-CT/cdiIYkris operon was amplified with (5΄-GGA GTA AAG ATA ATG GTG GAG AAT AAC TCC ATG AGT GGA GAT) and (5΄-GTG ATG GTG ATG ATG GTT CAT TTG TTT ATA TAT ATG TTT CAA CCA TTC CTC A), and introduced into pMCSG81 by ligation-independent cloning to generate expression plasmid pMCSG81-CPX300012. The final construct appends a non-cleavage His6 tag to the C-terminus of CdiIYkris. Protein expression was performed as described (19,20). Briefly, E. coli BL21-Gold (DE3) cells carrying expression plasmids were grown at 37°C, 200 rpm in enriched M9 medium. At OD600 ≈ 1, 0.01% (w/v) each of l-Leu, l-Ile, l-Lys, L-Phe, l-Thr and l-Val were added to inhibit methionine biosynthesis and promote l-selenomethionine (SeMet) incorporation. SeMet (90 mg) was added to 1 l of culture and protein expression was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The cells were incubated at 18°C overnight. The harvested cells containing SeMet-labeled CdiA-CT/CdiIYkris complex were re-suspended in lysis buffer [500 mM NaCl, 5% (w/v) glycerol, 50 mM HEPES–NaOH (pH 8.0), 20 mM imidazole, 10 mM β-mercaptoethanol, 1 tablet protease inhibitor/50 ml extract (Roche Diagnostic)] and stored at –80°C. The suspension was thawed, sonicated and centrifuged at 13 000  g for 30 min. Supernatant was collected and mixed with 4 ml of Ni2+-NTA Sepharose (GE Healthcare Life Sciences) equilibrated with lysis buffer. The initial Ni2+-affinity purification process was performed on a vacuum assisted purification system with 25 mm Kontes columns. The Ni2+-NTA resin suspension was loaded on the column and the unbound proteins eluted with 200 ml of lysis buffer. The CdiA-CT/CdiIYkris complex was eluted with 30 ml of lysis buffer supplemented with 500 mM imidazole (pH 8.0). The eluted protein was treated with 5 μg/ml subtilisin A at 4°C overnight. The following day the sample was concentrated on Amicon 10 kDa membrane and separated on a Superdex 200 (GE Healthcare Life Sciences) size-exclusion column equilibrated with lysis buffer. The fractions containing CdiA-CT/CdiIYkris were pooled and exchanged into crystallization buffer [250 mM NaCl, 20 mM HEPES–NaOH (pH 7.5), 5 mM dithiothreitol].
g for 30 min. Supernatant was collected and mixed with 4 ml of Ni2+-NTA Sepharose (GE Healthcare Life Sciences) equilibrated with lysis buffer. The initial Ni2+-affinity purification process was performed on a vacuum assisted purification system with 25 mm Kontes columns. The Ni2+-NTA resin suspension was loaded on the column and the unbound proteins eluted with 200 ml of lysis buffer. The CdiA-CT/CdiIYkris complex was eluted with 30 ml of lysis buffer supplemented with 500 mM imidazole (pH 8.0). The eluted protein was treated with 5 μg/ml subtilisin A at 4°C overnight. The following day the sample was concentrated on Amicon 10 kDa membrane and separated on a Superdex 200 (GE Healthcare Life Sciences) size-exclusion column equilibrated with lysis buffer. The fractions containing CdiA-CT/CdiIYkris were pooled and exchanged into crystallization buffer [250 mM NaCl, 20 mM HEPES–NaOH (pH 7.5), 5 mM dithiothreitol].
Crystallization and data collection
The SeMet-labeled CdiA-CT/CdiIYkris complex was crystallized using sitting drop vapor diffusion in a CrystalQuick96 well round-bottom plates (Greiner Bio-one, Monroe, NC, USA). A 400 nl droplet of the protein (71.5 mg/ml) was mixed with a 400 nl droplet of crystallization formulations and allowed to equilibrate over 135 μl of crystallization reagent. Nano-pipetting was performed using the Mosquito nanoliter liquid handling system (TTP LabTech). The plates were incubated at 4°C and 16°C. SeMet-labeled complex produced several crystals in different crystallization formulations. The best diffraction quality crystals were obtained from 0.1 M MgCl2, 0.1 M sodium citrate (pH 5.0), 15% polyethylene glycol 4000 at 16°C. Prior to flash-cooling in liquid nitrogen, CdiA-CT/CdiIYkris crystals were cryoprotected by a flash washing in their reservoir solution supplemented with 20% polyethylene glycol. Diffraction data were collected at the Structural Biology Center 19-ID beamline at the Advanced Photon Source, Argonne National Laboratory. The single-wavelength anomalous diffraction (SAD) dataset was collected at 100 K near the selenium K-absorption edge. Diffraction images were processed with the HKL3000 suite (21). Intensities were converted to structure factor amplitudes in Ctruncate (22,23) from the CCP4 package (24). The data collection and processing statistics are given in Table 1.
Table 1. Data processing and refinement statistics.
| Processing | |
| Wavelength (Å) | 0.9792 |
| Resolution range (Å)a | 30.0–1.70 (1.73–1.70) |
| Space group | P1 |
| Unit cell parameters (Å, °) | a = 48.89, b = 57.15, c = 64.30, α = 78.3, β = 90.2, γ = 79.1 |
| Unique reflections | 71 046 (3503) |
| Multiplicity | 2.2 (2.1) |
| Completeness (%) | 97.1 (95.5) |
| <I>/<σI> | 8.13 (2.26) |
| Wilson B factor (Å2) | 17.6 |
| R merge b | 0.082 (0.437) |
| Refinement | |
| Resolution (Å) | 30.0–1.70 |
| Reflections work/test set | 69 621/1423 |
| R work/Rfree | 0.174/0.202 |
| Average B factor (Å2) (no. of atoms) | |
| Macromolecules | 22.4 (5153) |
| Na+ | 28.4 (2) |
| Solvent | 28.1 (436) |
| Rmsd bond lengths (Å) | 0.011 |
| Rmsd bond angles (Å) | 1.368 |
| Ramachandran favoredd (%) | 97.8 |
| Ramachandran outliers (%) | 0 |
| Clashscored | 1.27 |
| PDB ID | 5E3E |
aValues in parentheses correspond to the highest resolution shell.
b R merge = ΣhΣj|Ihj – <Ih>|/ΣhΣjIhj, where Ihj is the intensity of observation j of reflection h.
c R = Σh|Fo| – |Fc|/Σh|Fo| for all reflections, where Fo and Fc are observed and calculated structure factors, respectively. Rfree is calculated analogously for the test reflections, randomly selected and excluded from the refinement.
dAs defined by Molprobity (74).
Structure solution and refinement
The structure was solved by SAD phasing of the selenium peak data using the HKL3000 software pipeline (21). The positions of 11 heavy atoms were determined in SHELXD and initial phases were obtained from SHELXE (25). Phases were further improved through iterations of MLPHARE (26) and DM (27). The initial protein model was built by HKL builder utilizing Buccaneer (28). The final model was obtained through alternating manual rebuilding in COOT (29) and crystallographic refinement in Phenix (30) and Refmac (24,31). Protocol refinement included optimization of TLS parameters with five groups per chain. Refinement statistics are shown in Table 1. The atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession code 5E3E.
