


Abstract
Current approaches to design efficient antisense RNAs (asRNAs) rely primarily on a thermodynamic understanding of RNA–RNA interactions. However, these approaches depend on structure predictions and have limited accuracy, arguably due to overlooking important cellular environment factors. In this work, we develop a biophysical model to describe asRNA–RNA hybridization that incorporates in vivo factors using large-scale experimental hybridization data for three model RNAs: a group I intron, CsrB and a tRNA. A unique element of our model is the estimation of the availability of the target region to interact with a given asRNA using a differential entropic consideration of suboptimal structures. We showcase the utility of this model by evaluating its prediction capabilities in four additional RNAs: a group II intron, Spinach II, 2-MS2 binding domain and glgC 5΄ UTR. Additionally, we demonstrate the applicability of this approach to other bacterial species by predicting sRNA–mRNA binding regions in two newly discovered, though uncharacterized, regulatory RNAs.
INTRODUCTION
In vivo RNA targeting via antisense base pairing provides an efficient mechanism to characterize RNA interactions as well as to post-transcriptionally regulate gene expression. In the native cellular environment, sequence-specific antisense RNAs (asRNAs) are ubiquitous in natural gene regulatory mechanisms, ranging from bacterial small RNAs (sRNAs) (both cis- and trans-encoded) (1–3) to more complex eukaryotic systems such as the RNA interference pathway and circular RNAs (4). Likewise, the common use of affinity-based purification assays to characterize in vivo RNA interactions (i.e. pulldown of a target RNA and its interacting partners from cellular extracts) relies on targeting the RNA of interest with an immobilized bait molecule, often an antisense RNA (5,6). Furthermore, the simplicity and universality of nucleic acid Watson–Crick complementarity makes antisense nucleic acids highly attractive for controlling gene expression (1,7–10) in biotechnological applications such as bacterial cellular engineering (11–14). Given the broad utility of RNA targeting via antisense binding, recent efforts to design effective synthetic asRNAs in bacteria have become more systematic, mimicking mechanisms of natural non-coding RNAs that downregulate their cognate messenger RNAs (mRNAs) by base-pairing (1,3,15). A more recent study in bacteria provided general guidelines for the design of asRNAs using large sets of gene-repression data (16). However, there remains a significant challenge in the asRNA applications described above: the design of effective antisense oligonucleotides for sequence-specific targeting of RNA in situ (3,5). This is particularly true in bacterial systems, since most design models for asRNAs have been developed in the context of eukaryotes.
Rational efforts to design asRNA have traditionally been aided by algorithms that predict RNA–RNA interactions (17,18). These approaches are numerous and stem from simple and fast surveying methods such as GUUGLe (19) and BLAST (20) that score potential target regions within an RNA of interest using the sole criterion of complementarity. These approaches have been followed by several methods that display varying degrees of accuracy and sophistication: from (i) those neglecting intramolecular structure (e.g. RNAduplex (21), RNAhybrid (22), TargetRNA (23) and RNAplex (24)) to (ii) those considering only one interaction site and intramolecular structure (e.g. Nupack (25), RNAup (26), AccessFold (27) and IntaRNA (28)), or even to (iii) those highly computationally complex tools that predict several interactions sites (e.g. IRIS (29)) and the joint RNA–RNA secondary structure using the energy partition function (e.g. PiRNA (30) and RIP (31)). For simplicity, some of these approaches for prediction of RNA–RNA interactions (e.g. RNAup (26) and IntaRNA (28)) rely on RNA ‘accessibility,’ based on the assumption that both interacting partners must be unfolded (i.e. accessible) prior to binding (17). In this context, accessibility is defined as the property of a given potential interaction site to be free of intramolecular base pairs. Target accessibility has been generally introduced in predictive algorithms as an energy penalty estimated from the ensemble of possible target structures with the corresponding target region unpaired. The specific role of target accessibility in the asRNA hybridization has been extensively studied with a particular focus on miRNAs and siRNAs (24,26,32–36). However, there are limitations with the aforementioned structure prediction approaches (e.g. high false positive rate (17) and limited accuracy (70% for molecules up to 500 nt and as low as 40% for longer RNAs (37)) due to simplifications in the energy model that overlook structural complexity and intracellular factors that affect hybridization. Furthermore, to our knowledge, very few works have shed light on how accessibility plays a role in antisense hybridization within living bacteria (38). This underscores the need for more realistic approaches that account for the in vivo environment, incorporating the influence of binding factors, ionic strength and molecular crowding (39).
Hereby, we propose a novel approach to predict and evaluate hybridization efficacy in bacteria that features the inclusion of large sets of experimental data collected in vivo. This model uniquely considers a regional availability factor. Regional characteristics of the target RNA have long been implicated in asRNA efficacy. For instance, Zhao and Lemke proposed a criterion that at least 4 highly accessible nucleotides are necessary for the initiation of asRNA-target RNA binding based on investigating correlations between predicted structure and asRNA efficacy (40). In addition, established RNA hybridization mechanisms further support this notion of ‘regionality’, e.g. the intermediate step in which a few nucleotides interact to initiate the binding (seeding interaction) (41) or recognition sequences that behave as first ‘points of contact,’ such as the YUNR motif (42). To derive a corresponding RNA molecular recognition model, we start from a common thermodynamic framework used in accessibility-based approaches (17) that considers the overall change of free energy of Gibbs 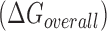 in the reaction system, a predictor of asRNA binding (19). We introduce a novel, semi-empirical measure of variable entropic contributions to asRNA binding of the targeted region, which we assume acts as a cohesive stretch of nucleotides within the larger structural context of folded RNA molecules. Lastly, we also optimize the considered models in vivo based on experimental data to account for crowding and other effects of the cellular environment. Hereafter, we refer to this predictive approach as the invivo-optimized Thermodynamic Accessibility-adjusted model, inTherAcc.
in the reaction system, a predictor of asRNA binding (19). We introduce a novel, semi-empirical measure of variable entropic contributions to asRNA binding of the targeted region, which we assume acts as a cohesive stretch of nucleotides within the larger structural context of folded RNA molecules. Lastly, we also optimize the considered models in vivo based on experimental data to account for crowding and other effects of the cellular environment. Hereafter, we refer to this predictive approach as the invivo-optimized Thermodynamic Accessibility-adjusted model, inTherAcc.
The inTherAcc model was developed using large data sets describing in vivo hybridization efficacy of asRNAs targeting approximately 80 regions within three well-studied RNA molecules: the autocatalytic group I (gI) intron from Tetrahymena, and the Escherichia coli noncoding RNAs CsrB and glutamate tRNA. Experimental characterization of asRNA hybridization efficacy was performed using a previously published assay that measures asRNA-target RNA hybridization via fluorescence, the in vivo RNA Structural Sensing System-(IRS3) (43).
Following model optimization, hybridization efficacy of numerous potential target regions within the 2-MS2 phage coat protein transcript (2-MS2), glgC 5΄ UTR (glgC), group II intron (gII) and the Spinach II (SpII) RNAs was predicted and experimentally assessed for accuracy. The performance of our model was benchmarked against a similarly optimized model lacking the proposed availability term and IntaRNA (28), an accessibility-based approach that also considers a regional adjustment by incorporating the existence of a user-definable seed.
Lastly we evaluated the ability of inTherAcc coupled to BLAST (20) to predict mRNA targets of recently discovered Z. mobilis sRNAs, Zms4 and Zms6 (44). Experimental confirmation using RIP-seq validated the ability of inTherAcc to aid prediction of potential mRNA targets for Zms4 and Zms6. Comparison of inTherAcc to IntaRNA predictions suggests complementarity between the prediction approaches. Finally, the demonstration of inTherAcc utility in another bacterial species underscores its broad applicability.
MATERIALS AND METHODS
Plasmids and strains
As previously described in (43), the fluorescence-based iRS3 system provides a measurement of asRNA–RNA hybridization by using various 8–27 nt sequences (asRNAs) that are complementary to a target RNA. In this system, a fluorescence shift is observed when an asRNA successfully binds the region of interest in the target RNA. A total of eighty asRNAs targeting unique regions in three target molecules (gI intron, CsrB and tRNA) were analyzed in this work for model optimization purposes. Forty-nine asRNAs targeting unique regions in four different target molecules (gII intron, SpinachII, glgC 5΄UTR and 2-MS2) were also used to assess model prediction capabilities. To construct these experimental asRNA systems, a modified Golden Gate cloning-based plasmid was introduced for high-throughput cloning that included the following changes to the previously published ‘Wild Type Intron Probe I reporter’ (43): a p-chlorophenylalanine negative selection cassette in place of the asRNA sequence (between EcoRI and the CB element flanked by two BsmbI restriction sites) (45,46). We termed this plasmid iRS3 Golden Gate (IRS3-GG) and it is illustrated in Supplementary Figure S1A. All target molecules, with the exception of the natively-targeted tRNA, were separately introduced in the iRS3-GG between the XbaI and SalI restriction sites (see plasmid map in Supplementary Figure S1A). In the case of gII, SpII, glgC and 2-MS2, Gibson assembly (47) (using Gibson Assembly mix from NEB) was performed. CsrB was introduced via traditional restriction cloning. All primers used for cloning of target molecule into iRS3-GG are listed in Supplementary Table S1.
All asRNA sequences within the plasmid (Supplementary Table S2), besides 11 asRNAs corresponding to regions within the gI intron that were previously synthesized and published (43), were either ordered from GenScript Inc., synthesized by a site-directed mutagenesis approach (QuikChange II Site-Directed Mutagenesis Kit, Agilent Technologies) by modifying a previously synthesized asRNA, synthesized via Gibson Assembly (47) or synthesized by using a high throughput Golden Gate approach as described in (48) on our iRS3-GG plasmid. For the Golden Gate approach, complementary primers (ordered from IDT) containing each asRNA sequence with the proper flanking overhangs were annealed and cloned after digestion with BsmbI (Thermo Scientific) to replace the p-chlorophenylalanine negative selection cassette. All specific cloning methods and primers used for cloning are included in Supplementary Table S2. To increase cloning throughput, two to five asRNAs were combined into a single reaction and later transformed into DH5α chemically-competent cells or NEB Turbo electro-competent cells and plated in Luria–Bertani (LB)/Agar media supplemented with p-cholorophenylalanine (p-Cl-Phe) to select for the clones harboring the appropriate asRNA. Once the asRNA sequences were confirmed by DNA sequencing, the newly synthesized plasmids were transformed into K-12 MG1655, or, in the case of CsrB, into a CsrB/CsrC-knockout K-12 MG1655 strain (CML 378) (49). An overview of the specifics of the asRNA synthesis strategy is included in Supplementary Figure S1B.
