


Abstract
Human GEN1 and yeast Yen1 are endonucleases with the ability to cleave Holliday junctions (HJs), which are proposed intermediates in recombination. In vivo, GEN1 and Yen1 function secondarily to Mus81, which has weak activity on intact HJs. We show that the genetic relationship is reversed in Drosophila, with Gen mutants having more severe defects than mus81 mutants. In vitro, DmGen, like HsGEN1, efficiently cleaves HJs, 5΄ flaps, splayed arms, and replication fork structures. We find that the cleavage rates for 5΄ flaps are significantly higher than those for HJs for both DmGen and HsGEN1, even in vast excess of enzyme over substrate. Kinetic studies suggest that the difference in cleavage rates results from a slow, rate-limiting conformational change prior to HJ cleavage: formation of a productive dimer on the HJ. Despite the stark difference in vivo that Drosophila uses Gen over Mus81 and humans use MUS81 over GEN1, we find the in vitro activities of DmGen and HsGEN1 to be strikingly similar. These findings suggest that simpler branched structures may be more important substrates for Gen orthologs in vivo, and highlight the utility of using the Drosophila model system to further understand these enzymes.
INTRODUCTION
Recombination is an integral process for DNA damage repair as well as for horizontal gene transfer during conjugation or transduction (1). Robin Holliday proposed a molecular model for recombination that included a four-stranded intermediate linking two DNA helices (2). Support for the existence of this intermediate, now termed a Holliday junction (HJ), came from electron microscopy of phage lambda DNA undergoing recombination in Escherichia coli (3). In vitro synthetic HJ cleavage assays using prokaryotic enzymes, like RuvC, provided the first direct evidence for the existence of structure-selective endonucleases (SSEs) capable of HJ cleavage, termed HJ resolvases (4). Prokaryotic resolvases show preference for HJs over other DNA branched structures and cleave symmetrically about the HJ axis, creating duplex DNA products with a nick that can be ligated without additional processing (5). Their in vivo resolvase function was evident in that mutations in the genes encoding these enzymes led to reduced recombination and sensitivity to DNA damaging agents (6). These properties became the benchmark for defining canonical resolvases.
The in vivo and in vitro evidence for resolvase function in bacteria led to the search for similar activities and genes in eukaryotes. Electron microscopy of yeast recombination intermediates provided visual evidence that eukaryotic recombination can involve Holliday junction intermediates (7). Extensive in vitro studies of HJ cleavage activity identified Yen1 in budding yeast and GEN1 in human cells (8,9), as well as the MUS81–EME1–SLX1–SLX4 complex (hereafter called MUS–SLX complex) in humans and mice (10–12). HsGEN1 dimerizes on HJs to coordinate symmetrical cleavage across the HJ, whereas MUS–SLX resolves HJs through the coordination of a nick by SLX1 with a counter-nick by MUS81–EME1.
Although GEN1 and the MUS–SLX complex display canonical resolvase activity similar to that of prokaryotic resolvases, their biochemical properties differ from those of prokaryotic resolvases in that the eukaryotic enzymes are not obligate homodimers and they cleave other branched DNA structures such as flaps, replication forks, and nicked HJs (8–12). Furthermore, mutations in the genes encoding these enzymes do not cause the same recombination defects and DNA damage sensitivities that occur when bacterial resolvases are knocked out. Loss of Mus81 results in hypersensitivity to a broad range of DNA damaging agents (13–15), but hypersensitivities resulting from loss of Slx1 are weaker to a non-overlapping set of damaging agents (10,16). These findings suggest that many of Mus81's repair functions lie outside of a MUS–SLX complex. GEN1 resolvase is genetically less complicated because it only involves one gene. However, mutations in S. cerevisiae YEN1 or murine Gen1 do not cause any apparent DNA repair defects on their own, but they do increase the severity of mus81 mutant phenotypes in double mutants, suggesting that Yen1/Gen1 act primarily as backups to Mus81 (17–20). Consequently, one of the primary challenges to characterizing Yen1/GEN1 has been the need to study the null effect in the background of null mutations in other endonucleases.
Drosophila provides a unique platform for understanding functions of Yen1/GEN1 because the hierarchical relationship between DmGen and Mus81 appears to be reversed. Unlike in other organisms, Drosophila mus81 mutants are hypersensitive to only a few DNA damaging agents, and then only mildly (21). Furthermore, in the absence of the DNA repair helicase Blm, loss of DmGen causes a much more severe phenotype (death early in larval development) than loss of Mus81 (death late in pupal development) (22).