Expression and purification for biochemical characterization
CdiA-CT/CdiIYkris complexes were overexpressed in E. coli BL21-Gold (DE3) cells (Novagen). Cells were grown aerobically at 37°C in LB medium containing 50 μg/ml ampicillin and 40 mM potassium phosphate. CdiA-CT/CdiIYkris protein expression was induced by the addition of 1 mM IPTG at an OD600 ∼ 1 and incubated at 18°C for 16 h. Cells were collected by centrifugation at 5100 rpm for 30 min and washed with resuspension buffer [20 mM Tris–HCl (pH 7.4), 500 mM NaCl]. Cells were broken by sonication on ice in resuspension buffer supplemented with 10 mg/ml lysozyme and 40 μM phenylmethylsulfonyl fluoride. Unbroken cells and debris were removed by centrifugation at 14 000 rpm for 30 min followed by filtration through a 0.45 μM membrane. Clarified lysates were loaded onto a 5 ml Ni2+-charged HiTrap column (GE Healthcare) and the column washed with resuspension buffer supplemented with 10 mM imidazole. The CdiA-CT/CdiIYkris complex was eluted with a linear gradient of imidazole (10–500 mM) in resuspension buffer. Fractions were collected, combined and concentrated to a volume of ∼1 ml using a 10-kDa centrifugal concentrator.
His6-tagged CdiIYkris immunity protein was overexpressed and purified as described above and concentrations were determined by absorbance at 280 nm. CdiA-CTYkris proteins were isolated from CdiIYkris-His6 by Ni2+-affinity chromatography under denaturing conditions. CdiA-CT/CdiIYkris-His6 complexes were denatured in 6 M urea, 150 mM NaCl, 20 mM Tris–HCl (pH 7.4) overnight, then subjected to Ni2+-affinity chromatography in the same buffer. Denatured CdiA-CTYkris was collected in the flow-through fractions and dialyzed against refolding buffer [0.1 M Tris–HCl (pH 7.4), 0.1 M glycine] with progressively lower urea concentrations (4 M, then 2 M and finally no urea) for 4 h at 4°C, followed by dialysis into 20 mM Tris–HCl (pH 7.4), 150 mM NaCl. The same protocol was used for all CdiA-CTYkris toxin variants. Circular dichroism spectroscopy was used to confirm that the toxins were refolded.
Enzyme activity assays
cCMP hydrolysis reactions were performed in 20 mM Tris–HCl (pH 7.4), 150 mM NaCl. Reactions were initiated by the addition of 20 μl of 80 μM CdiA-CTYkris or bovine pancreatic RNase A to 60 μl solution of 4 mM cCMP. The production of 3΄-CMP was monitored by measuring the absorbance at 296 nm (Δε296nm = 506.4 M−1 cm−1 (32)). Under these conditions, RNase A and wild-type CdiA-CTYkris hydrolyzed approximately 27% and 13% (respectively) of the cCMP substrate within 4 min. To confirm that CdiIYkris inhibits CdiA-CTYkris activity, 1 mg of purified CdiIYkris-His6 was incubated with 0.5 mg of CdiA-CTYkris at 4°C overnight, and the complex was purified by Ni2+-affinity chromatography prior to use in activity assays. The eluted fractions containing a near 1:1 ratio of CdiIYkris-His6 to CdiA-CTYkris was determined by SDS-PAGE, and the concentration of the complex was determined using the extinction coefficient of both proteins. The same experiment was performed with RNase A and CdiIYkris and though RNase A has low affinity for CdiIYkris, some fractions contained a 1:1 ratio of RNase A:CdiIYkris. RNase assays were performed using total RNA isolated from E. coli strain X90 as described previously (33). E. coli RNA (100 μg/ml) was digested with 20 nM purified CdiA-CTYkris in 20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 100 μg/ml bovine serum albumin (30 μl final volume) at room temperature. Where indicated, reactions were supplemented with 100 nM purified CdiIYkris for 30 min prior to the addition of RNA substrate. Reactions were quenched in denaturing gel-loading buffer and run on 8 M urea 10% polyacrylamide gels buffered with 1 Tris–borate–EDTA. Gels were stained with ethidium bromide to visualize nucleic acids.
Tris–borate–EDTA. Gels were stained with ethidium bromide to visualize nucleic acids.
Wild-type CdiA-CTYkris and mutant variants were activated inside E. coli X90 cells using controlled proteolysis of ssrA(DAS)-tagged CdiIYkris immunity protein as described (12,17). Cells were grown in tetracycline-supplemented LB media to OD600 ∼ 0.1, then the expression of cdiA-CT/cdiI-DAS constructs was induced with 0.4% l-arabinose. Induced cultures were incubated at 37°C with shaking for 3 h, then harvested into an equal volume of ice-cold methanol. Cells were collected by centrifugation and the pellets frozen at –80°C prior to extraction of total cellular RNA as described (33). Isolated RNA (10 μg) was run on 8 M urea 10% polyacrylamide gels and visualized by staining with ethidium bromide.
Competition co-cultures
Escherichia coli EPI100 inhibitor strains carrying cosmids pCH12241 (wild-type CdiA-CTYkris), pCH13071 (His175Ala), pCH13157 (Arg186Ala), pCH13155 (Thr276Ala) or pCH13156 (Tyr278Ala) were co-cultured at a 1:1 ratio with E. coli MC4100 target cells carrying plasmid pTrc99A (cdiI–) or pCH12558 (cdiIYkris) in LB media. Viable inhibitor and target cells were enumerated on selective media at the beginning of co-culture and after 3 h. The competitive index was calculated as the ratio of target cells to inhibitor cells at 3 h divided by the initial inhibitor to target cell ratio. The competitive indices for three independent experiments are reported ± standard errors.
Biolayer interferometry
CdiIYkris binding affinities for CdiA-CTYkris and RNase A were determined by biolayer interferometry. All binding reactions were performed at 25°C in 20 mM Tris–HCl (pH 7.4), 150 mM NaCl. For toxin-immunity binding, CdiIYkris-His6 was immobilized on Ni2+-NTA biosensors and exposed to 3.125–50 μM CdiA-CTYkris. NHS-PEG4-Biotin (ThermoFischer Scientific) was added to RNase A at a 1:1 ratio for 30 min at room temperature. Then the biotinylated protein was purified on a Zeba™ spin desalting column (ThermoFisher Scientific). RNase A was immobilized on streptavidin biosensors and exposed to 6.25–50 μM CdiIYkris. A buffer reference was subtracted from all binding curves before curve fitting. Curve fitting and data processing were performed using BLItz Pro Software (ForteBio Inc.).
Thermal denaturation
Circular dichroism (CD) spectra were collected in duplicate on 0.5 mg/ml protein solutions in water. Ellipticity was measured every 0.5 nm with a response of 8 s and a scanning speed of 10 nm/min. The temperature was increased from 20°C to 90°C and spectra were recorded every 10°C. The global unfolding curve was obtained using Equation (1)
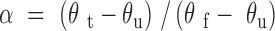 |
(1) |
where α is the fraction of folded protein at any temperature, θt the ellipticity value for a specific temperature, θυ represents 100% denatured protein (90°C) and θf represents 100% native protein (20°C).