For the evaluation of sRNA target prediction as aided by inTherAcc and IntaRNA, we utilized pBBR1MCS2-pgap vector (50) for constitutive expression. Each sRNA was synthesized by GenScript® and then cloned into pBBR1MCS2-pgap vector between NheI and SalI, resulting in pBBR1MCS2-pgap-sRNA. For 2MS2BD-Zms4/Zms6/control constructs, gBlock® (IDT) of 2MS2BD-Zms4/Zm6/control was used for cloning into pBBR1MCS2-pgap vector, resulting in plasmids abbreviated 2MS2-Zms4/2MS2-Zms6/2MS2-control. These plasmids were transformed by electroporation into Zymomonas mobilis 8b.
Selection of target RNAs
Rationale for target molecule selection was based on molecule complexity, size and functional interactions. For instance, the gI intron is a relatively large (393 nucleotides), well-studied RNA model (51,52) whose many structurally significant regions have been previously probed with the iRS3 system (43). These studies have shown that this autocatalytic molecule may well-represent the complexity of structural features present in most RNAs targeted for regulation (e.g. UTRs of mRNAs (53)). On the contrary, the 76-nucleotide long glutamate–tRNA has a wide assortment of interactions with intracellular factors, including mRNAs, rRNAs, various modification enzymes and other proteins despite exhibiting tight tertiary structure comparable to that of the gI intron (54). The third molecule chosen for model optimization, CsrB, is a non-coding RNA whose multiple protein binding motifs contribute to the translational regulation of a large number of mRNAs (55). Compared to the previously described molecules, CsrB (369 nucleotides) is less structurally sophisticated than the gI intron and the tRNA.
For assessing the prediction capabilities of our model, four alternative RNA molecules were used: the 2-MS2 coat protein binding domain, the model LtrB group II intron, the Spinach II RNA and the glgC messenger RNA 5΄UTR. This set of RNAs covers a wide array of types, functions, structures and sizes. MS2 and Spinach II are commonly used to investigate RNA interactions, more specifically, to isolate RNAs to determine specific RNA interacting complexes (5) and track RNA movement (56), respectively. Similarly, 5΄ UTRs often use their structure to regulate the translation of their associated mRNA. The gII intron was selected given the interest in targeting ribozymes for understanding the molecular mechanisms for catalytic activity, largely regulated by their complex folding (57).
Fluorescence measurements and calculations of asRNA hybridization using the in vivo RNA structural sensing system (iRS3)
In general, flow cytometry experiments were carried out as previously reported (43). All target molecules (except for the glutamate tRNA) were evaluated under overexpression conditions in which the hybridization efficacy is evaluated as the ratio between the fluorescence in the presence of the target RNA with baseline fluorescence (in the absence of the target RNA) subtracted out (FLon-FLoff) to the baseline fluorescence (FLoff). For all hybridization calculations, FLoff was scaled by an adjustment factor of 0.65 to account for the excess abundance of the reporter probe relative to the target RNA, as approximated by recently obtained RNA-sequencing data (unpublished). In the case of the tRNA, the target was evaluated using native levels given its natural presence and abundance in E. coli cells using plasmid in Supplementary Figure S1C. In this case, FLon and FLoff represent the fluorescence in the presence of the asRNA (iRS3+specific oligonucleotide) and the fluorescence in the absence of the asRNA, respectively. FLoff fluorescence was measured right before induction (at time ‘0’) and FLon was collected 45 min after induction (See Supplementary Figure S2A for a correlation between uninduced and time ‘0’). Seeding cultures originated from independent overnights and uninduced and induced samples proceeded from the same initial seeding culture. Specifically, seeding was done in LB (40 ml + 50 μg/ml of kanamycin) and split up into two 20 ml cultures at the time of induction (1–2 h of growth upon seeding) for the collection of model optimization data. When testing model predictions, seeding was done in LB (200 μl + 50 μg/ml of kanamycin) and split up into two 100 μl cultures in 96-well plates at the time of induction.
In vivo DMS footprinting and calculation of target availability (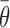 )
)
The DMS reactivity of the gI intron was obtained using a previously published protocol (43). In this work, we published the in vivo DMS reactivity profile for the full gI intron (Supplementary Figure S3). Previously, the reactivity for only select regions had been published (43). The nucleotide indexing for the gI intron follows the established consensus for this well-known molecule. These data were filtered and normalized using specialized software, the capillary automated footprinting analysis (58). The reactivity values for the untreated sample were subtracted from the average reactivity value of two independent DMS treated samples. The DMS reactivity for Gs and Us was estimated by assuming the same reactivity as their pairing partners (if paired), and, when unpaired, an average reactivity value for ‘exposed’ nucleotides was assigned. Special cases were those Gs and Us exposed in loops (G58, U59, G92, G112, G119, U120, G126, U179, U185, G200, G201, U202, U225, G227, U247, G254, G279, U300, G303, U322, U323, G331, U340, G341, G357, G358, G368 and U372) that were assigned values more similar to their neighbors and other As and Cs present in loops. The regional target availability factor was then calculated using the average of the individual reactivity values of each nucleotide in the given target region over the length of the target region.
Derivation of the accessibility-based thermodynamic model
The quantity 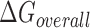 is the overall free energy change related to the different mechanistic steps associated with asRNA binding to the target RNA region; the folding and binding processes considered are depicted in Figure 1. This quantity is represented as the combined contribution of the free energies of: (i) the Watson–Crick base-pairing of the asRNA to the target RNA region
is the overall free energy change related to the different mechanistic steps associated with asRNA binding to the target RNA region; the folding and binding processes considered are depicted in Figure 1. This quantity is represented as the combined contribution of the free energies of: (i) the Watson–Crick base-pairing of the asRNA to the target RNA region 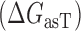 , (ii) the local unfolding of the target RNA region required for asRNA binding
, (ii) the local unfolding of the target RNA region required for asRNA binding 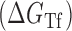 and (iii) the unfolding of the asRNA required for binding
and (iii) the unfolding of the asRNA required for binding 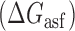 . The sum of these terms comprises the total energy of hybridization,
. The sum of these terms comprises the total energy of hybridization, 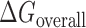 :
:
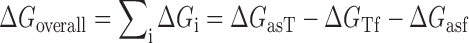 |
(1) |
Figure 1.

Proposed accessibility-based mechanism of antisense hybridization in living cells. (A) Example target region with color-coded local availability (estimated by base-pairing probabilities) is shown in canonical conformation, as would be expected in the native state. In antisense RNAs (asRNA) targeting by the iRS3, the targeted region must unbind from P3 to become single stranded as shown in (B) with a free energy change of –ΔGTf. The iRS3 consists of four main elements: a cis-blocking strand (CB, orange), a ribosome binding site (RBS, blue), the sequence encoding green fluorescence protein (GFP, green) and the probe (pink and black) of 8–27 nucleotides targeting a specific region shown in (B). The expected native state of the iRS3 is shown in (C), and it must also unfold to bind the target region as shown in (D) with a free energy change of –ΔGasf. Finally, the two unfolded structures bind as in (E) with a free energy change of ΔGasT to stabilize the unfolded iRS3 and allow translation of GFP. Effective asRNA targeting results in a high fluorescence response.
In Equation ((1)), subscripts asT, Tf and asf denote the asRNA-target RNA hybridization, the target RNA folding and the asRNA folding, respectively.
Calculation of free energy of hybridization 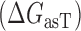
To calculate the Gibbs free energy of binding between the perfectly complementary stretch of nucleotides (asRNA) within the iRS3-asRNA system and the target region, the energy parameters for the nearest neighbor model published in (59) were used. Only canonical base-pairing (Watson–Crick base pairs), penalties for self-complementarity within the asRNA and AU ending were considered for the calculation of the stacking energies.
Calculation of free energy of the target region 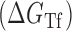
To calculate the Gibbs free energy of target region folding, the energy parameters for the nearest neighbor model previously published (59) were used. The target region plus one extra nucleotide at each end was considered to account for stacking contributions of neighboring base pairs. The folding of the target RNA was considered to be a local event due to the tight coupling of prokaryotic transcription and translation. Such assumptions of local folding have previously been used in a structural study of bacterial genes (60). To calculate the stacking energy contributions, the consensus secondary structure of the gI intron was considered (61). For all the other target molecules, a secondary structure prediction from the RNAStructure webserver was used (62). Since GU base pairs are somewhat extensively found in the structure of our target RNAs, they were treated as nearest neighbor stacks, similar to Watson–Crick helices. In addition, the penalty for ending in a GU was the same as an AU ending. In our treatment of GU pairs we followed the parameters reported by Mathews et al. (63). No energy parameters for other structural motifs such as loops, bulges, etc. were taken into account.
Calculation of the availability factor
To support high-throughput estimations of regional availability ( ), without involving experimental structural studies, local availability (θk) was estimated by one minus base pairing probabilities determined by Boltzmann-distributed structural variations provided by the Nupack webserver (64). This structural accessibility estimation was shown to be captured by in vivo experimental DMS reactivity at the regional level, supporting the use of base pairing probabilities as an estimator of experimentally determined regional availability (Supplementary Figure S4).
), without involving experimental structural studies, local availability (θk) was estimated by one minus base pairing probabilities determined by Boltzmann-distributed structural variations provided by the Nupack webserver (64). This structural accessibility estimation was shown to be captured by in vivo experimental DMS reactivity at the regional level, supporting the use of base pairing probabilities as an estimator of experimentally determined regional availability (Supplementary Figure S4).