Here, we confirm that DmGen is important in DNA damage repair by showing that, unlike in yeast and mammalian cells, Gen single mutants are severely hypersensitive to several different DNA damaging agents. We show that, like its human ortholog, DmGen efficiently cleaves HJs, 5΄ flaps, splayed arms, and replication fork structures. We find that the cleavage rate for 5΄ flaps is significantly higher than the cleavage rate for HJs. Kinetic studies suggest that the difference in cleavage rates results from a slow, rate-limiting conformational change prior to HJ cleavage: formation of a productive dimer on the HJ. We compared DmGen to human GEN1 in side-by-side experiments. While slight differences such as the propensity to dimerize do exist between DmGen and HsGEN1, we find the activities of the orthologs to be strikingly similar, including the higher cleavage rate on flaps than on HJs. These findings suggest that simpler branched structures may be more important substrates for Gen orthologs in vivo, and they highlight the utility of using the Drosophila model system to further understand this class of enzymes.
MATERIALS AND METHODS
Drosophila stocks and genetics
All stocks were maintained at 25°C on standard media. The following null mutations were described previously: mus81NheI (21) and GenZ5997 (22), which was made hemizygous with Df(3L)6103. Sensitivity to DNA damaging agents was done as described previously (23). For nitrogen mustard (HN2), hydroxyurea (HU), and methylmethane sulfonate (MMS), 250 μl water containing the agent at the indicated concentration were added to each vial containing feeding larvae. Camptothecin (CPT) was dissolved in DMSO and diluted in 10% ethanol and 0.2% Tween; control larvae were treated with DMSO only. For IR, vials with third instar larvae were irradiated with 20 Grays from a 137Cs source (GammaCell GG10). Progeny were scored for 5 days after eclosion began. Relative survival was calculated as the ratio of mutant to control flies per vial and was normalized to the ratio in untreated vials. Statistical analyses were done using Prism (GraphPad, San Diego, CA, USA).
Purification of full-length and truncated Gen (1–518) from E. coli
DmGen cDNA was codon-optimized by GenScript. Full-length DmGen (1–726 aa) and truncated DmGen (1-518 aa) were cloned into the NdeI and XhoI sites of pET21b (Novagen, Madison, WI), which carries a C-terminal hexahistidine tag (His). The nuclease-dead mutations E143A and E145A, previously described by (24), were made by QuikChange site-directed mutagenesis (Agilent Technologies, Santa Clara, CA, USA). DmGen-His was expressed in RDK cells (gift of Dr. Steve Matson) with 0.4 mM IPTG, and DmGen (1–518)-His was expressed in Rosetta II pLysS (Novagen) with 1.0 mM IPTG. All proteins were expressed at 18°C for 18 h. The DmGen (1–518)-His and DmGen (1–518)Dead-His pellets were lysed in NiA buffer (20 mM KH2PO4 pH 7.0, 100 mM ammonium acetate, 1 mM TCEP, 0.02% sodium azide, 500 mM NaCl, 50 mM imidazole), sonicated, pelleted, and the clarified supernatant was loaded onto a 5 ml HisTrap HP column (GE Healthcare Life Sciences, Pittsburgh, PA) and eluted with NiB (20 mM KH2PO4 pH 7.0, 100 mM ammonium acetate, 1 mM TCEP, 0.02% sodium azide, 500 mM NaCl, 500 mM imidazole). Peak fractions were diluted in NiA minus salt to 50 mM NaCl and loaded onto a 6 ml Resource S column (GE Healthcare Life Sciences) pre-equilibrated with MonoSA (20 mM KH2PO4 pH 7.0, 100 mM ammonium acetate, 1 mM TCEP, 0.02% sodium azide, 50 mM NaCl) and gradient eluted with MonoSB (20 mM KH2PO4 pH 7.0, 100 mM ammonium acetate, 1 mM TCEP, 0.02% sodium azide, 1 M NaCl). Peak fractions were concentrated to 5 ml and loaded onto a Superdex S200 column (GE Healthcare Life Sciences) and eluted with S200 buffer (50 mM HEPES pH 7.0, 400 mM NaCl, 100 mM ammonium acetate, 1 mM TCEP, 0.02% sodium azide). Full-length DmGen-His and DmGenDead-His were purified over HisTrap and S200 columns. Following elution from the S200 column, purity was assessed by dynamic light scattering and SDS-PAGE.