RESULTS
Structure of the CdiA-CT/CdiIYkris complex
The structure of the CdiA-CT/CdiIYkris complex from Y. kristensenii ATCC 33638 was solved to 1.7 Å resolution with three toxin/immunity protein complexes in the asymmetric unit (Figure 1A and Table 1). As we have found with all previous CdiA-CT structures, the final model only contains the C-terminal domain, corresponding to residues Gly168–Pro281 (as numbered from Val1 of the VENN motif). The entirety of the CdiIYkris immunity protein (residues Met1–Asn98) is resolved, though chain C is missing four N-terminal residues. Two of the CdiA-CTYkris domains (chains B and D) are very similar to one another (rmsd of 0.40 Å), whereas the third toxin domain (chain F) superimposes upon chain B and D with rmsd values of 0.51 Å over all α-carbon atoms. These differences arise from a slight shift of helices α1 and α2 in toxin chain F. In contrast, all three CdiIYkris chains adopt very similar conformations with rmsd values between 0.28 and 0.33 Å over all α-carbon atoms.
Figure 1.

Structure of CdiA-CT/CdiIYkris complex. (A) The CdiA-CT/CdiIYkris complex is depicted in ribbon representation. Secondary structure elements are indicated, with those of CdiIYkris denoted with a prime symbol (΄). (B) The CdiA-CT/CdiIYkris complex interface is mediated by an extensive network of ion pairs and H-bonds. Interacting residues are represented as sticks with oxygen, nitrogen, toxin carbon and immunity carbon atoms are colored red, blue, green and cyan, respectively. Electrostatics and H-bonds are represented by orange and black dashed lines, respectively.
The C-terminal domain of CdiA-CTYkris consists of an extended β-sheet system and four α-helices. The toxin has a kidney-shaped structure, in which the two antiparallel β-sheets form lobes that are connected through a hinge region. The short α1 helix is positioned within the hinge between the two β-sheet lobes. Helix α2 runs parallel to the back side of the first β-sheet (β1, β3 and β4). The second β-sheet (β2, β5 and β6) curves toward the outside of the structure, with α3 helix running through its central cavity. Helix α4 follows α3 directly and lies antiparallel to strand β2. The CdiIYkris immunity protein is composed of eight α-helices (α1΄ – α8΄) packed together to form a nearly spherical structure with weak structural homology to a putative TetR family transcriptional repressor (Table 2). The CdiIYkris protein fits into the curved cavity of the CdiA-CTYkris toxin domain. The primary interactions involve helix α1 and the second β-sheet (β2, β5 and β6) of the toxin and helix α6΄ of the immunity protein and its surrounding loops. The toxin/immunity protein interface is mediated largely by shape and electrostatic potential complementation and includes 14 direct hydrogen-bonds (H-bonds) and eight salt-bridges (Figure 1B and Table 3). The toxin-immunity protein interface buries ∼1010 Å2, corresponding to 15.0% and 17.1% of the total surface area of CdiA-CTYkris and CdiIYkris, respectively.
Table 2. Structural homologs of CdiA-CTYkris (Chain B) and CdiIYkris (Chain A).
| Protein | Homolog | PDB identifier | Z-Score | rmsd (Å)a | Lali/tnresb | nresc | %idd |
|---|---|---|---|---|---|---|---|
| CdiA-CTYkris | Angiogenin | 4B36 | 5.1 | 3.4 | 83/113 | 112 | 4 |
| CdiA-CTYkris | Protein P-30 (onconase) | 3FD7 | 5.1 | 3.3 | 79/113 | 103 | 11 |
| CdiA-CTYkris | Mus musculus pancreatic RNase 1 | 3TSR | 4.9 | 3.6 | 89/113 | 117 | 6 |
| CdiIYkris | Shew_3104 (putative TetR-family member) | 3PPB | 4.9 | 3.3 | 82/98 | 188 | 0 |
armsd over aligned α-carbon atoms.
bLali/tnres is the number of residues in the structural alignment/total number of residues of CdiA-CTYkris or CdiIYkris.
cnres is the number of total residues in the homologous protein.
d%id is the percent sequence identity between the aligned proteins.
Table 3. Hydrogen bonds and salt bridges between CdiIYkris (Chain A) and CdiA-CTYkris (Chain B).
| CdiA-CTYkris | CdiIYkris | Distance (Å) |
|---|---|---|
| Hydrogen bonds | ||
| ASN 191 O | TYR 78 N | 2.88 |
| ASN 191 ND2 | GLU 23 OD2 | 2.82 |
| LYS 193 NZ | CYS 75 O | 2.76 |
| LYS 193 N | SER 76 O | 2.80 |
| SER 196 OG | GLN 20 NE2 | 3.17 |
| SER 196 N | GLN 20 OE1 | 3.06 |
| GLY 229 N | GLU 40 OE1 | 3.17 |
| GLY 229 N | GLU 40 OE2 | 3.07 |
| GLY 229 N | TYR 12 OH | 3.21 |
| TYR 230 N | TYR 12 OH | 2.90 |
| TYR 230 O | TYR 12 OH | 3.55 |
| GLU 232 N | CYS 16 SG | 3.89 |
| THR 276 OG1 | ASP 21 OD2 | 2.54 |
| TYR 278 OH | GLN 20 N | 3.01 |
| Salt bridges | ||
| ARG 174 NH2 | ASP 21 OD1 | 3.22 |
| ARG 174 NE | ASP 21 OD1 | 2.74 |
| HIS 175 NE2 | ASP 21 OD1 | 2.60 |
| HIS 175 NE2 | ASP 21 OD2 | 2.60 |
| LYS 193 NZ | ASP 74 OD1 | 2.85 |
| GLU 232 OE2 | ARG 15 NH1 | 2.98 |
| ARG 259 NHI | ASP 74 OD2 | 2.74 |
| ARG 259 NH2 | ASP 74 OD2 | 3.31 |
CdiA-CTYkris is structurally homologous to the RNase A family
Using the DALI server (34), we identified human angiogenin, Rana pipiens protein P-30 (onconase) and mouse pancreatic ribonuclease (RNase 1) as the closest structural homologs of CdiA-CTYkris (Table 2). Though CdiA-CTYkris shares little sequence identity with these RNase A family members (Figure 2A), angiogenin and RNase 1 adopt similar structures with two β-sheets forming an overall kidney shape (Figure 2C and D) (35,36). All RNase A paralogs contain three or four disulfide bonds, but the CdiA-CTYkris toxin domain lacks cysteine residues (Figure 2A). The nuclease active site is conserved in all RNase A family members and consists of catalytic residues His13, Lys40 and His116 (angiogenin numbering) (37). RNA cleavage is initiated by His13, which acts as a general base to abstract a proton from the 2΄-hydroxyl of substrate RNA. Deprotonation precipitates an intra-molecular nucleophilic attack on the adjacent phosphodiester bond. Lys40 is thought to stabilize the pentavalent transition-state intermediate (38), and His116 donates a proton to the 5΄ leaving group. This phosphotransferase reaction cleaves the RNA backbone, producing fragments with cyclic 2΄,3΄-phosphodiester and 5΄-hydroxyl termini. Structural alignment shows that toxin residues His175, Val192 and Thr276/Tyr278 superimpose onto His13, Lys40 and His116 (respectively) of angiogenin (Figure 3A). Though Val192 is unlikely to stabilize the transition state, toxin residues Arg186, Asn191 and Lys193 are in the same vicinity (Figure 3B) and have side-chains that could participate in catalysis. Further, the backbone carbonyl and side-chain of angiogenin residue Thr44 contribute to substrate specificity by forming direct H-bonds with pyrimidine bases (39). CdiA-CTYkris residue Ser196 occupies a similar position as Thr44 (Figure 3B), suggesting that it may also interact with substrate nucleobases. We note that several of the predicted active-site residues interact directly with the immunity protein and contribute to the formation of the CdiA-CT/CdiIYkris complex (Figure 3C). Thus, CdiIYkris most likely neutralizes toxin activity by blocking access to RNA substrates.