Calculation of free energy of folding for the asRNA 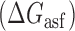
The ‘allSub’ subroutine of the RNAStructure webserver (62) was used to predict the secondary structure of the asRNA+iRS3 transcript (5΄-6 nt + asRNA + 56 nt-3΄). The Gibbs free energy of the minimum free energy structure was used to represent the asRNA folding energy 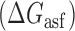 . For the purpose of this analysis, the transcript considered 62 nucleotides in addition to the 8–27 nucleotides of the asRNA (for a total of 70–89 nucleotides). Additionally, six nucleotides upstream of the asRNA were included as part of the transcript to account for imprecision of transcriptional start sites. In this way, any potential interactions between the asRNA and the segment downstream from the ribosomal binding site (RBS) site were accounted for. The specific sequence is as follows: 5΄GAA UUC -asRNA- UAC CAU UCA CCU CUU GGA UUU GGG UAU UAA AGA GGA GAA AGG UAC CAU GAG UAA AG 3΄.
. For the purpose of this analysis, the transcript considered 62 nucleotides in addition to the 8–27 nucleotides of the asRNA (for a total of 70–89 nucleotides). Additionally, six nucleotides upstream of the asRNA were included as part of the transcript to account for imprecision of transcriptional start sites. In this way, any potential interactions between the asRNA and the segment downstream from the ribosomal binding site (RBS) site were accounted for. The specific sequence is as follows: 5΄GAA UUC -asRNA- UAC CAU UCA CCU CUU GGA UUU GGG UAU UAA AGA GGA GAA AGG UAC CAU GAG UAA AG 3΄.
Model optimization via regression analysis using experimental hybridization data
Regression analysis was used to statistically evaluate the contributions of the proposed biophysical factors in the derived models. Briefly, a linear model relating the experimental response variable υ (defined as the logarithm of the ratio of (FLon-Floff) to Floff measurements) to the previously described factors 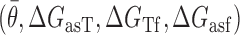 was composed and the coefficients for the various factors were fit by ordinary least squares regression. Coefficient fitting and statistical analysis of parameter contributions to the overall model were performed using MatLab Math, Statistics and Optimization package (specifically ‘fitlm’ function). A total of 383 independent fluorescence measurements (representing asRNA hybridization efficiency) across all three optimization molecules were used for regression analysis.
was composed and the coefficients for the various factors were fit by ordinary least squares regression. Coefficient fitting and statistical analysis of parameter contributions to the overall model were performed using MatLab Math, Statistics and Optimization package (specifically ‘fitlm’ function). A total of 383 independent fluorescence measurements (representing asRNA hybridization efficiency) across all three optimization molecules were used for regression analysis. 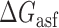 was constrained to an interval between −19.3 kCal/mol and −17.8 kCal/mol where its influence became statistically insignificant (P-value > 0.05) allowing the other more relevant factors
was constrained to an interval between −19.3 kCal/mol and −17.8 kCal/mol where its influence became statistically insignificant (P-value > 0.05) allowing the other more relevant factors 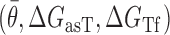 to be studied in isolation (see Supplementary Figure S5). In addition, the predictors in Equation ((8)) were normalized by the length of the asRNA to decrease linear dependency on this design parameter. A 3-fold cross-validation was performed to test for prediction ability for both optimized models (groups were randomly selected and all replicates were kept together based on their corresponding region). Each cross-validated R2 was calculated as the adjusted coefficient of determination of the linear regression fit between the experimental data of each independent group and corresponding predicted values from a model derived from the remaining 2 independent groups.
to be studied in isolation (see Supplementary Figure S5). In addition, the predictors in Equation ((8)) were normalized by the length of the asRNA to decrease linear dependency on this design parameter. A 3-fold cross-validation was performed to test for prediction ability for both optimized models (groups were randomly selected and all replicates were kept together based on their corresponding region). Each cross-validated R2 was calculated as the adjusted coefficient of determination of the linear regression fit between the experimental data of each independent group and corresponding predicted values from a model derived from the remaining 2 independent groups.
For all regression analyses conducted in this work, factors and their potential interactions were considered statistically meaningful if their P-value (t-test) was lower than 0.005. Additionally, the quality of the regression was qualitatively evaluated by visual inspection, ensuring that the residuals showed a strong normal distribution (see Supplementary Figure S6).
Selection of target regions for evaluation of model prediction ability
About 1300 potential target regions within each molecule (gII intron, SpinachII, glgC 5΄UTR, 2-MS2) were randomly generated. Starting from the first nucleotide of the molecule, regions of random length between 9 and 17 nucleotides were designed sequentially with one nucleotide overlap between each region. This process was iterated 7 additional times with respect to integer-increasing nucleotide overlap between regions, ultimately producing 8 sets of target regions with 1–8 nucleotide overlaps. To ensure that every nucleotide of each molecule was included within each set of target regions, the last region within each set was not of random length. Instead, if the first nucleotide of a prospective region was within 9–17 nt of the last nucleotide, the final probe of the respective iteration was established as the region from the first nucleotide of the prospective region to the last nucleotide of the molecule. The full set of asRNAs targeting these regions was then filtered by their calculated folding free energies to select a subset of 366 asRNAs with 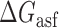 ranging from −19.3 to −17.8 kcal/mol.
ranging from −19.3 to −17.8 kcal/mol.
Lastly, this filtered set of asRNA designs was used to predict hybridization efficacies via the optimized model. The total number and sequence of asRNAs for experimental validation for each RNA were chosen based on molecule length, biophysical model hybridization prediction and targeting region. Six asRNAs were chosen for the smallest target molecule (2-MS2), 13 for the ‘mid-sized’ molecules (glgC 5΄UTR and Spinach II) and 17 for the largest (gII intron). Approximately 40% of asRNAs for each molecule were selected for their low predicted hybridization values, defined as a predicted hybridization efficacy equal to or less than the median of the asRNA pool within a molecule. The remaining asRNAs selected were within the predicted high hybridization efficacy pool, specifically, with predicted hybridization greater than the median. Precautions were taken to avoid selection of asRNAs targeting highly similar regions (>5 shared nucleotides); however, exceptions were made when two asRNAs targeting similar regions showed interesting differences in terms of predicted hybridization efficacy (differences greater than the standard error of the pool).
Statistical evaluation of model prediction ability
For each target region designed for experimental validation (above), hybridization efficacies as predicted by benchmark software IntaRNA were also estimated (28). First, the hybridization energy of each region was calculated using the pre-set seed, folding and output parameters with inputs of target RNA sequence and asRNA sequence. The hybridization energy was then normalized by the length of the asRNA oligonucleotide. The lowest (most negative) normalized energy values indicated a higher predicted hybridization potential. Predicted (by both in vivo-optimized models and IntaRNA) and experimental hybridization efficacies for each of the 4 molecules were then linearly scaled to fall between 0 and 1. To statistically evaluate the prediction potential of our models, experimental and predicted ‘high’ hybridization efficacy was defined as any hybridization efficacy greater than one standard deviation above the hybridization efficacy mean of points below the median within experimental and predicted subsets, respectively. Any points below these thresholds were considered to have ‘low’ hybridization efficacies within their categories. To evaluate the performance of our models, we also calculated the positive predictive value (PPV) of regions with high hybridization potential and the false negative rate (FNR) of regions with low hybridization efficacy defined in this specific context as follows:
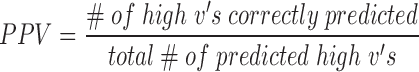 |
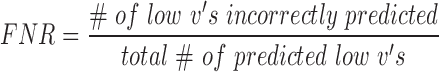 |
Evaluation of prediction of sRNA–mRNA Binding Regions
Approximately 150 potential binding regions within Zms4 and Zms6 (sRNAs recently discovered in Z. mobilis (44) but not fully characterized) were randomly generated following the process described in ‘Selection of target regions for evaluation of model prediction ability’. Hybridization efficacy of each region was predicted using the inTherAcc model. Ten total regions were selected for further target prediction analysis for each sRNA: 5 regions that exhibited the highest and 5 regions that exhibited the lowest predicted hybridization efficacy. During the selection process, regions with any overlap to a prior selected region were not considered in an attempt to select for unique regions. The reverse complement of the selected regions was inputted to nucleotide BLAST (20) to identify potential target mRNAs of these two sRNAs in Zymomonas mobilis subsp. mobilis ZM4 (taxid:264203). For selected regions encompassing less than or equal to 10 or 12 nucleotides, 2 or 1 nucleotides of the neighboring sRNA sequence were added onto both ends, respectively, to increase sequence specificity of the hits obtained by BLAST. Five potential targeting arrangements were chosen for each region from BLAST results with the constraints: (i) Minimization of E-value, (ii) Correct orientation of gene sequence, and (iii) Location of sequence at most 400 nucleotides upstream of a TSS or 200 nucleotides downstream of a TTS. For each target region designed for experimental target validation (above), hybridization efficacies as predicted by benchmark software IntaRNA were also estimated (28). First, the hybridization energy of regions within each sRNA with target mRNAs was calculated using the pre-set seed, folding and output parameters with inputs of sRNA sequence and Z. mobilis genome, target NCBI reference sequence NC_006526, within both −300 to +300 nucleotides around the start codon and stop codon, the maximum consideration window offered by the IntaRNA software. Results from both start and stop codon were consolidated within each sRNA and ranked according to energy values. An equal number of target genes to those of inTherAcc, harboring the lowest energy of interaction with the sRNA were ultimately chosen as IntaRNA predictions for Zms4 and Zms6.
RIP-seq of sRNAs Zms4 and Zms6 to identify physically associated mRNA targets
Z. mobilis 8b strain (65) was cultured in RMG media (Glucose, 20.0 g/l; Yeast Extract, 10.0 g/l; KH2PO4, 2.0 g/l; pH 6.0) at 33°C. Strains containing 2MS2-Zms4/Zms6/control plasmids were cultured in 5 ml RMG overnight with 350 μg/ml of kanamycin then transferred into 50 ml RMG to initial OD600nm of 0.1. Cells were grown anaerobically at 33°C for 12 h then pelleted by centrifugation.