DmGen (1–518)-His was dialyzed into high salt buffer (25 mM HEPES pH 7.5, 100 mM sodium acetate, 10 mM magnesium acetate, 5% glycerol, 1 mM DTT) before injection onto a Superdex 200 column (GE Healthcare Life Sciences) connected to a DAWN HELEOS-II (Wyatt Technology Corporation, Santa Barbara, CA, USA) light scattering instrument and a Optilab T-rEX refractometer (Wyatt Technology Corporation). The Astra V software package (Wyatt Technology Corporation) was used to determine the molar mass of the sample.
Nuclease assays
Synthetic DNA substrates were prepared by annealing oligonucleotides, shown in Supplementary Table S1. Oligonucleotides (oligos) 888, 891, 892, 893, 894, 895, 897, 992 were described previously (25); 940, 994, 888+10, 990+10 were modified from these. These oligos were used to form the majority of substrates used in this study, termed ‘Bellendir substrates’. Oligos 1, 2, 3, 4 and 7 were described previously (26) and were used to form the ‘Rass substrates’ used in Figure 5. All substrates were prepared as previously described (27). Briefly, one oligonucleotide was 5΄ end-labeled using T4 polynucleotide kinase and γ-32P ATP. Substrates were annealed in annealing buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 50 mM NaCl, 5 mM DTT), PAGE-purified, and quantified by A260.
Figure 5.

DmGen and HsGEN1 cut 5΄ flaps faster than HJs in a variety of reaction conditions. (A) Schematic showing the substrates and reaction buffers tested. The substrates shown in red and dark blue are those used in our experiments (‘Bellendir oligos’), and those substrates in magenta and cyan are from the previous published study on HsGEN1 (‘Rass oligos’) (9). The sequences of central nucleotides near the branch points are shown in black. The buffer components of the Bellendir and Rass buffers are listed below the respective oligos. (B) A bar graph showing the first order rates of cleavage on either Bellendir substrates (5΄ flap-B and HJ-B) or Rass substrates (5΄ flap-R and HJ-R) in either the Bellendir Buffer or Rass Buffer by DmGen (at 25°C) or HsGEN1 (at 37°C). Colors for each substrate correspond to (A). Note that the final two bars are from experiments done in the same conditions of previously published data in (9). Each rate was determined from the average of ≥3 time courses (0 s–10 min or 30 min). Error bars are for the standard deviation of each mean rate. For rate values, see Supplementary Table S3.
For nuclease assays, tr-DmGen-His (1–518-His) or tr-HsGEN1-His (1–527) (gift of Dr. Steve West) was incubated with the 32P-labeled structures in a 10 μl reaction mixture containing 50 mM Tris pH 8, 100 μg/ml BSA, 1 mM DTT, 10% glycerol, 50 mM KCl, and 5 mM MgCl2 (Bellendir buffer—used in all kinetic experiments unless otherwise stated) or 60 mM sodium phosphate pH 7.4, 100 μg/ml BSA, 1 mM DTT, and 5 mM Mg(OAc)2 (Rass buffer) at either room temperature for reactions containing DmGen or 37°C for reactions containing HsGEN1. For fixed end-point assays, unless otherwise indicated, 20 nM protein was incubated with 1 nM substrate. For ligation of products, 1 U of T4 DNA Ligase (NEB) was incubated at 22°C for 30 min. The reaction was stopped by adding an equal volume of formamide loading dye (85% formamide, 50 mM ethylenediaminetetraacetic acid (EDTA), 1% bromophenol blue, 1% xylene cyanol), heated at 95°C for 5 min, and a fraction was loaded onto a polyacrylamide gel. After electrophoresis, gels were dried and imaged on a Typhoon Trio+ (GE Healthcare Life Sciences). Bands were quantified using ImageQuant (GE HealthCare Life Sciences).