Figure 2.

The CdiA-CTYkris nuclease domain adopts the RNase A fold. (A) Structure-based alignment of the CdiA-CTYkris nuclease domain with human angiogenin (ANG) and mouse RNase 1. Secondary structure elements are indicated above the alignment. Putative catalytic and substrate-binding residues are boxed in red and blue, respectively. (B) CdiA-CTYkris nuclease domain. (C) Human ANG (PDB ID: 4B36 (35)). (D) Mouse pancreatic RNase 1 (PDB ID: 3TSR (36)). Structures in panels B–D are shown in cartoon representation with catalytic residues depicted in stick representation. (E) Superimposition of CdiA-CTYkris with ANG and RNase 1 in ribbon representation.
Figure 3.

Active sites of angiogenin and CdiA-CTYkris. (A and B) Superimposition of the active sites of ANG and the CdiA-CTYkris nuclease domain. The ANG active site is composed of a conserved catalytic triad of His13, Lys40 and His116, which overlay onto CdiA-CTYkris residues His175, Val192 and Thr276/Tyr278, respectively. Panel B shows a clockwise 90° rotation along the z-axis with respect to panel A. (C) Proposed CdiA-CTYkris active site residues interact with the immunity protein. All panels depict structures in cartoon representations with interacting residues shown in stick representation.
CdiA-CTYkris toxin degrades RNA in vitro and in vivo
The structural similarity between CdiA-CTYkris and the RNase A family suggests that the toxin has RNase activity. Indeed, we found that purified CdiA-CTYkris toxin rapidly degrades E. coli RNA in vitro, and the activity was neutralized when CdiIYkris was included in the reaction (Figure 4A). We note that the reactions were not supplemented with divalent cation, suggesting that CdiA-CTYkris-mediated nuclease activity is metal-independent like RNase A (40). Toxin domains carrying mutations in putative active-site residues showed significantly reduced RNase activity, though the Arg186Ala and Thr276Ala mutants were able to degrade a significant proportion of the substrate upon prolonged incubation (Figure 4A). To assess toxin activity in vivo, we used controlled proteolysis of the CdiIYkris immunity protein to activate wild-type and mutant toxin domains inside E. coli cells (12,17). The resulting constructs were expressed under the control of an arabinose-inducible promoter and total RNA was extracted for analysis by gel electrophoresis. Induction of the empty vector had no apparent effect on cellular RNA (Figure 4B). By contrast, activation of the wild-type toxin resulted in nearly complete degradation of tRNAs and the appearance of high-molecular weight fragments that presumably represent 23S and 16S rRNA cleavage fragments (Figure 4B). As expected from the in vitro RNase assays, expression of the Thr276Ala and Arg186Ala mutant toxins resulted in detectable tRNA degradation, whereas cells expressing the His175Ala and Tyr278Ala variant toxins showed little evidence of nuclease activity (Figure 4B).
Figure 4.

CdiA-CTYkris cleaves bacterial RNAs. (A) CdiA-CTYkris RNase activity in vitro. Purified CdiA-CTYkris variants were incubated with total E. coli RNA as described in the Methods. Reactions were quenched at the indicated times and run on denaturing polyacrylamide gels and visualized by staining with ethidium bromide. Where indicated (+CdiI), reactions were supplemented with purified CdiIYkris immunity protein. (B) CdiA-CTYkris RNase activity in vivo. The indicated CdiA-CTYkris variants were activated inside E. coli cells, and total RNA isolated for denaturing polyacrylamide gel analysis. (C) Competition co-culture assays. Inhibitor cells expressing the indicated CdiA-CTYkris toxins were co-cultured at a 1:1 ratio with target cells that either lack (–) or carry (+) the cdiIYkris immunity gene. The competitive index is the ratio of target to inhibitor cells at 3 h divided by the initial inhibitor to target cell ratio. Data for three independent experiments are presented ± the standard error of the mean.
We then tested whether the mutant toxin domains exhibit growth inhibition activity during cell-mediated CDI competition. We fused wild-type and mutant CdiA-CTYkris domains onto CdiAEC93 at the VENN peptide motif to generate chimeric CdiA effector proteins. Inhibitor cells expressing these chimeras were co-cultured with target bacteria at a 1:1 ratio, and viable inhibitor and target bacteria were enumerated after 3 h. Target-cell fitness was expressed as the competitive index, which is the ratio of target to inhibitor cells at the end of the co-culture. Target bacteria were effectively outcompeted by inhibitor cells that deploy the wild-type CdiA-CTYkris domain (Figure 4C), indicating that the toxin is delivered into E. coli cells. Moreover, target bacteria proliferated in co-cultures when they expressed the CdiIYkris immunity protein (Figure 4C), demonstrating that the inhibitory effect is due to CdiA-CTYkris nuclease activity. By contrast, inhibitor cells that deploy mutated CdiA-CTYkris toxin domains were unable to inhibit the growth of target bacteria (Figure 4C). Thus, although the Arg186Ala and Thr276Ala variants retain residual RNase activity, these toxins are not sufficiently potent to inhibit target-cell growth.
CdiA-CTYkris hydrolyzes cCMP
In addition to the phosphotransferase reaction described above, RNase A also hydrolyzes 2΄,3΄-cyclic phosphodiesters. Though the same catalytic residues activate water for hydrolysis, this reaction is much slower and independent of phosphotransferase activity (40). To determine whether CdiA-CTYkris possesses activity, we monitored the conversion of cytidine 2΄,3΄-cyclic monophosphate (cCMP) to cytidine 3΄-monophosphate (3΄-CMP), which absorbs light at 296 nm. Like RNase A, wild-type CdiA-CTYkris hydrolyzed cCMP to 3΄-CMP, though the rate of hydrolysis was ∼4-fold lower than that observed for RNase A (Figure 5A). Inclusion of CdiIYkris attenuated hydrolytic activity by at least 80%, but the immunity protein had no effect on the hydrolytic activity of bovine pancreatic RNase A (Figure 5A). CdiA-CTYkris toxins carrying the His175Ala, Thr276Ala, Tyr278Ala and Arg186Ala mutations showed significantly reduced cCMP hydrolytic activity (Figure 5B), suggesting that these residues play critical roles in catalysis. By contrast, toxin domains with Asn191Ala, Lys193Ala and Ser196Ala substitutions showed no decrease in hydrolytic activity (Figure 5B).
Figure 5.