Total RNA of 2MS2-Zms4/2MS2-Zms6/2MS2-control strains was prepared according to previously published methods (66). The RNA was incubated with isopropanol and GlycoBlue™ (ThermoScientific) at −20°C overnight. After centrifugation, pelleted RNA was washed with 95% cold ethanol and centrifuged. RNA was resuspended in 50 μl RNase-free water (Ambion) and stored at −80°C for sequencing.
For use as an affinity tag, MS2 coat protein fused with maltose binding protein (MS2-MBP) (67) was expressed in and harvested from E. coli.
Two micrograms of purified MS2-MBP protein was incubated with 100 μl of total RNA (500 ng/μl) extracted from the cells containing 2MS2BD-Zms4/Zms6/control for 1 h at 4°C. Washed amylose beads were incubated with 2MS2BD-Zms4/Zms6/control+MS2-MBP complex for 2 h at 4°C. Supernatants were removed from the beads by applying a magnet. Beads were washed three times with wash buffer and incubated with 50 μl of elution buffer for 15 min. The elution step was repeated so that total 100 μl were collected for each sample. For RNA precipitation, equal volume of isopropanol and 10 μl of GlycoBlue™ was added to elution sample and incubated overnight at −20°C. RNA was pelleted at 15 000 rpm for 15 min at 4°C and washed with 1 ml ethanol. The air-dried RNA pellet was resuspended in 50 μl RNase-free water, quantified and checked for quality using a Bioanalyzer before sequencing. NEBNext® Multiplex RNA Library Prep Set for Illumina® (New England Biolabs Inc.) was used for generating RNA libraries. Sequencing was performed using Illumina® NextSeq technology with paired-end 2 × 150 nt run (Genomic Sequencing and Analysis Facility at the University of Texas at Austin). All sequenced libraries were mapped to the Z. mobilis 8b complete genome (pending publication) using BWA (0.7.12-r1039) (68). DESeq2 (69) was used to identify transcripts enriched in 2MS2-Zms4 and 2MS2-Zms6 samples compared to the 2MS2 only control.
RESULTS
Description of asRNA hybridization efficacy by a thermodynamic model that includes a regional measure of interaction availability
In the context of this work, hybridization efficacy is defined as the ability of a given oligonucleotide to establish base-pairing interactions as a cohesive unit with its corresponding target region within an RNA molecule. To quantitatively estimate asRNA hybridization efficacy, we assume that it is directly proportional to the ratio of the concentration of asRNA-target RNA in the bound state (B) over the concentration of the asRNA in the unbound state (U). Starting with the standard equation for the equilibrium constant for asRNA-target RNA binding:
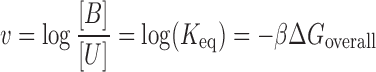 |
(2) |
Here, 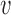 , termed hybridization efficacy, provides a measure of the asRNA-target RNA hybridization. This parameter is estimated experimentally using the logarithm of the ratio of the fluorescence measurements representative of the asRNA-target interaction to the fluorescence background levels [(FLon − FLoff)/FLoff] obtained from the iRS3 reporter (see Materials and Methods for more details). Briefly, this reporter system is composed of an asRNA that targets a specific region within the target RNA and a cis-blocking element (CB) that sequesters a RBS and controls the expression of a downstream green fluorescent protein. Therefore, fluorescence is observed upon asRNA-target RNA hybridization (FLon) as the CB-RBS interaction is disrupted and green fluorescent protein is expressed due to interaction of the asRNA with the target RNA region (Figure 1). FLoff is the fluorescence measured in the absence of the target RNA.
, termed hybridization efficacy, provides a measure of the asRNA-target RNA hybridization. This parameter is estimated experimentally using the logarithm of the ratio of the fluorescence measurements representative of the asRNA-target interaction to the fluorescence background levels [(FLon − FLoff)/FLoff] obtained from the iRS3 reporter (see Materials and Methods for more details). Briefly, this reporter system is composed of an asRNA that targets a specific region within the target RNA and a cis-blocking element (CB) that sequesters a RBS and controls the expression of a downstream green fluorescent protein. Therefore, fluorescence is observed upon asRNA-target RNA hybridization (FLon) as the CB-RBS interaction is disrupted and green fluorescent protein is expressed due to interaction of the asRNA with the target RNA region (Figure 1). FLoff is the fluorescence measured in the absence of the target RNA.
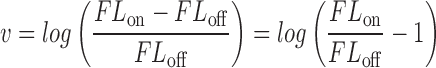 |
(3) |
The model for accessibility-based  depicted in Figure 1 is comprised of the changes in free energy due to asRNA-target binding
depicted in Figure 1 is comprised of the changes in free energy due to asRNA-target binding  , target region unfolding
, target region unfolding 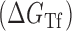 and folding of the asRNA
and folding of the asRNA 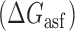 . This model captures the thermodynamic driving force of intermolecular base-pairing and the penalties for breaking the structures of the asRNA and target regions and is obtained by combining Equations ((1)), ((2)) and ((3)):
. This model captures the thermodynamic driving force of intermolecular base-pairing and the penalties for breaking the structures of the asRNA and target regions and is obtained by combining Equations ((1)), ((2)) and ((3)):
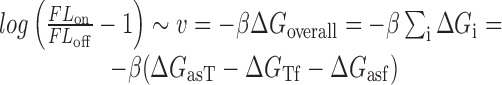 |
(4) |
Hereafter, Equation ((4)) represents the baseline thermodynamic model from which we depart for further optimization. It is worth noting that similar thermodynamic derivations have been previously used to describe accessibility-based antisense hybridization (26,28). Other approaches (i.e. AccessFold, RNAplex) have also considered an additional term for an energy penalty due to availability, which influences the transition state energy barrier (initiation energy) that the system is required to overcome to produce the bound complex (24,27). The novelty of this work lies in our two-sided treatment of target accessibility, which is incorporated in a modified free energy of target folding 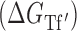 . We consider target accessibility a combination of the energy penalty for the local disruption of the target region using only the minimum free energy structure
. We consider target accessibility a combination of the energy penalty for the local disruption of the target region using only the minimum free energy structure 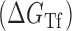 as well as the regional availability factor (
as well as the regional availability factor (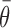 ), interpreted as a variable entropic contribution (unaccounted for in the nearest neighbor model) that depends on the specific structural context of the targeted region:
), interpreted as a variable entropic contribution (unaccounted for in the nearest neighbor model) that depends on the specific structural context of the targeted region:
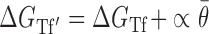 |
(5) |
In the nearest neighbor model, parameter values were obtained from short (4–10 basepairs), unstructured RNA oligomers, where the entire RNA was involved in the duplex formation. In this case, configurational entropy contributions are absent as there is no change in any structural flexibility or configurational degrees of freedom outside of the region forming the duplex. However, in the case of unfolding larger and more structured RNAs such as the gI intron, changes in target RNA folding outside of the targeted region can have a significant positive effect on the overall flexibility and configurational entropy of the target RNA. This configurational entropy can reduce the free energy penalty of target unfolding to make the overall process more favorable. Note, the sign of 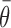 is positive in Equation ((5)) to match that of the entropy penalty of folding a large structured RNA, such that a high regional availability makes unfolding more favorable than estimated by the baseline thermodynamic model
is positive in Equation ((5)) to match that of the entropy penalty of folding a large structured RNA, such that a high regional availability makes unfolding more favorable than estimated by the baseline thermodynamic model 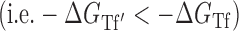 . Note also that we have assumed temperature-independence of the accessibility factor since all parameters were fitted from data at the same temperature.
. Note also that we have assumed temperature-independence of the accessibility factor since all parameters were fitted from data at the same temperature.
One example of the regional availability factor concept is shown in Figure 2. In this example, the unbinding of the colored target region from the P3 helix allows the entire domain of P7-P3–P8 helices significantly greater flexibility (Figure 2B) beyond what may be accounted for in the target region and its complement alone (Figure 2A). Here, we propose that the complex changes in RNA folding associated with asRNA-target RNA binding can be approximated by the regional availability factor. In part, the use of a regional availability factor is intended to account for structural fluctuations in dynamic regions that have a differential influence on hybridization efficacy, which we hypothesize can inform targeting by antisense RNAs. Furthermore, we propose that these regions involved in metastable alternative structures are hallmarked by the ensemble of suboptimal structures obtained from RNA folding algorithms such as Nupack (64). Thus, as a starting point before in vivo optimization, we estimate regional availability for a target region as a cohesive unit by summation of each nucleotide's local availability over the length of the target region:
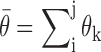 |
(6) |
Figure 2.

Proposed regional availability factor in novel biophysical model. The influence of structural availability (as captured by the ensemble of suboptimal structures) on intermolecular interactions can be attributed to variable entropic contributions to asRNA-target binding free energies that depend on structural context of targeted regions. The f →u subscript denotes the change from folded to unfolded states and the apostrophe represents the adjusted free energy accounting for regional availability. The init, AU and NNk subscripts indicate the standard free energy terms associated with initiation, terminal AU pairs and stacking free energies (respectively) in the standard nearest neighbor model. (A) The thermodynamic model from Figure 1 captures the nearest neighbor free energy of unfolding the target region based on the minimum free energy (MFE) structure. (B) The proposed regional availability factor (estimated as the sum of one minus the base pairing probabilities in the target region calculated from the ensemble of suboptimal structures in Nupack (64)) captures entropic effects of target RNA distal structural variations that occur upon target unfolding. As an example, there is a potential for a significant portion of the gI intron (regions P7, P3, P8) to have increased vibrational and translational degrees of freedom upon breaking of the long-range tertiary base pairs in the highlighted target region of the P3 helix, an effect which is captured by this example suboptimal structure. Furthermore, with the use of in vivo optimization, the effects of entropy contributions from interactions with unknown cellular factors in these dynamic regions may also be captured by the ensemble of suboptimal structures from Nupack.