For the kinetic analysis, the experiments were conducted by two methods: (i) simultaneous addition, in which the reaction was initiated by the simultaneous addition of DmGen or HsGEN1, Mg2+ and DNA, and (ii) prebinding analysis, in which, DmGen and either 5΄ flap or HJ0 were incubated together for a few minutes before the reactions were initialized with the addition of MgCl2. For time points, 1 μl aliquots were removed and quenched in 2.5 mg/ml Proteinase K, 2.5% SDS, and 125 mM EDTA. The amounts of protein and substrate used in the kinetics assays are given in the figure legend. To determine the percentage substrate cleaved, the amount of product was calculated as a fraction of the total radioactivity per lane. For the HJ0, only half the cleavage products (those in which the labeled strand is cut) are detected. Conversely, the HJ-Rass structure displayed propensity to adopt a specific orientation that leads to biased cleavage orientation (28). To account for this, rates were determined from native gels. For the 5΄ flap, a fraction of the substrate was unproductive or degraded. To account for this non-functional substrate, data were normalized to the expected amount of detectable product. The apparent pseudo first-order rate constant, kapp, for each concentration was determined by fitting the full reaction curves to a single-exponential function [(y = A*exp(kappt) + C] using KaleidaGraph software (Synergy Software, Reading, PA, USA). To examine the concentration dependence of the rates of cleavage of the flap an HJ0, kapp was plotted as a function of DmGen concentration, and these plots were fit to hyperbolic binding curves to determine the apparent binding affinity, K1/2, of DmGen to the flap or HJ0, using KaleidaGraph.
DNA-binding assays
DmGen (1–518)-His was incubated in a 10 μl reaction with 50 pM 32P-labeled DNA in binding buffer (10 mM HEPES pH 7.5, 100 μg/ml BSA, 1 mM DTT, 5% glycerol, 60 mM KCl) containing 5 mM EDTA. Incubation was at RT for 30 min. Reactions were immediately analyzed by 4% neutral PAGE at 4°C. After running, gels were either dried or exposed overnight at –80°C and imaged on a Typhoon Trio+ (GE Healthcare Life Sciences). Bands corresponding to unbound and bound DNA were quantified using ImageQuant (GE HealthCare Life Sciences). The K1/2 of binding and the Hill co-efficient for the binding of the HJ0 or the 5΄ flap was determined by fitting the data using KaleidaGraph software (Synergy Software, Reading, PA) and applying the following equation:
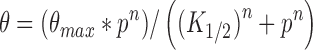 |
where θ is the fraction of total substrate that is bound, θmax is the maximum fraction of substrate bound, p is the protein concentration, and n is the Hill coefficient. K1/2 is the concentration at which half of the substrate is bound by protein and KD = (K1/2)n.
Expression of Gen in S. pombe and sensitivity analysis
Strains, RusA plasmids, and pREP41 plasmids are listed in Supplementary Table S2. Transformations were performed using the lithium acetate-based method described previously (29). For spot tests, strains containing plasmids were grown to saturation in EMM2−leucine dropout medium, washed twice with water, diluted to OD600 = 1, and 10-fold serially diluted to 10−4 cells/ml. Ten microliters aliquots from each dilution were spotted onto minimal medium plates containing MMS, CPT, HU or BLEO, then incubated at 32°C for 4 days before being photographed.
Immunofluorescence microscopy
Polyclonal antibodies were raised to a peptide spanning residues 236–335 of DmGen and affinity-purified by Genomic Antibody Technology (SDIX, Newark, DE, USA). All imaging was done with a laser-scanning confocal microscope (710, Carl Zeiss) and analyzed with ImageJ.
For embryo staining, 2–3 h old embryos were dechorionated, fixed in equal volumes 7% formaldehyde:heptane, devittelinized, and then stained. The primary antibody was rabbit anti-DmGen-N (1:1000), which was visualized with goat anti-rabbit IgG (H+L)-Alexa Fluor 488 (1:500, Life Technologies). DAPI (1:1000) staining was done for 2 min at room temperature.
For Drosophila S2 cells, DmGen cDNA was cloned into the pMT-V5-HisA vector (Life Technologies), which contains the CuSO4-inducible metallothionein promoter and a C-terminal His tag. The construct was stably transfected into S2 cells. Cells were plated at 1×106 cells/ml on poly-l-lysine-treated coverslips. DmGen-His expression was induced for 3 days before staining. Staining was performed as in (30). The primary antibodies were rabbit anti-DmGen-N (1:10 000) and mouse anti-His (1:500). The primary antibodies were visualized with goat anti-rabbit IgG (H+L)-Alexa Fluor 488 (1:10,000) and goat anti-mouse IgG (H+L)-Alexa Fluor 555 (1:10 000, Life Technologies). DNA was detected by staining with DAPI (1:5000, Thermo Fisher Scientific, Carlsbad, CA, USA) for 1 min at room temperature.