Phosphodiester hydrolytic activity of CdiA-CTYkris. (A) cCMP hydrolysis was monitored by measuring the absorbance of 3΄-CMP at 296 nm over time. Reactions were supplemented with CdiIYkris where indicated. (B) Phosphodiester hydrolytic activities of CdiA-CTYkris variants were monitored as described for panel A. The same wild-type CdiA-CTYkris data in panel A is also used for panel B. All experiments were performed in triplicate.
CdiA-CT/CdiIYkris complex binding affinity
We next used biolayer interferometry to determine the binding affinity of the CdiA-CT/CdiIYkris complex. Increasing concentrations of CdiA-CTYkris were titrated against His6-tagged CdiIYkris immobilized on a Ni2+-NTA biosensor, and toxin association and dissociation was assessed by a shift in wavelength (Figure 6A). A dissociation constant of 40.9 ± 4.9 nM was calculated from the rates of complex association and dissociation (Figure 6A). We also examined the affinity of bovine pancreatic RNase A for CdiIYkris. Because RNase A showed non-specific binding to the Ni2+-NTA biosensor, we biotinylated RNase A and immobilized it on streptavidin biosensors to determine its binding affinity for CdiIYkris. The calculated dissociation constant RNase A binding to CdiIYkris is 1.95 ± 0.90 μM (Figure 6B), corresponding to ∼50-fold lower affinity compared the CdiA-CTYkris toxin domain. The considerably lower affinity of CdiIYkris for RNase A is consistent with the observation that CdiIYkris does not neutralize RNase A activity under the conditions tested (Figure 5A).
Figure 6.

Toxin-immunity binding interactions. (A) Immobilized His6-tagged CdiIYkris was exposed to CdiA-CTYkris (3.125–50 μM), and association and dissociation kinetics were assessed by a shift in wavelength (nm). (B) Biotinylated RNase A was immobilized and exposed to CdiIYkris (6.25–50 μM) as described above. All experiments were performed in triplicate and the calculated global Kd values are reported for both interactions.
Thermal stability of RNase domains
All RNase A family members contain either three or four conserved disulfide linkages, which impart significant structural stability (40). By contrast, cysteine residues are conspicuously absent from the CdiA-CTYkris nuclease domain. Therefore, we compared the structures of CdiA-CTYkris and mouse RNase 1 to identify elements that may compensate for the absent disulfide bonds. RNase 1 helix α2 is linked to β2 through the Cys27–Cys85 linkage (Figure 7A, left), which is substituted with hydrophobic interactions between α2 (Leu183, Leu184) and β3 (Val245) in CdiA-CTYkris (Figure 7A, right). Notably, helix α2 of CdiA-CTYkris is perpendicular to the corresponding helix in RNase 1. The Cys41-Cys95 disulfide links extended loops 2 and 6, and these loops are substituted with the perpendicular helix α2 and two shorter loops in CdiA-CTYkris (Figure 7B). Cys59–Cys111 connects helix α3 to the C-terminus of β5, and loop 4 connecting α3 and β5 is tethered by the fourth linkage between Cys66 and Cys73 (Figure 7C, left). Again, CdiA-CTYkris lacks this extended loop region and instead has an additional helix (α4) in this position (Figure 7C, right). To assess the relative stabilities of RNase A and CdiA-CTYkris, we monitored the thermal denaturation of each enzyme by circular dichroism spectroscopy. CdiA-CTYkris showed a gradual loss in secondary structure content over a broad temperature range of 40–80°C (Figure. 8A and C). By contrast, bovine RNase A exhibited a cooperative unfolding transition with a calculated melting temperature of 60 ± 2°C (Figure 8B and C), consistent with previously reported values (41,42). These data suggest that the CdiA-CTYkris domain undergoes gradual thermal denaturation and consequently does not conform to a two-state folding model.
Figure 7.

Structural comparison of mouse RNase 1 and CdiA-CTYkris. For the three panels, Cartoon representation depicts CdiA-CTYkris (green) and mouse RNase 1 (white, PDB code: 3TSR) with disulfide bonds represented in sticks with carbon and sulfur atoms colored white and yellow, respectively. (A) The Cys27–Cys85 bond in RNase 1 (left) is substituted by hydrophobic interactions in CdiA-CTYkris (right). (B) Superimposition of RNase 1 and CdiA-CTYkris. The disulfide bond between Cys41 and Cys96 in RNase 1 is replaced by an α-helix in CdiA-CTYkris. (C) RNase 1 has an extended loop that is stabilized by Cys58–Cys111 and Cys66–Cys73 linkages (left). This loop region is substituted with a fourth α-helix in the CdiA-CTYkris structure (right).
Figure 8.

Thermal stability of CdiA-CTYkris and RNase A. The secondary structure content of CdiA-CTYkris (A) and RNase A (B) was monitored by circular dichroism spectroscopy at temperatures between 20 and 90°C. Spectra colored in gray-scale progress from dark to light with increasing temperature. In both panels, the arrow represents the increase in temperature that correlates to a decrease of the spectral ellipticity (θ). (C) Comparison of CdiA-CTYkris and RNase A stability using global unfolding curves. The Tm for each protein is estimated to be the temperature at which there is 50% folded and unfolded protein (represented dashed grey line). All experiments were performed in triplicate.
DISCUSSION
These results show that the CDI system of Y. kristensenii ATCC 33638 deploys a nuclease toxin that shares the RNase A superfamily fold. RNase A/RNase 1 was first isolated from exocrine pancreas, and the enzyme family was initially thought to function solely in the digestion of dietary nucleic acids (37,43). Subsequently, it became clear that RNase A is but one of several paralogs that are secreted from various tissues and have important non-digestive functions in vertebrates. Angiogenin/RNase 5 was identified as a potent tumor-derived angiogenesis factor (44,45), thus providing the first indication that these enzymes have diverse functions. Additionally, many RNase A superfamily members mediate innate immune responses against viral, bacterial and helminthic pathogens (43,46). Though CdiA-CTYkris shares the RNase A fold and exhibits nuclease activity, these structural similarities most likely reflect convergent evolution. Firstly, there are no identifiable sequence homologs in invertebrates (or lower eukaryotes) to link the bacterial and vertebrate enzymes. Secondly, bacterial toxins are almost invariably encoded adjacent to cognate immunity genes, which are essential to prevent auto-inhibition. This genetic organization necessitates that toxin and immunity genes evolve reciprocally as pairs in cis. We note that several RNase A paralogs are potent cytotoxins, and mammalian cells produce an abundant cytosolic RNase inhibitor protein to counteract this toxicity. Though the RNase A inhibitor is functionally analogous to an immunity protein, it is a leucine-rich repeat protein with an unusual horseshoe-shaped structure and is quite distinct from CdiIYkris (47,48). Moreover, the inhibitor gene (RNH1) is located on a different chromosome than the RNase gene cluster (49,50), further distinguishing this system from typical bacterial toxin-immunity modules.