In Equation ((6)), i and j represent the start and end of each region correspondingly and θk is the local availability of nucleotide k. The local availabilities (θk) can be estimated by base-paired probabilities based on the ensemble of suboptimal structures of the target RNA (unlike energy of target unfolding that is based on the minimum free energy structure alone), allowing inclusion of regional equilibrium structural fluctuations that often facilitate intermolecular interactions (70,71). Therefore, our proposed biophysical model, considers the following four predictors:
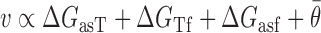 |
(7) |
Model optimization using in vivo experimental profiling of asRNA hybridization efficacy
While there is novelty in considering ensemble base-pairing probabilities as a regional availability factor, the most notable aspect of this study lies in the in vivo optimization of the above models using experimental hybridization data for a diversity of RNA targets. Conceivably, one of the greatest challenges in prediction of hybridization efficacy is the ability to account for asRNA-target interactions in vivo, where interactions with other molecules are prevalent due to molecular crowding and complex patterns of ionic strength that vary across different organisms (39). Additionally, the entropy changes of binding may vary also with the cellular environment since unanticipated intermolecular interactions (often involving these metastable structural regions) can have significant effects on system entropy. We therefore hypothesize that the proposed models can be experimentally optimized in vivo to capture regional fluctuations and cellular effects to improve estimation of asRNA hybridization efficiency. To this end, our baseline thermodynamic (Equation (4)) and proposed biophysical (Equation 7) models were optimized by taking into account in vivo hybridization patterns collected directly within cells.
For this work, we have collected large sets of antisense hybridization data using a recently published fluorescence-based assay (iRS3) for in vivo RNA profiling (43). Specifically, we interrogated 80 regions within three diverse target RNAs: the gI intron (393 nt, in which 35 regions were probed), the csrB regulator (369 nt, in which 27 regions were probed), and the glutamate tRNA (76 nt, in which 18 regions were probed). Figure 3 illustrates all the collected hybridization profiles for these three target molecules, where the heat maps depict differential levels of asRNA-target binding. A list of all 80 asRNAs designed for these molecules is included in Supplementary Table S2. These molecules make appropriate targets for this study given their complex structural features that challenge the ability to predict hybridization. For instance, in the gI intron, secondary structure domains that are essential for catalysis such as P4–P6 and P3–P9 (Figure 3A) (72,73) contain tertiary contacts (gray boxes in 3A) that are connected to each other via pseudoknots (covered by regions 8–10) (74). Predicting hybridization efficacy in pseudoknot domains is extremely challenging and most current secondary structure prediction approaches fail to predict these complex structures. In addition, these interactions are capable of disrupting the folding pathway, e.g. from misfolded to the native state, generating low abundance intermediates in which certain regions are rendered single-stranded (52,75–76). As discussed in a previous work (43), our experimental probing system bears the potential to sense transient states present in vivo, which is consistent with the relatively high hybridization efficacy of regions 8–10. In the case of CsrB, six of the regions with the lowest hybridization efficacies (regions 5, 7, 10, 11, 20 and 22 in Figure 3B) contain the binding recognition motif (GGA) for its major target, the CsrA protein (77,78,79); specifically the GGA motif in the stem loop of region 22, has been recently suggested as a strong binding site (80). Our ability to see these patterns reflected in the level of hybridization potential of these regions indicates that our data set captures the effect of in vivo interactions with other cellular factors. Lastly, our in vivo experimental data also captures expected high hybridization efficacy within the tRNA at the highly flexible anticodon arm (corresponding to region 8 in Figure 3C), consistent with molecular dynamic simulations and crystallographic B-factors for various tRNA models (81). Collectively, these observations validate the experimental data collected and used for model optimization.
Figure 3.

asRNA hybridization map as measured by in vivo oligonucleotide hybridization. Heat maps of the asRNA hybridization efficacies for (A) The Tetrahymema group I intron (35 target regions). Numbers with a dash right next to a nucleotide indicate the standard indexing of the gI intron. Stems (domains) have been named by the convention in our previous work (42) using the letter ‘P’ followed by a number for the gI intron. Tertiary contacts are indicated with a gray double-headed arrow. (B) The small RNA CsrB (27 target regions). (C) The glutamate tRNA (18 target regions), in which tertiary contacts are indicated with dashed lines. For all three heat maps, color-coded lines represent length and location of a region targeted by the iRS3 asRNA and color represents hybridization efficacies that can be decoded using the bar scale at the bottom. The target regions/asRNAs were numbered in ascending order from 5΄ to 3΄ and labels were colored in accordance with relative hybridization efficacies.
Optimization of the baseline thermodynamic (Equation (4)) and biophysical (Equation (7)) models was performed from collected experimental data by: (i) setting an interval constraint on 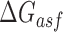 (−19.3 kCal/mol <
(−19.3 kCal/mol < 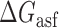 < −17.8 kCal/mol) wherein this factor is negligible, (ii) scaling all parameters to adjust for their relative importance (e.g. determination of parameter coefficients) and (iii) incorporating the interplay between prediction parameters suggested by strong statistical interactions (see Materials and Methods section). All parameters resulting from this optimization are included in Supplementary Table S3. As shown in Figure 4, we observe that the regional availability factor (
< −17.8 kCal/mol) wherein this factor is negligible, (ii) scaling all parameters to adjust for their relative importance (e.g. determination of parameter coefficients) and (iii) incorporating the interplay between prediction parameters suggested by strong statistical interactions (see Materials and Methods section). All parameters resulting from this optimization are included in Supplementary Table S3. As shown in Figure 4, we observe that the regional availability factor (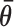 ) by itself and in relation with the energy of target unfolding
) by itself and in relation with the energy of target unfolding 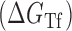 is prominent in its influence as a predictor of hybridization efficacy. This observation underscores the importance of the differential relationship between the two target accessibility measures. Interestingly, this statistically-derived mathematical form marginally resembles the scaling of the stacking energies by base-pairing probabilities used by Sfold in siRNA design (36). Importantly, the optimization of the baseline thermodynamic (Equation (4)) and biophysical (Equation 7) models led to the development of the inTher (in vivo optimized Thermodynamic), Equation ((8)) and in vivo optimized Thermodynamic Accessibility-adjusted (inTherAcc), Equation ((9)) models, respectively.
is prominent in its influence as a predictor of hybridization efficacy. This observation underscores the importance of the differential relationship between the two target accessibility measures. Interestingly, this statistically-derived mathematical form marginally resembles the scaling of the stacking energies by base-pairing probabilities used by Sfold in siRNA design (36). Importantly, the optimization of the baseline thermodynamic (Equation (4)) and biophysical (Equation 7) models led to the development of the inTher (in vivo optimized Thermodynamic), Equation ((8)) and in vivo optimized Thermodynamic Accessibility-adjusted (inTherAcc), Equation ((9)) models, respectively.
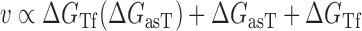 |
(8) |
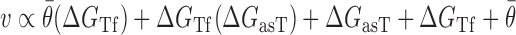 |
(9) |
Figure 4.

Relative significance of each term in the (A) inTherAcc and (B) inTher models. Optimization of baseline thermodynamic (Equation (4)) and biophysical models (Equation (7)) with in vivo data produces significant models (P-values < 1E-4 and 1E-10, respectively). The addition of the availability term (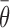 ) and its statistically significant interaction with the unfolding energy of the target region (ΔGtf) to the inTherAcc model increases the significance of common parameters seen in (B).
) and its statistically significant interaction with the unfolding energy of the target region (ΔGtf) to the inTherAcc model increases the significance of common parameters seen in (B).
Importantly, as presented in Figure 5, regression analyses of the ability of these models to capture in vivo hybridization data shows that the proposed optimized versions of both, thermodynamic and biophysical models (in Equations (8) and 9, Figure 5A and B, respectively) exhibits improved performance relative to the non-optimized models (Equations (4) and (7), Figure 5C and D). Furthermore, the use of the regional availability factor in the inTherAcc model (Equation (9), Figure 5B) shows an additional enhancement relative to its counterpart inTher in its ability to capture in vivo asRNA hybridization data, making it the best model developed in this work. These findings set the grounds for a final test case in which inTherAcc predictions of highly ‘hybridizable’ regions in four structurally diverse RNAs were experimentally validated. Collectively, these results suggest that consideration of physical intracellular interactions (as captured by the collected data) is vital to improve the accuracy of hybridization behavior predictions.
Figure 5.

Improvement in performance for in vivo optimized models underscores the influence of intracellular factors. Comparing linear correlations of (A) the in vivo optimized Thermodynamic model (Equation (8)), (B) the in vivo optimized Thermodynamic Accessibility-adjusted model (Equation 9), (C) the un-optimized thermodynamics-only method (Equation (4)) and (D) the un-optimized biophysical model (Equation (7)) shows the ability of the inTher model family to capture the in vivo collected data. This improved performance can be attributed to the incorporation of statistical interactions between prediction parameters that likely well-represent the cellular environment.
The inTherAcc model proves effective in predicting extreme asRNA hybridization regions in other RNA targets
Initial evaluation of the predictive capabilities of the inTherAcc model was performed by a 3-fold cross validation analysis using the same in vivo data set used for optimization. Our evaluation shows that the cross-validated R2 is 0.37 and 0.09 for inTherAcc and inTher models, respectively, confirming the increased predictive potential of the inTherAcc model. Given these results, we further tested the prediction capabilities of the inTherAcc model using four additional unique RNA targets: the 2-MS2 RNA tag (2-MS2), the model RNA LtrB group II intron (gII), the Spinach II RNA (SpnII) in a tRNA scaffold (82) and the glgC mRNA 5΄UTR (glgC).