Atomic force microscopy
50 μM DmGen (1–518)-His was diluted to 2 μM in storage buffer (50 mM HEPES pH 7.0, 400 mM NaCl, 100 mM ammonium acetate, 1 mM TCEP, 10% glycerol) and then to 20 and 37 nM in high salt buffer (25 mM HEPES pH 7.5, 100 mM sodium acetate, 10 mM magnesium acetate, 5% glycerol, 1 mM DTT) and 20 μl was immediately deposited onto freshly-cleaved mica. The mica surface was then immediately washed with water, and a stream of nitrogen gas was used to dry the surface. Images were acquired with a Nanoscope IIIA atomic force microscope (Veeco, Santa Barbara, CA, USA) in tapping mode with a resolution of 512 × 512 pixels at a scan rate of 1.97 Hz and over a 1 × 1 μm scan size. AFM tips were from NanoSensors (Neuchatel, Switzerland) with a spring constant between 21 and 98 N/m and resonance frequencies between 146 and 236 kHz. AFM images for the samples were consistent over two depositions and multiple tips (at least two for each deposition). Poor images resulting from blunted tips were excluded from analysis. At least seven representative images of each sample were second order plane-fitted and flattened, and three-dimensional images were generated using NanoScope Analysis version 1.53r1 (Bruker Corporation, Billerica, MA, USA). Volume analysis of protein peaks was conducted with Image SXM 195-1 (Steve Barrett, University of Liverpool, UK) as described in (31). Volumes corresponding to protein aggregates were excluded from analysis. Volume plots were generated using KaleidaGraph 4.1.3 (Synergy Software). Protein molecular mass was converted into predicted AFM volume using the following equation:
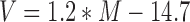 |
where V is AFM volume in nm3, and M is molecular mass in kDa (32).
Sequence alignments
Sequence alignments were performed using ClustalX 2.1 (33) and edited in GeneDoc version 2.7.000 (34).
RESULTS
DmGen mutants are more sensitive to DNA damage than mus81 mutants
In yeast and humans, Yen1 and GEN1 seem to act in DNA damage repair secondarily to Mus81 (17,19,20,35). This relationship seems to be switched in Drosophila as previous studies show that flies mutant in Gen and Blm, which encodes a helicase that can participate in dissolution of double-HJs, die earlier in development than Mus81 Blm double mutants (22). To more thoroughly assess the relationship between DmGen and Mus81, we examined the sensitivity of single and double mutants to a variety of DNA damaging agents (Figure 1). To investigate effects on replication-associated damage, we used (a) camptothecin (CPT), a topoisomerase I poison that results in replication-associated DSBs (36); (b) methyl methanesulfonate (MMS), an alkylating agent that induces lesions that can block replication forks (37) and (c) hydroxyurea (HU), which inhibits ribonucleotide reductase, leading to decreased dNTP pools and fork slowing and stalling (38). Wild-type flies are not sensitive to these agents at the doses tested (Figure 1). Gen mutants show significant sensitivity to each agent, indicating an important role in responding to replication-associated damage (Figure 1A–C). Conversely, mus81 mutants do not show sensitivity to CPT or MMS; however, mus81 Gen double mutants have more severe sensitivity than Gen single mutants, indicating a secondary role for Mus81 in repairing damage during replication (Figure 1A and B). Interestingly, mus81 mutants are healthier than wild-type flies following HU treatment (Figure 1C) (21), and the mus81 Gen double mutant does not show increased sensitivity compared to the Gen single mutant. These data indicate that in flies, DmGen facilitates repair of HU-induced stalled or slowed replication forks, whereas Mus81 may exacerbate problems caused by HU.
Figure 1.