RNase A is one of the preeminent models to explore protein structure and function, and it is the first enzyme for which the catalytic mechanism was deduced (37,40). Superfamily members contain invariant His–Lys–His triads that catalyze independent phosphotransfer and phosphodiester hydrolysis reactions. Inspection of the CdiA-CTYkris nuclease active site reveals that its catalytic mechanism is similar, yet distinct, from that of the RNase A superfamily. CdiA-CTYkris residue His175 superimposes onto His13 of angiogenin, suggesting that the imidazole side-chain also acts as a general base during catalysis. Toxin residue Arg186 could stabilize the transition state in the same manner as RNase A Lys41. Notably, RNase T1 family nucleases use a highly conserved Arg residue to stabilize the reaction intermediate (51). However, the toxin active site lacks a second His residue, and instead contains Thr276 and Tyr278 in the structurally equivalent position as the only residues capable of proton donation. Site-directed mutagenesis shows that Tyr278 has a greater impact on nuclease activity, suggesting it may have a direct role in catalysis. According to this model, His175 and Tyr278 mediate proton transfers, and Arg186 stabilizes the pentavalent phosphoryl transition-state intermediate. The proposed active-site arrangement and catalytic mechanism are reminiscent of tRNA-splicing endonucleases, which are thought to use Tyr as a general base to initiate the reaction and His to protonate the 5΄-leaving group (52,53). Two other eukaryotic RNases, Ire1 (54) and RNase L (55), use the same catalytic strategy to cleave RNA, but their nuclease domains are not related to tRNA-splicing endonucleases. tRNA splicing endonucleases are built from the same core α1β1β2β3α2β4 fold as restriction endonucleases and other PD-(D/E)XK phosphodiesterases (56,57). Remarkably, the catalytic residues of tRNA-splicing and restriction endonucleases emanate from opposite edges of the central β-sheet. Thus, the restriction endonuclease core fold has been co-opted to support metal-independent RNase activity, in stark contrast with the metal-dependent DNA hydrolysis activity that characterizes most PD-(D/E)XK superfamily enzymes. Our results suggest that an analogous His-Arg-Tyr catalytic triad has arisen independently using the RNase A fold as a scaffold.
Homologs of the CdiA-CTYkris RNase domain are distributed throughout Gram-negative and Gram-positive bacteria. All of the homologs are encoded adjacent to probable immunity genes, and most are associated with secretion systems known to mediate bacterial competition. For example, a handful form the C-terminal toxin domains of CdiA proteins from Serratia proteomaculans, Photorhabdus luminescens, Pseudomonas citronellolis and Bordetella species N (Figure 9). The domain is also found at the C-terminus of several rearrangement hotspot (Rhs) proteins, mostly from Pseudomonas species. In γ-proteobacteria, Rhs proteins are exported by type VI secretion systems (T6SS) and have been shown to have anti-bacterial activity (58–61). Though Rhs effectors have not been characterized in Flavobacteria, the homologs from Chryseobacterium gallinarum and C. gregarium could be deployed in a T6SS-dependent manner as these systems are clearly active in other Bacteriodetes (62,63). We also recovered an unusual Rhs-repeat protein from Streptomyces malaysiense (Figure 9). Because Gram-positive bacteria do not possess T6SS, this putative effector is presumably exported through another mechanism, perhaps in a Sec-dependent manner analogous to the WapA YD-repeat proteins from Bacillus species (58). In addition to the Rhs proteins, the RNase domain is also found within a group of putative T6SS effectors distributed sporadically through the Burkholderiales and Enterobacteriacae. Bacterial RNase A-like toxins are also well-represented at the C-terminus of putative type VII secretion system (T7SS) effectors (Figure 9). These proteins carry N-terminal ESAT-6/WXG100 motifs, which presumably guide the toxin domains through the secretion apparatus (64,65). After a few years of conjecture about their role in competition (8,66,67), T7SS was recently shown to export nuclease toxins from Staphylococcus aureus (68,69). The final class of CdiA-CTYkris nuclease homologs are clade-specific lipoproteins from Bacillus cereus and Bacillus anthracis. These lipoproteins are encoded with probable immunity proteins, but two observations argue that these nucleases do not mediate competition. First, the predicted lipid modifications should anchor the nucleases in the membrane and presumably preclude transfer to other bacteria. Second, the RNase domains show little sequence variation between isolates. Diversification between strains is a hallmark of toxins that mediate inter-strain competition (14,70,71). Thus, this group of lipoprotein RNases probably scavenges nucleotides from RNA in the environment, analogous to the assimilatory function of pancreatic RNase 1 in digestion (37,43).
Figure 9.

CdiA-CTYkris nuclease domain homologs. Predicted bacterial RNase A-like domains were aligned using Clustal omega and rendered using the ESPript 3.0 server (73). Sequences are from the following proteins (NCBI reference identifiers in parentheses): Y. kristensenii ATCC 33638, CdiA (EEP92680.1); S. proteomaculans 568, CdiA (WP_012146779.1); Bordetella sp. N, CdiA (WP_057651960.1); P. citronellolis TTU2014-008ASC, CdiA (WP_058071149.1); P. fluorescens BBc6R8, Rhs (WP_023659002.1); P. temperata Hm, putative T6SS effector (WP_051477485.1); Oscillatoriales cyanobacterium USR001, SMC domain protein (OCQ92111.1); P. prosekii LMG 26867, Rhs (SDS72132.1); B. anthracis Ames, predicted lipoprotein (WP_000822712.1); B. cereus W, ESAT-6 protein (WP_001985713.1); B. subtilis W23, ESAT-6 protein (EFG91422.1); P. brassicacearum DF41, Rhs (WP_025215693.1); P. luminescens PB45.5, CdiA (WP_065389372.1); C. gallinarum DSM 27622, Rhs (WP_053328404.1); Caenimonas sp. SL110, Rhs (WP_048438936.1); Mycobacterium sp. UM_WWY, hypothetical (WP_036426273.1); Promicromonosporaceae bacterium W15, PT-HINT domain protein (WP_051136513.1); Nocardioides sp. Root224, hypothetical (WP_056152939.1); Vibrio nigripulchritudo AM115, Rhs (WP_022598889.1); S. malaysiense MUSC 136, Rhs (WP_071387441.1). Residue numbers correspond to CdiA-CTYkris (from Val1 of the VENN motif), and red-highlighted positions are invariant between homologs.
Finally, we note that the alignment of the CdiA-CTYkris nuclease homologs shows that His175 and Thr276 are completely conserved, but the position corresponding to Tyr278 is frequently substituted with a Phe residue (Figure 9). This observation is somewhat surprising given the importance of Tyr278 for toxin nuclease activity. The side-chain of Tyr278 occupies a position similar to angiogenin Leu117 and RNase A Phe120, and the latter residue has been shown to stack onto pyrimidine nucleobases and contribute to substrate binding (72). Therefore, it is possible that the Tyr278Ala substitution interferes with nuclease activity by reducing toxin affinity for substrate, rather than directly disrupting catalysis. This model indicates that another non-conserved residue must assume responsibility for proton transfers and suggests a degree of plasticity in the bacterial RNase A-like active site. Further detailed studies of CdiA-CTYkris nuclease activity are required to fully understand its distinct mechanism of action.
ACKNOWLEDGEMENTS
CD data were acquired at the Laser Spectroscopy Facility at the Department of Chemistry at UC Irvine.