To interrogate highly ‘hybridizable’ regions within these RNA molecules, 1300 target regions across the entirety of these four molecules were randomly compiled. The regions were randomly varied in length between 9–17 nucleotides (see Materials and Methods section). The hybridization efficacies of these regions were calculated using the inTherAcc model. Following predictions with the inTherAcc model, 49 regions were selected for experimental validation; 6 regions for the 2-MS2, 13 regions for Spinach, 13 regions for glgC and 17 regions for the larger gII were experimentally tested. In general, regions representing a wide range of predicted hybridization efficacy were selected, with a particular interest in those with highest ranked predicted efficacy (Figure 6A). The heat maps illustrated in Figure 6A depict relative levels of asRNA hybridization efficacy that were detected for each target molecule using the iRS3 high throughput plasmid (iRS3-GG) (see Supplementary Figure S1A) as described in the Materials and Methods section. It is worth noting that two of the top predicted regions (regions 2 and 17) for asRNA hybridization efficacy in the gII intron correspond to well-studied regions that contain one and two tertiary structure contacts, respectively (83). These contacts are known to be involved in long-range interactions (generally weaker than secondary structure interactions). In the case of the regions with the highest hybridization efficacy for 2-MS2, regions 5 and 6 both overlap with a 2-MS2 coat protein binding site (84) located in a loop. Likewise, it is noteworthy that region 2 within the GlgC 5΄UTR, targeting its preferred CsrA interacting site (85), appears to be one of the regions with the lowest hybridization efficacies. On the other hand, regions 6 and 7 in the glgC 5΄UTR overlap with the relatively more single-stranded (86,87) SD and start codon regions, respectively, and show one of the highest hybridization efficacies. Lastly, in the Spinach molecule, region 4 covers the binding site for DFHBI (88,89), the target molecule of this aptamer. Overall, these observations indicated that our predictions of extreme hybridization potential captured important structural-functional features of these molecules.
Figure 6.

Experimental evaluation of hybridization efficacy in four RNAs shows inTherAcc model prediction accuracy comparable to that of benchmark IntaRNA. (A) Relative hybridization efficacy of each tested region is indicated on the predicted secondary structure (64) of respective molecules via color-coded lines, in which green and red represent highest and lowest hybridization efficacy, respectively, per scale bar (bottom). Each nucleotide is colored based on equilibrium probability (bar on the right) according to Nupack (64) output. Regions which were correctly predicted by inTherAcc to be high or low are denoted by an asterisk. (B) Comparison of positive Predictive Value-PPV (top) for high hybridization efficacies and False Negative Ratio-FNR (bottom) for low hybridization efficacies, for inTher (red), inTherAcc (blue) and IntaRNA (green).
Importantly, when calculating the PPV of regions with high hybridization efficacy and the FNR of regions with low hybridization efficacy for all the data collected, inTherAcc (but not inTher) performed overall comparably to IntaRNA predictions in terms of PPV and FNR. We chose to benchmark against IntaRNA since it is an accessibility-based approach, uses a seed interaction that resembles our regional interaction notion and has been tested for bacterial systems (28). However, inTherAcc displays improved prediction performance, relative to IntaRNA, particularly for the gII intron (R2 = 0.13 versus 0.08) and 2-MS2 (R2 = 0.949 versus 0.014) as shown in Figure 7. No difference in performance was observed when considering the linear correlations for glgC and SpinachII RNAs (R2 < 5% for all three models). In summary, these findings support the potential prospects of considering both, IntaRNA and inTherAcc, complementary approaches in the prediction of hybridization efficacy (see Supplementary Figure S7 for a summary of all the prediction versus experimental results).
Figure 7.

Regression analysis on experimental versus inTherAcc-(top), inTher-(center) and IntaRNA-(bottom) predicted hybridization efficacy for (A) 2-MS2 and (B) gII intron. inTherAcc exhibits superior performance in predicting hybridization potential in (A) 2-MS2 and (B) group II intron compared to both inTher and IntaRNA models when considering linear regression fits. (B) Higher performance accuracy of hybridization efficacy in gII intron is achieved by inTherAcc due to its capability to predict extreme lows and highs. Error bars indicate standard error of the mean. Both predicted and experimental hybridization efficacies were linearly scaled from 0 to 1.
inTherAcc aids in prediction of target mRNAs
As a final model validation, we evaluated the ability of inTherAcc to aid in prediction of target mRNAs of newly-identified sRNAs in a different bacterium. We selected two sRNAs responsive in expression level to ethanol stress, Zms4 (280 nt) and Zms6 (304 nt) of Z. mobilis (44). A RIP-seq experiment was performed by tagging each sRNA with 2-MS2 RNA. Following purification of the sRNAs with the MS2-binding protein and sequencing the physically associated RNAs, the most likely targets were identified as those that showed the greatest transcript enrichment compared to a control (2-MS2 with no sRNA attached). Because of the response of Zms4 and Zms6 to ethanol stress, we expect their mRNA targets to include stress-related genes. Indeed, as expected, many potential targets discovered by RIP-seq for both Zms4 and Zms6 (Supplementary Table S4) were related to stress responses, including global stress response regulators, heat shock proteins, protein folding chaperones and DNA repair proteins. Because the inTherAcc model is well-suited to help narrow the large pool of potential targets by predicting those with most favorable hybridization efficacies, potential regions of interest in both sRNAs were randomly compiled and ranked by hybridization efficacies using our inTherAcc model (Figure 8A), as described in the Materials and Methods section. As observed in Figure 8A, interesting ‘hot spots,’ defined as regions exhibiting predicted extreme (high or low) hybridization efficacies were identified and considered for further analysis. The rationale behind using regions with predicted high and low hybridization efficacies is based on the hypothesis that these regions are likely to be functional sites either highly available or unavailable based on active binding to in vivo factors. The reverse complement sequences of the five highest and five lowest predicted hybridization efficacies were selected for BLAST analysis to identify potential ‘top’ likely interacting mRNA targets (for a total of 52–54 unique genes considered). Comparisons of these results with data obtained from RIP-seq experiments supported the target prediction capability of the inTherAcc model. As shown in Figure 8B, inTherAcc predicted about 28 and 22 potential targets, respectively for Zms4 and Zms6, found in the set of RIP-seq-determined enriched transcripts. Importantly, about 8 and 7 potential targets respectively for Zms4 and Zms6 were found within the top approximately 20% pulled-down targets (ranked by fold change enrichment relative to the 2-MS2-only control). In all cases for each region predicted to be an mRNA binding site, multiple potential targets were found suggesting the ability of these sRNAs to exert multiplex regulation (Supplementary Table S4). As expected, a considerable portion of enriched transcript associations of Zms4 and Zms6 correctly predicted by inTherAcc code for proteins involved in ethanol tolerance mechanisms, specifically those that facilitate (i) protein folding and transport, (ii) redox metabolism and (iii) stress response (90,91), further validating our results. In addition, inTherAcc showed a comparable performance to benchmark IntaRNA (Supplementary Table S4 and Figure 8B). The limited number of matches in target prediction (Figure 8B) between both approaches underscores the potential complementarity between them. Collectively, these results show the potential of the model to aid in gene target prediction and, more specifically, to identify potential functional regions that act via base-pairing.
Figure 8.

inTherAcc aids in prediction of mRNA targets for Z. mobilis (A) Zms4 and (B) Zms6. Ten regions evenly distributed at the top (green) and bottom (red) of the hybridization efficacy scale were selected as potential mRNA–sRNA binding sites for further prediction of target mRNA candidates and comparison with RIP-seq data. The regions that matched with the 18% of top enriched candidates (log2 of fold change sRNA/only MS2) are marked with blue arrows. (C) Overview of the prediction performance for IntaRNA (green) and inTherAcc (blue). Venn diagram showing the total enriched candidates (log2(sRNA/only MS2) > 0). A total of 52 and 54 candidates respectively for Zms4 and Zms6 were predicted using both approaches. Darker green and darker blue circles represent the top 18% enriched candidates that each approach predicted correctly.
DISCUSSION AND CONCLUSIONS
The inTherAcc model incorporates a series of thermodynamic terms to account for energetics of intramolecular folding, intermolecular binding and the target region availability using the Boltzmann distribution of possible structural configurations. The novelty of this approach lies in the integration of large scale in vivo data as well as the interplay between the components of target accessibility as understood by (i) an availability factor based on suboptimal structures and (ii) thermodynamic consideration of RNA unfolding, identified during model optimization. Our results suggest that the family of inTher models that we have developed could assist current asRNA predictions to capture ‘hybridizability’ in vivo. Our work also highlights the potential of using in vivo experimental data sets to increase prediction accuracy for effective selection of sites for asRNA targeting and provides a methodology to do so. The observed relationship between target RNA folding energy and regional target availability as estimated by a summation of local base-pairing probabilities was shown via statistical model optimization (Figure 4 and Supplementary Table S3) and suggests that scaling this free energy by its availability factor plays a significant role in determining efficacy of RNA hybridization in vivo. Other research groups have used similar scaling approaches with significant improvements in the performance of siRNA design and predictions (60,92–93). The main difference of our scaling scheme relative to these previous efforts is its regional nature. While previous works scaled the stacking energies of interacting nucleotides one by one according to nucleotide-specific base-pairing probabilities, this approach assumes that any given asRNA behaves as an indivisible unit. In addition, this in vivo optimization has brought about coefficients for our model that are meaningful in capturing intracellular behavior. For instance, as expected, we observed a strong influence of the intramolecular structure of the target region on the hybridization efficacy (Figure 4). Moreover, the estimated coefficients could be indicators of the presence of binding factors, the effect of molecular crowding or even the presence of ionic species in the cellular milieu. For example, divalent ion influence on ribozyme active site structural arrangement (94) was likely to an extent accounted for by optimizing inTher models with gI intron data. It is therefore not surprising that the optimized inTherAcc model was an improved predictor of gII intron hybridization efficacy. To the best of our knowledge, no approach in the past has attempted to consider the in vivo environment by optimizing a current biophysical model using large sets of in vivo data collected in bacteria and applying it to predict other RNA molecules, while simultaneously studying the influence of target accessibility.