Drosophila Gen mutants are more sensitive to DNA damaging agents than mus81 mutants. Graphs show survival of mutants relative to control siblings (see Materials and Methods). (A) 0.025 mM camptothecin (CPT); (B) 0.04% methyl-methane sulfonate (MMS); (C) 70 mM hydroxyurea (HU); (D) 0.004% nitrogen mustard (HN2); (E) 2000 rads ionizing radiation (IR). Each point corresponds to one vial; means and 95% confidence intervals are shown. Dotted lines indicate 100% relative survival (note that Y axes differ between treatments). Paired t-tests between mutant and control individuals were done to evaluate sensitivity of mutants to each treatment; statistical significance of sensitivity is indicated below each genotype. Differences between genotypes were assessed by one-way ANOVA and are indicated above each graph. n.s. = not significant (P > 0.05); **P < 0.01; ***P < 0.001; ****P < 0.0001.
We treated larvae with mechlorethamine (HN2) and ionizing radiation (IR) to investigate sensitivity to interstand crosslinks and DSBs, respectively. Gen mutants were significantly more sensitive to both agents, with mus81 mutants showing sensitivity only to HN2 (Figure 1D and E); however mus81 Gen double mutants are significantly more sensitive to IR (Gen single mutants were already completely inviable at the tested dose of HN2), suggesting that Mus81 may play a backup role to DmGen in DSB repair (Figure 1D and E).
In summary, Gen mutants are more sensitive than mus81 mutants to all of the DNA damaging agents tested (Figure 1). The increased sensitivity of mus81 Gen double mutants indicates that in flies, Mus81 plays a secondary role to DmGen. These findings contrast with data from yeast and human cells that show that Yen1/GEN1 is secondary to MUS81 (17,19,20,35).
DmGen rescues the DNA-damage sensitivity of S. pombe mus81 mutants
The sensitivity of DmGen mutants to DNA damaging agents could indicate that DmGen functions to cut recombination intermediates. To test this possibility, we took advantage of fission yeast Schizosaccharomyces pombe, which has a mus81 ortholog but no Yen1/GEN1 ortholog (13,14). Truncated HsGEN1 (residues 1–527) expressed in S. pombe mus81 mutants rescues sensitivity to DNA damaging agents (39). Similarly, we find that expression of a truncated form of DmGen (residues 1–518, similar to truncated HsGEN1 (39)) rescues sensitivity of mus81Δ mutants to MMS, CPT, HU and the radiomimetic drug bleomycin (BLEO) (Supplementary Figure S1A). This rescue is dependent on DmGen nuclease activity, as expression of nuclease-dead DmGen has a dominant-negative effect, resulting in less growth than seen in the negative control (Supplementary Figure S1A). This effect, which is also seen with nuclease-dead HsGEN1 or budding yeast Yen1 (17,39), strongly suggests that the catalytically inactive enzymes bind repair intermediates and block alternative repair pathways. We conclude that DmGen (1–518) is functional in vivo in the repair of DNA damage in mus81Δ mutants, suggesting that despite their different genetic phenotypes, HsGEN1 and DmGen share one or more critical activities that can compensate for loss of Mus81 activity.
DmGen localizes to the cytoplasm of early embryos and S2 cells
The reversed roles of DmGen and Mus81 in Drosophila relative to humans and yeast could indicate differences in access to the damaged DNA as a result of different cellular localizations. The activity of human GEN1 and yeast Yen1 is limited to cells undergoing mitosis by protein localization and/or activation. Specifically, HsGEN1 is sequestered in the cytoplasm until nuclear membrane breakdown, and yeast Yen1's activity and access to the nucleus are controlled by dephosphorylation (40–42). A previous study using a polyclonal antibody to DmGen suggested that it localizes to the nucleus of 0–3 h old embryos (24). To further assess DmGen localization, we generated a polyclonal antibody to a different epitope (residues 236–335). Immunofluorescence using this antibody reveals that DmGen localizes largely or exclusively to the cytoplasm in wild-type embryos (Supplementary Figure S2A). Because this result contrasts with the previous study, we further confirmed cytoplasmic localization in cultured cells by overexpression of DmGen carrying a C-terminal hexahistadine (His) tag under control of an inducible promoter (Supplementary Figure S2B). A small fraction of uninduced cells show leaky expression of His-tagged DmGen, as evidenced from the strong staining by the His antibody; most of the uninduced cells showed only background staining with the His antibody but show significant staining throughout the cytoplasm with our polycolonal antibody to DmGen, suggesting that this cytoplasmic protein is endogenous DmGen (Supplementary Figure S2B, top panels). After induction, both anti-DmGen and anti-His antibodies detect high levels of a cytoplasmic protein, with no detectable signal in interphase nuclei (Supplementary Figure S2B, bottom panels). These results strongly suggest that our antibody binds to DmGen in cells and that DmGen, like Yen1 and HsGEN1, is primarily or exclusively cytoplasmic during interphase. While we cannot exclude the possibility that a low level of nuclear DmGen escaped our detection, our results imply that the genetic differences between DmGen and its orthologs are not simply due to differences in gross protein localization.