FUNDING
National Institutes of Health (NIH) [GM102318 to C.W.G., C.S.H. & subcontract to A.J., GM117373 to C.W.G. & C.S.H., GM094585 to A.J., GM115586 to A.J.]; U.S. Department of Energy, Office of Biological and Environmental Research [DE-AC02-06CH11357 to A.J.]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Aoki S.K., Diner E.J., de Roodenbeke C.T., Burgess B.R., Poole S.J., Braaten B.A., Jones A.M., Webb J.S., Hayes C.S., Cotter P.A. et al. . A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010; 468:439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruhe Z.C., Low D.A., Hayes C.S.. Bacterial contact-dependent growth inhibition. Trends Microbiol.. 2013; 21:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Willett J.L., Ruhe Z.C., Goulding C.W., Low D.A., Hayes C.S.. Contact-dependent growth inhibition (CDI) and CdiB/CdiA two-partner secretion proteins. J. Mol. Biol. 2015; 427:3754–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kajava A.V., Cheng N., Cleaver R., Kessel M., Simon M.N., Willery E., Jacob-Dubuisson F., Locht C., Steven A.C.. Beta-helix model for the filamentous haemagglutinin adhesin of Bordetella pertussis and related bacterial secretory proteins. Mol. Microbiol. 2001; 42:279–292. [DOI] [PubMed] [Google Scholar]
- 5. Makhov A.M., Hannah J.H., Brennan M.J., Trus B.L., Kocsis E., Conway J.F., Wingfield P.T., Simon M.N., Steven A.C.. Filamentous hemagglutinin of Bordetella pertussis. A bacterial adhesin formed as a 50-nm monomeric rigid rod based on a 19-residue repeat motif rich in beta strands and turns. J. Mol. Biol. 1994; 241:110–124. [DOI] [PubMed] [Google Scholar]
- 6. Ruhe Z.C., Nguyen J.Y., Beck C.M., Low D.A., Hayes C.S.. The proton-motive force is required for translocation of CDI toxins across the inner membrane of target bacteria. Mol. Microbiol. 2014; 94:466–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willett J.L., Gucinski G.C., Fatherree J.P., Low D.A., Hayes C.S.. Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:11341–11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang D., Iyer L.M., Aravind L.. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011; 39:4532–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nikolakakis K., Amber S., Wilbur J.S., Diner E.J., Aoki S.K., Poole S.J., Tuanyok A., Keim P.S., Peacock S., Hayes C.S. et al. . The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol. Microbiol. 2012; 84:516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beck C.M., Morse R.P., Cunningham D.A., Iniguez A., Low D.A., Goulding C.W., Hayes C.S.. CdiA from Enterobacter cloacae delivers a toxic ribosomal RNase into target bacteria. Structure. 2014; 22:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morse R.P., Nikolakakis K.C., Willett J.L., Gerrick E., Low D.A., Hayes C.S., Goulding C.W.. Structural basis of toxicity and immunity in contact-dependent growth inhibition (CDI) systems. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:21480–21485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aoki S.K., Poole S.J., Hayes C.S., Low D.A.. Toxin on a stick: modular CDI toxin delivery systems play roles in bacterial competition. Virulence. 2011; 2:356–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson P.M., Beck C.M., Morse R.P., Garza-Sanchez F., Low D.A., Hayes C.S., Goulding C.W.. Unraveling the essential role of CysK in CDI toxin activation. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:9792–9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morse R.P., Willett J.L., Johnson P.M., Zheng J., Credali A., Iniguez A., Nowick J.S., Hayes C.S., Goulding C.W.. Diversification of beta-augmentation interactions between CDI toxin/immunity proteins. J. Mol. Biol. 2015; 427:3766–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson P.M., Gucinski G.C., Garza-Sanchez F., Wong T., Hung L.W., Hayes C.S., Goulding C.W.. Functional diversity of cytotoxic tRNase/immunity protein complexes from Burkholderia pseudomallei. J. Biol. Chem. 2016; 291:19387–19400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diner E.J., Beck C.M., Webb J.S., Low D.A., Hayes C.S.. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev.. 2012; 26:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGinness K.E., Baker T.A., Sauer R.T.. Engineering controllable protein degradation. Mol. Cell. 2006; 22:701–707. [DOI] [PubMed] [Google Scholar]
- 18. Thomason L., Court D.L., Bubunenko M., Costantino N., Wilson H., Datta S., Oppenheim A.. Recombineering: genetic engineering in bacteria using homologous recombination. Curr. Protoc. Mol. Biol. 2007; 16. [DOI] [PubMed] [Google Scholar]
- 19. Donnelly M.I., Zhou M., Millard C.S., Clancy S., Stols L., Eschenfeldt W.H., Collart F.R., Joachimiak A.. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr. Purif. 2006; 47:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim Y., Dementieva I., Zhou M., Wu R., Lezondra L., Quartey P., Joachimiak G., Korolev O., Li H., Joachimiak A.. Automation of protein purification for structural genomics. J. Struct. Funct. Genomics. 2004; 5:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M.. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 22. French S., Wilson K.. On the treatment of negative intensity observations. Acta Crystallogr. A. 1978; A34:517–525. [Google Scholar]
- 23. Padilla J.E., Yeates T.O.. A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr. D Biol. Crystallogr. 2003; 59:1124–1130. [DOI] [PubMed] [Google Scholar]
- 24. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. . Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheldrick G.M. A short history of SHELX. Acta Crystallogr. A. 2008; 64:112–122. [DOI] [PubMed] [Google Scholar]
- 26. Otwinowski Z. Proceedings of the CCP4 Study Weekend 25–26 January 1991. 1991; Warrington: Daresbury Laboratory; 80–85. [Google Scholar]
- 27. Cowtan K. DM: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM newsletter on protein crystallography. 1994; 31:34–38. [Google Scholar]
- 28. Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006; 62:1002–1011. [DOI] [PubMed] [Google Scholar]
- 29. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 30. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G.N., Vagin A.A., Dodson E.J.. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997; 53:240–255. [DOI] [PubMed] [Google Scholar]
- 32. Tanimizu N., Ueno H., Hayashi R.. Role of Phe120 in the activity and structure of bovine pancreatic ribonuclease A. J. Biochem. 1998; 124:410–416. [DOI] [PubMed] [Google Scholar]
- 33. Garza-Sanchez F., Janssen B.D., Hayes C.S.. Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J. Biol. Chem. 2006; 281:34258–34268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holm L., Rosenstrom P.. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010; 38:W545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thiyagarajan N., Acharya K.R.. Crystal structure of human angiogenin with an engineered loop exhibits conformational flexibility at the functional regions of the molecule. FEBS Open Biol. 2013; 3:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lomax J.E., Bianchetti C.M., Chang A., Phillips G.N. Jr, Fox B.G., Raines R.T.. Functional evolution of ribonuclease inhibitor: insights from birds and reptiles. J. Mol. Biol. 2014; 426:3041–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cuchillo C.M., Nogues M.V., Raines R.T.. Bovine pancreatic ribonuclease: fifty years of the first enzymatic reaction mechanism. Biochemistry. 2011; 50:7835–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chatani E., Hayashi R.. Functional and structural roles of constituent amino acid residues of bovine pancreatic ribonuclease A. J. Biosci. Bioeng. 2001; 92:98–107. [DOI] [PubMed] [Google Scholar]