Through in vivo optimization of model parameters, we achieved a highly reliable qualitative prediction of highly ‘hybridizable’ regions in a wide array of RNA molecules. Overall, the inTherAcc model performs at levels above 63% and below 60% in PPV and FNR, correspondingly. Furthermore, inTherAcc predictions are sensitive to known protein and small molecule binding sites in 2-MS2 coat protein binding domain and SpinachII, showcasing its potential to recognize regulatory features within RNAs. It also is at least comparable to benchmark IntaRNA, bearing an advantage in specific cases likely due to the incorporation of in vivo factors during model optimization. Interestingly, some of the observed discrepancies between experimental and predicted hybridization efficacy in the glgC 5΄UTR can be attributed to competitive binding between the asRNA and factors that naturally interact with this RNA that are not fully accounted for by the collected data set. In many of these cases, we suspect that even our experimentally collected data sets fail to capture the full set of molecular interactions (e.g. with other intracellular factors) given that only limited environmental conditions were tested where the full range of these interactions does not occur. This is likely the case for regulatory RNA regions like the glgC UTR, in which different interactions are observed in vivo under nutritional stresses (not tested in this work). As a result, we hypothesize that further prediction accuracy can be achieved for these models by expanding the collected data sets to include a variety of environmental conditions (e.g. cellular stresses) to capture a broader range of interactions.
Remarkably, the inTherAcc approach provides the following general strategies in asRNA design: (i) the suggestion of a free energy interval within which the thermodynamic stability of the asRNA does not seem to influence hybridization efficacy, (ii) the realization that both low and high inTherAcc-predicted hybridization efficacies could indicate functional sites that may be interesting targets for asRNAs and (iii) evidence of the potential influence of suboptimal structures in hybridization efficacy that aids in identification of target dynamic regions. Overall, we envision that the inTherAcc approach will assist in the characterization of newly-identified regulatory RNAs and the design of synthetic elements that require RNA binding through complementarity by improving reliability of RNA targeting performance in vivo, particularly in bacteria.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Randall Hughes (Center for Systems and Synthetic Biology of The University of Texas at Austin) for his help in building our master Golden Gate cloning-ready plasmid. The authors would also like to acknowledge Victoria Fry and Praveen Vilamathas for their assistance in synthesizing the asRNAs and in collecting fluorescence data used in this work as well as Samuel Mihelic (The University of Texas at Austin Department of Biomedical Engineering) for his kind help in optimizing various Matlab codes.
Author contributions: Designed Research: J.V.A., M.K.M., K.C.B., K.G.R., W.B.P. and L.M.C.; Performed experiments: J.V.A., M.K.M., K.C.B., K.H., S.H.C. and P.A.; Analyzed data: J.V.A., M.K.M., K.C.B., K.G.R., K.H., S.H.C., P.A. and L.M.C. Wrote paper: J.V.A., M.K.M., K.C.B., P.A., K.H. and L.M.C.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Welch Foundation [F-1756]; Consejo Nacional de Ciencia y Tecnología for the graduate fellowship [CONACYT- 194638 to J.V.A]; Natural Materials and Systems [AFOSR grant contract FA9550-12-1-0200]; Air Force Office of Scientific Research (AFOSR) Young Investigator Program [FA9550-13-1-0160]; Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program (to K.C.B.); National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) [DGE-1110007 to K.H.]. Funding for open access charge: Air Force Office of Scientific Research (AFOSR) Young Investigator Program [FA9550-13-1-0160].
Conflict of interest statement. None declared.
REFERENCES
- 1. Vazquez-Anderson J., Contreras L.M.. Regulatory RNAs: charming gene management styles for synthetic biology applications. RNA Biol. 2013; 10:1778–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Georg J., Hess W.R.. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011; 75:286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cho S.H., Haning K., Contreras L.M.. Strain engineering via regulatory noncoding RNAs: not a one-blueprint-fits-all. Curr. Opin. Chem. Eng. 2015; 10:25–34. [Google Scholar]
- 4. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 5. Faoro C., Ataide S.F.. Ribonomic approaches to study the RNA-binding proteome. FEBS Lett. 2014; 588:3649–3664. [DOI] [PubMed] [Google Scholar]
- 6. Srisawat C., Engelke D.R.. RNA affinity tags for purification of RNAs and ribonucleoprotein complexes. Methods. 2002; 26:156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan J.H., Lim S., Wong W.S.. Antisense oligonucleotides: from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006; 33:533–540. [DOI] [PubMed] [Google Scholar]
- 8. Bennett C.F., Swayze E.E.. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010; 50:259–293. [DOI] [PubMed] [Google Scholar]
- 9. Coleman J., Green P.J., Inouye M.. The use of RNAs complementary to specific mRNAs to regulate the expression of individual bacterial genes. Cell. 1984; 37:429–436. [DOI] [PubMed] [Google Scholar]
- 10. Haning K., Cho S.H., Contreras L.M.. Small RNAs in mycobacteria: an unfolding story. Front. Cell. Infect. Microbiol. 2014; 4:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakashima N., Miyazaki K.. Bacterial Cellular Engineering by Genome Editing and Gene Silencing. Int. J. Mol. Sci. 2014; 15:2773–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakashima N., Tamura T.. Conditional gene silencing of multiple genes with antisense RNAs and generation of a mutator strain of Escherichia coli. Nucleic Acids Res. 2009; 37:e103–e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chae T.U., Kim W.J., Choi S., Park S.J., Lee S.Y.. Metabolic engineering of Escherichia coli for the production of 1,3-diaminopropane, a three carbon diamine. Sci. Rep. 2015; 5:13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoo S.M., Na D., Lee S.Y.. Design and use of synthetic regulatory small RNAs to control gene expression in Escherichia coli. Nat. Protoc. 2013; 8:1694–1707. [DOI] [PubMed] [Google Scholar]
- 15. Chaudhary A.K., Na D., Lee E.Y.. Rapid and high-throughput construction of microbial cell-factories with regulatory noncoding RNAs. Biotechnol. Adv. 2015; 33:914–930. [DOI] [PubMed] [Google Scholar]
- 16. Hoynes-O’Connor A., Moon T.S.. Development of design rules for reliable antisense RNA behavior in E. coli. ACS Synthetic Biol. 2016; 5:1441–1454. [DOI] [PubMed] [Google Scholar]
- 17. Backofen R. Gorodkin J, Ruzzo LW. RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods. 2014; Totowa, NJ: Humana Press; 417–435. [Google Scholar]
- 18. Lorenz R., Wolfinger M.T., Tanzer A., Hofacker I.L.. Predicting RNA secondary structures from sequence and probing data. Methods. 2016; 103:86–98. [DOI] [PubMed] [Google Scholar]
- 19. Gerlach W., Giegerich R.. GUUGle: a utility for fast exact matching under RNA complementary rules including G-U base pairing. Bioinformatics. 2006; 22:762–764. [DOI] [PubMed] [Google Scholar]
- 20. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 21. Gruber A.R., Lorenz R., Bernhart S.H., Neuböck R., Hofacker I.L.. The Vienna RNA Websuite. Nucleic Acids Res. 2008; 36:W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R.. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tjaden B., Goodwin S.S., Opdyke J.A., Guillier M., Fu D.X., Gottesman S., Storz G.. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006; 34:2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tafer H., Hofacker I.L.. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics. 2008; 24:2657–2663. [DOI] [PubMed] [Google Scholar]
- 25. Dirks R.M., Bois J.S., Schaeffer J.M., Winfree E., Pierce N.A.. Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev. 2007; 49:65–88. [Google Scholar]
- 26. Muckstein U., Tafer H., Hackermuller J., Bernhart S.H., Stadler P.F., Hofacker I.L.. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006; 22:1177–1182. [DOI] [PubMed] [Google Scholar]
- 27. DiChiacchio L., Sloma M.F., Mathews D.H.. AccessFold: predicting RNA-RNA interactions with consideration for competing self-structure. Bioinformatics. 2016; 32:1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Busch A., Richter A.S., Backofen R.. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008; 24:2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pervouchine D.D. IRIS: intermolecular RNA interaction search. Genome Inform. 2004; 15:92–101. [PubMed] [Google Scholar]
- 30. Chitsaz H., Salari R., Sahinalp S.C., Backofen R.. A partition function algorithm for interacting nucleic acid strands. Bioinformatics. 2009; 25:i365–i373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang F.W.D., Qin J., Reidys C.M., Stadler P.F.. Partition function and base pairing probabilities for RNA–RNA interaction prediction. Bioinformatics. 2009; 25:2646–2654. [DOI] [PubMed] [Google Scholar]
- 32. Lu Z.J., Mathews D.H.. Efficient siRNA selection using hybridization thermodynamics. Nucleic Acids Res. 2008; 36:640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ding Y., Lawrence C.E.. Statistical prediction of single-stranded regions in RNA secondary structure and application to predicting effective antisense target sites and beyond. Nucleic Acids Res. 2001; 29:1034–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bernhart S.H., Muckstein U., Hofacker I.L.. RNA Accessibility in cubic time. Algorithms Mol. Biol. 2011; 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tafer H. Gorodkin J, Ruzzo LW. RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods. 2014; Totowa, NJ: Humana Press; 477–490. [Google Scholar]
- 36. Ding Y., Chan C.Y., Lawrence C.E.. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004; 32:W135–W141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mathews D.H., Burkard M.E., Freier S.M., Wyatt J.R., Turner D.H.. Predicting oligonucleotide affinity to nucleic acid targets. RNA. 1999; 5:1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vickers T.A., Wyatt J.R., Freier S.M.. Effects of RNA secondary structure on cellular antisense activity. Nucleic Acids Res. 2000; 28:1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leamy K.A., Assmann S.M., Mathews D.H., Bevilacqua P.C.. Bridging the gap between in vitro and in vivo RNA folding. Q. Rev. Biophys. 2016; 49:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao J.J., Lemke G.. Rules for ribozymes. Mol. Cell. Neurosci. 1998; 11:92–97. [DOI] [PubMed] [Google Scholar]