DmGen cuts 5΄ flaps, replication forks, splayed arms and Holliday junctions
The in vivo data strongly suggest that DmGen, like its orthologs, is a structure-specific endonuclease and might have Holliday junction resolvase activity. In previous in vitro experiments, N-terminal 6xHis-tagged DmGen exhibited weak activity on 5΄ flaps, double flaps, and replication forks, but no activity on HJs (24). These results contrast with the strong activity seen with recombinant HsGEN1 and Yen1 which were tagged on the C-terminus, and the recent crystal structures of GEN1 from the thermophilic yeast C. thermophilum (CtGEN1) and humans (HsGEN1) implicate the N-terminal region in cleavage (8,9,43–46). We considered the possibility that a tag near the N-terminal nuclease domain might impact cleavage activity of DmGen and sought to compare N-terminally and C-terminally tagged proteins.
We expressed and purified both N- and C-terminal tagged DmGen in full-length and truncated (1–518) forms (Supplementary Figure S3A and B). To examine substrate specificity, we incubated DmGen with radiolabeled DNA substrates. The nuclease activity of the N-terminal-tagged proteins on the 5΄ flap is weak (∼3% cut), but the C-terminal-tagged versions of DmGen show high activity (92% cut), with no evidence of contaminating nuclease activity (Supplementary Figure S3C and S4C). Both full-length and truncated C-terminal His-tagged DmGen exhibit robust cleavage of 5΄ flaps, replication fork-like structures (RFs), splayed arms (SAs), and fixed, mobile, and nicked HJs (Figure 2A; Supplementary Figure S4). We did not detect cleavage of unbranched dsDNA, nicked duplex DNA, or 3΄ flaps (Figure 2A; Supplementary Figure S4). Yen1 and HsGEN1 have not been shown to cleave SAs (8,40). To determine whether DmGen's cleavage of the SA is a novel substrate specificity, we tested whether our substrates were cleaved differently by DmGen than by HsGEN1 (Supplementary Figure S5). We see that like DmGen, HsGEN1 cleaves SAs, and cuts our 5΄ flap and HJ substrates at the same sites (Supplementary Figure S5).
Figure 2.

DmGen is a structure-specific endonuclease and a resolvase. (A) Substrates radiolabelled at the 5΄ end of one strand (asterisks) were incubated with full-length (FL) DmGen or truncated (518) DmGen. Arrows indicate sites of cleavage determined by denaturing PAGE, as shown below. The bracket indicates the expected size range of the cleavage products for the HJ12 substrate. (B) Schematic illustrating the cleavage and ligation experiment of the HJ0+10nt substrate formed by annealing one labeled strand of ∼50nt, one unlabeled strand of ∼50nt, and two unlabeled strands of ∼60nt. The addition of ∼10nt on one arm are indicated in green and the asterisk indicates the location of the radiolabel. Cleavage with DmGen creates nicked duplex DNA that can be ligated to create duplex DNA. When visualized on a denaturing PAGE, the presence of a newly ligated longer strand can be observed. (C) Denaturing PAGE of HJ0+10nt cleavage and ligation experiment depicted in (B). (0) indicates substrate only and (+) indicate the addition of DmGen and/ or T4 ligase. Cartoons on the right of gel indicate the various products formed. Analysis of the cleavage and ligation products of the HJ0+10nt substrate reveals ∼55% detectable product after cleavage. Incubation of cleavage products with T4 ligase shows a new 60nt band (30% of total) and a reduction of the cleaved product (25% of total).