- 39. delCardayre S.B., Raines R.T.. A residue to residue hydrogen bond mediates the nucleotide specificity of ribonuclease A. J. Mol. Biol. 1995; 252:328–336. [DOI] [PubMed] [Google Scholar]
- 40. Raines R.T. Ribonuclease A. Chem. Rev. 1998; 98:1045–1066. [DOI] [PubMed] [Google Scholar]
- 41. Laity J.H., Shimotakahara S., Scheraga H.A.. Expression of wild-type and mutant bovine pancreatic ribonuclease A in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klink T.A., Woycechowsky K.J., Taylor K.M., Raines R.T.. Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A. Eur. J. Biochem. 2000; 267:566–572. [DOI] [PubMed] [Google Scholar]
- 43. Rosenberg H.F. RNase A ribonucleases and host defense: an evolving story. J. Leukoc. Biol. 2008; 83:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kurachi K., Davie E.W., Strydom D.J., Riordan J.F., Vallee B.L.. Sequence of the cDNA and gene for angiogenin, a human angiogenesis factor. Biochemistry. 1985; 24:5494–5499. [DOI] [PubMed] [Google Scholar]
- 45. Strydom D.J., Fett J.W., Lobb R.R., Alderman E.M., Bethune J.L., Riordan J.F., Vallee B.L.. Amino acid sequence of human tumor derived angiogenin. Biochemistry. 1985; 24:5486–5494. [DOI] [PubMed] [Google Scholar]
- 46. Koczera P., Martin L., Marx G., Schuerholz T.. The Ribonuclease A Superfamily in Humans: Canonical RNases as the Buttress of Innate Immunity. Int. J. Mol. Sci. 2016; 17:E1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kobe B., Deisenhofer J.. Mechanism of ribonuclease inhibition by ribonuclease inhibitor protein based on the crystal structure of its complex with ribonuclease A. J. Mol. Biol. 1996; 264:1028–1043. [DOI] [PubMed] [Google Scholar]
- 48. Kobe B., Deisenhofer J.. Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature. 1993; 366:751–756. [DOI] [PubMed] [Google Scholar]
- 49. Weremowicz S., Fox E.A., Morton C.C., Vallee B.L.. The placental ribonuclease inhibitor (RNH) gene is located on chromosome subband 11p15.5. Genomics. 1990; 8:717–721. [DOI] [PubMed] [Google Scholar]
- 50. Weremowicz S., Fox E.A., Morton C.C., Vallee B.L.. Localization of the human angiogenin gene to chromosome band 14q11, proximal to the T cell receptor alpha/delta locus. Am. J. Hum. Genet. 1990; 47:973–981. [PMC free article] [PubMed] [Google Scholar]
- 51. Yoshida H. The ribonuclease T1 family. Methods Enzymol. 2001; 341:28–41. [DOI] [PubMed] [Google Scholar]
- 52. Xue S., Calvin K., Li H.. RNA recognition and cleavage by a splicing endonuclease. Science. 2006; 312:906–910. [DOI] [PubMed] [Google Scholar]
- 53. Li H., Trotta C.R., Abelson J.. Crystal structure and evolution of a transfer RNA splicing enzyme. Science. 1998; 280:279–284. [DOI] [PubMed] [Google Scholar]
- 54. Lee K.P., Dey M., Neculai D., Cao C., Dever T.E., Sicheri F.. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell. 2008; 132:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang H., Zeqiraj E., Dong B., Jha B.K., Duffy N.M., Orlicky S., Thevakumaran N., Talukdar M., Pillon M.C., Ceccarelli D.F. et al. . Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol. Cell. 2014; 53:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bujnicki J.M., Rychlewski L.. Unusual evolutionary history of the tRNA splicing endonuclease EndA: relationship to the LAGLIDADG and PD-(D/E)XK deoxyribonucleases. Protein Sci. 2001; 10:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Steczkiewicz K., Muszewska A., Knizewski L., Rychlewski L., Ginalski K.. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012; 40:7016–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Koskiniemi S., Lamoureux J.G., Nikolakakis K.C., t’Kint de Roodenbeke C., Kaplan M.D., Low D.A., Hayes C.S.. Rhs proteins from diverse bacteria mediate intercellular competition. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7032–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Whitney J.C., Beck C.M., Goo Y.A., Russell A.B., Harding B.N., De Leon J.A., Cunningham D.A., Tran B.Q., Low D.A., Goodlett D.R. et al. . Genetically distinct pathways guide effector export through the type VI secretion system. Mol. Microbiol. 2014; 92:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hachani A., Allsopp L.P., Oduko Y., Filloux A.. The VgrG proteins are "a la carte" delivery systems for bacterial type VI effectors. J. Biol. Chem. 2014; 289:17872–17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alcoforado Diniz J., Coulthurst S.J.. Intraspecies competition in Serratia marcescens is mediated by type VI-secreted Rhs effectors and a conserved effector-associated accessory protein. J. Bacteriol. 2015; 197:2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Russell A.B., Wexler A.G., Harding B.N., Whitney J.C., Bohn A.J., Goo Y.A., Tran B.Q., Barry N.A., Zheng H., Peterson S.B. et al. . A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe. 2014; 16:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wexler A.G., Bao Y., Whitney J.C., Bobay L.M., Xavier J.B., Schofield W.B., Barry N.A., Russell A.B., Tran B.Q., Goo Y.A. et al. . Human symbionts inject and neutralize antibacterial toxins to persist in the gut. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garufi G., Butler E., Missiakas D.. ESAT-6-like protein secretion in Bacillus anthracis. J. Bacteriol. 2008; 190:7004–7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fan Y., Tan K., Chhor G., Butler E.K., Jedrzejczak R.P., Missiakas D., Joachimiak A.. EsxB, a secreted protein from Bacillus anthracis forms two distinct helical bundles. Protein Sci. 2015; 24:1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang D., de Souza R.F., Anantharaman V., Iyer L.M., Aravind L.. Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Direct. 2012; 7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Holberger L.E., Garza-Sanchez F., Lamoureux J., Low D.A., Hayes C.S.. A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett. 2012; 586:132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cao Z., Casabona M.G., Kneuper H., Chalmers J.D., Palmer T.. The type VII secretion system of Staphylococcus aureus secretes a nuclease toxin that targets competitor bacteria. Nat. Microbiol. 2016; 2:16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ohr R.J., Anderson M., Shi M., Schneewind O., Missiakas D.. EssD, a nuclease effector of the Staphylococcus aureus ESS pathway. J. Bacteriol. 2017; 199:e00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Riley M.A. Positive selection for colicin diversity in bacteria. Mol. Biol. Evol. 1993; 10:1048–1059. [DOI] [PubMed] [Google Scholar]
- 71. Kleanthous C., Walker D.. Immunity proteins: enzyme inhibitors that avoid the active site. Trends Biochem Sci. 2001; 26:624–631. [DOI] [PubMed] [Google Scholar]
- 72. Wlodawer A., Miller M., Sjolin L.. Active site of RNase: neutron diffraction study of a complex with uridine vanadate, a transition-state analog. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:3628–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Davis I.W., Murray L.W., Richardson J.S., Richardson D.C.. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004; 32:W615–W619. [DOI] [PMC free article] [PubMed] [Google Scholar]