- 41. Rodrigo G., Landrain T.E., Jaramillo A.. De novo automated design of small RNA circuits for engineering synthetic riboregulation in living cells. Proc. Natl. Acad. Sci. 2012; 109:15271–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lucks J.B., Qi L., Mutalik V.K., Wang D., Arkin A.P.. Versatile RNA-sensing transcriptional regulators for engineering genetic networks. Proc. Natl. Acad. Sci. 2011; 108:8617–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sowa S.W., Vazquez-Anderson J., Clark C.A., De La Pena R., Dunn K., Fung E.K., Khoury M.J., Contreras L.M.. Exploiting post-transcriptional regulation to probe RNA structures in vivo via fluorescence. Nucleic Acids Res. 2015; 43:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cho S.H., Lei R., Henninger T.D., Contreras L.M.. Discovery of ethanol-responsive small RNAs in Zymomonas mobilis. Appl. Environ. Microbiol. 2014; 80:4189–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kast P., Hennecke H.. Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations. J. Mol. Biol. 1991; 222:99–124. [DOI] [PubMed] [Google Scholar]
- 46. Kast P. pKSS–a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene. 1994; 138:109–114. [DOI] [PubMed] [Google Scholar]
- 47. Gibson D.G. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011; 498:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Engler C., Marillonnet S.. Valla S, Lale R. DNA Cloning and Assembly Methods. 2014; Totowa, NJ: Humana Press; 119–131. [Google Scholar]
- 49. Sowa S.W., Gelderman G., Leistra A.N., Buvanendiran A., Lipp S., Pitaktong A., Vakulskas C.A., Romeo T., Baldea M., Contreras L.M.. Integrative FourD omics approach profiles the target network of the carbon storage regulatory system. Nucleic Acids Res. 2017; doi:10.1093/nar/gkx048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zou S.-l., Zhang K., You L., Zhao X.-m., Jing X., Zhang M.-h.. Enhanced electrotransformation of the ethanologen Zymomonas mobilis ZM4 with plasmids. Eng. Life Sci. 2012; 12:152–161. [Google Scholar]
- 51. Wan Y., Suh H., Russell R., Herschlag D.. Multiple Unfolding Events during Native Folding of the Tetrahymena Group I Ribozyme. J. Mol. Biol. 2010; 400:1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Russell R., Das R., Suh H., Travers K.J., Laederach A., Engelhardt M.A., Herschlag D.. The paradoxical behavior of a highly structured misfolded intermediate in RNA folding. J. Mol. Biol. 2006; 363:531–544. [DOI] [PubMed] [Google Scholar]
- 53. Ding Y., Tang Y., Kwok C.K., Zhang Y., Bevilacqua P.C., Assmann S.M.. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014; 505:696–700. [DOI] [PubMed] [Google Scholar]
- 54. Brion P., Westhof E.. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 1997; 26:113–137. [DOI] [PubMed] [Google Scholar]
- 55. Babitzke P., Romeo T.. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr. Opin. Microbiol. 2007; 10:156–163. [DOI] [PubMed] [Google Scholar]
- 56. Paige J.S., Wu K.Y., Jaffrey S.R.. RNA mimics of green fluorescent protein. Science. 2011; 333:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frommer J., Appel B., Muller S.. Ribozymes that can be regulated by external stimuli. Curr. Opin. Biotechnol. 2015; 31:35–41. [DOI] [PubMed] [Google Scholar]
- 58. Mitra S., Shcherbakova I.V., Altman R.B., Brenowitz M., Laederach A.. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res. 2008; 36:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xia T., SantaLucia J. Jr, Burkard M.E., Kierzek R., Schroeder S.J., Jiao X., Cox C., Turner D.H.. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998; 37:14719–14735. [DOI] [PubMed] [Google Scholar]
- 60. Shao Y., Wu Y., Chan C.Y., McDonough K., Ding Y.. Rational design and rapid screening of antisense oligonucleotides for prokaryotic gene modulation. Nucleic Acids Res. 2006; 34:5660–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tijerina P., Mohr S., Russell R.. DMS footprinting of structured RNAs and RNA-protein complexes. Nat. Protoc. 2007; 2:2608–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Reuter J., Mathews D.. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010; 11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mathews D.H., Sabina J., Zuker M., Turner D.H.. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999; 288:911–940. [DOI] [PubMed] [Google Scholar]
- 64. Zadeh J.N., Steenberg C.D., Bois J.S., Wolfe B.R., Pierce M.B., Khan A.R., Dirks R.M., Pierce N.A.. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2011; 32:170–173. [DOI] [PubMed] [Google Scholar]
- 65. Zhang M., Eddy C., Deanda K., Finkelstein M., Picataggio S.. Metabolic engineering of a Pentose metabolism pathway in Ethanologenic Zymomonas mobilis. Science. 1995; 267:240–243. [DOI] [PubMed] [Google Scholar]
- 66. DiChiara J.M., Contreras-Martinez L.M., Livny J., Smith D., McDonough K.A., Belfort M.. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res. 2010; 38:4067–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Said N., Rieder R., Hurwitz R., Deckert J., Urlaub H., Vogel J.. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009; 37:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lai D., Proctor J.R., Meyer I.M.. On the importance of cotranscriptional RNA structure formation. RNA. 2013; 19:1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grohman J.K., Gorelick R.J., Kottegoda S., Allbritton N.L., Rein A., Weeks K.M.. An immature retroviral RNA genome resembles a kinetically trapped intermediate state. J. Virol. 2014; 88:6061–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Beaudry A.A., Joyce G.F.. Minimum secondary structure requirements for catalytic activity of a self-splicing group I intron. Biochemistry. 1990; 29:6534–6539. [DOI] [PubMed] [Google Scholar]
- 73. Jaeger L., Michel F., Westhof E.. Eckstein F, Lilley DMJ. Catalytic RNA. 1997; Berlin, Heidelberg: Springer Berlin Heidelberg; 33–51. [Google Scholar]
- 74. Ikawa Y., Yoshioka W., Ohki Y., Shiraishi H., Inoue T.. Self-splicing of the Tetrahymena group I ribozyme without conserved base-triples. Genes Cells. 2001; 6:411–420. [DOI] [PubMed] [Google Scholar]
- 75. Mitchell D. 3rd, Jarmoskaite I., Seval N., Seifert S., Russell R.. The long-range P3 helix of the Tetrahymena ribozyme is disrupted during folding between the native and misfolded conformations. J. Mol. Biol. 2013; 425:2670–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xue Y., Gracia B., Herschlag D., Russell R., Al-Hashimi H.M.. Visualizing the formation of an RNA folding intermediate through a fast highly modular secondary structure switch. Nature Commun. 2016; 7:doi:10.1038/ncomms11768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Romeo T., Vakulskas C.A., Babitzke P.. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ. Microbiol. 2013; 15:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lapouge K., Perozzo R., Iwaszkiewicz J., Bertelli C., Zoete V., Michielin O., Scapozza L., Haas D.. RNA pentaloop structures as effective targets of regulators belonging to the RsmA/CsrA protein family. RNA Biol. 2013; 10:1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Holmqvist E., Wright P.R., Li L., Bischler T., Barquist L., Reinhardt R., Backofen R., Vogel J.. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 2016; 35:991–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vakulskas C.A., Leng Y., Abe H., Amaki T., Okayama A., Babitzke P., Suzuki K., Romeo T.. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res. 2016; 44:7896–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bahar I., Jernigan R.L.. Vibrational dynamics of transfer RNAs: comparison of the free and synthetase-bound forms1. J. Mol. Biol. 1998; 281:871–884. [DOI] [PubMed] [Google Scholar]
- 82. Ponchon L., Dardel F.. Recombinant RNA technology: the tRNA scaffold. Nat. Methods. 2007; 4:571–576. [DOI] [PubMed] [Google Scholar]
- 83. Cui X., Matsuura M., Wang Q., Ma H., Lambowitz A.M.. A group II intron-encoded maturase functions preferentially in cis and requires both the reverse transcriptase and X domains to promote RNA splicing. J. Mol. Biol. 2004; 340:211–231. [DOI] [PubMed] [Google Scholar]
- 84. Shtatland T., Gill S.C., Javornik B.E., Johansson H.E., Singer B.S., Uhlenbeck O.C., Zichi D.A., Gold L.. Interactions of Escherichia coli RNA with bacteriophage MS2 coat protein: genomic SELEX. Nucleic Acids Res. 2000; 28:E93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Baker C.S., Morozov I., Suzuki K., Romeo T., Babitzke P.. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol. Microbiol. 2002; 44:1599–1610. [DOI] [PubMed] [Google Scholar]
- 86. Wan Y., Qu K., Zhang Q.C., Flynn R.A., Manor O., Ouyang Z., Zhang J., Spitale R.C., Snyder M.P., Segal E. et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014; 505:706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E.. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010; 467:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Strack R.L., Disney M.D., Jaffrey S.R.. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat. Methods. 2013; 10:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Warner K.D., Chen M.C., Song W., Strack R.L., Thorn A., Jaffrey S.R., Ferré-D’Amaré A.R.. Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat. Struct. Mol. Biol. 2014; 21:658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cray J.A., Stevenson A., Ball P., Bankar S.B., Eleutherio E.C.A., Ezeji T.C., Singhal R.S., Thevelein J.M., Timson D.J., Hallsworth J.E.. Chaotropicity: a key factor in product tolerance of biofuel-producing microorganisms. Curr. Opin. Biotechnol. 2015; 33:228–259. [DOI] [PubMed] [Google Scholar]
- 91. Ingram L.O. Ethanol Tolerance in Bacteria. Crit. Rev. Biotechnol. 1989; 9:305–319. [DOI] [PubMed] [Google Scholar]
- 92. Ding Y., Chan C.Y., Lawrence C.E.. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005; 11:1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shao Y., Chan C.Y., Maliyekkel A., Lawrence C.E., Roninson I.B., Ding Y.. Effect of target secondary structure on RNAi efficiency. RNA. 2007; 13:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Woodson S.A. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr. Opin. Chem. Biol. 2005; 9:104–109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.