Analysis of the cleavage products by denaturing polyacrylamide gel electrophoresis (PAGE) reveals that the predominant cut sites are approximately at or one nucleotide (nt) 3΄ of the junction branch point on the 5΄ flap, RF and SA (Figure 2A; Supplementary Figures S4 and S5). On the mobile HJ12 substrate, which contains a 12 bp homologous core within which the junction can migrate, we observe multiple cut sites, all within the 12 bp core (Figure 2A; Supplementary Figure S4). To map HJ cleavage sites on all four strands of the HJ0, we alternately labeled each strand of the immobile HJ0 structure (Supplementary Figure S6). The major cut sites appear to be at the junction and/or one to two nt 3΄ to the junction branch point (Supplementary Figure S6). To further analyze the cleavage of the HJ0, we created a derivative of the HJ0 termed HJ0+10nt by annealing one labeled strand of ∼50 nt, one unlabeled strand of ∼50 nt, and two unlabeled strands of ∼60 nt (Figure 2B and C). This junction allows us to determine if DmGen cuts symmetrically about the axis to produce nicked duplex products that can be ligated. If the HJ cleavage products can be ligated, a new band corresponding to a ∼60 nt DNA will appear on the gel in the presence of ligase but not its absence (Figure 2B). Analysis of the cleavage and ligation products of the HJ0+10nt substrate reveals ∼55% detectable product upon cleavage alone. Incubation of cleavage products with T4 ligase shows a new 60 nt band (30% of total) and a reduction of the cleaved product (25% of total) (Figure 2C). These data show that DmGen, like HsGEN1, exhibits canonical resolvase activity in addition to robust endonuclease activity on 5΄ flaps, RFs and SAs.
Because full-length and truncated C-terminally-tagged DmGen have similar substrate specificities and activities, and the truncated protein shows activity in S. pombe (Figure 2; Supplementary Figures S1–S4), we used the more stable truncated protein in subsequent in vitro experiments; for simplicity, we refer to this truncated protein as DmGen.
DmGen can dimerize on DNA substrates
HsGEN1 and CtGEN1 are monomers in solution and dimerize on HJs (8,9,43). We analyzed DmGen by size-exclusion chromatography with multi-angle light scattering (SEC-MALS) after affinity and ion-exchange chromatography steps and compared it to a BSA standard (Supplementary Figure S7). BSA eluted from the column in three distinct peaks, with the monomer (66 kDa) eluting between 14 and 15 min. The majority of DmGen elutes as a monomer, exhibiting an average molecular weight of ∼58 kDa (predicted 60 kDa), however there is a small amount of dimer present. (Supplementary Figure S7B). To further explore possible dimerization of DmGen, we used atomic force microscopy (AFM), which allows us to directly observe the oligomerization state of the protein (Figure 3A). Previous studies showed that there is a linear relationship between the molecular mass of a protein and its observed volume in AFM images, allowing oligomerization state and association constants of protein–protein complexes to be determined (31,32). Plots of the distribution of volumes of DmGen deposited at 20 and 37 nM DmGen show a major peak consistent with the volume expected for a DmGen monomer and a smaller peak consistent with the volume expected for a DmGen dimer (Figure 3A and Supplemental Figure S8). Due to the low population of dimer and crowding of the protein on the surface, accurate determination of the protein dimerization constant is not possible; however, based on the population of protein contained within the monomer and dimer peaks, we estimate the protein dissociation constant to be in the hundred nanomolar range (Supplementary Figure S7).
Figure 3.

DmGen can dimerize. (A) Topographical AFM images of truncated DmGen showing monomers and dimers. 20 nM truncated DmGen was deposited onto naked mica and imaged with tapping mode AFM in air. The gradient bar represents 0–1.2 nm height above the mica surface. Yellow arrows denote DmGen dimers. 1 μm x 1 μm images and volume analyses can be found in Supplementary Figure S8. (B) EMSA analysis of DmGen with HJ0. Truncated DmGen (0.1, 0.5, 1, 5, 10, 50, 100, 250, 500, 1000, 10 000 pM) was incubated at room temperature with 50 pM radiolabeled DNA in the presence of EDTA and bound products were separated using 4% native PAGE at 4°C. The HJ0 cartoon indicates the position of the DNA alone, whereas the positions of the HJ0 bound by monomer and dimer are indicated by a solid arrow and a dashed arrow, respectively. (C) The same as in (B), except with the 5΄ flap. Plots graphing the fraction of DNA bound as a function of DmGen concentration for three to four independent EMSAs can be found in Supplementary Figure S9.
To determine the affinity and stoichiometry of DmGen binding to DNA, we used electrophoretic mobility shift assays (EMSAs) (Figure 3B and C). We incubated increasing concentrations of DmGen (0.1 pM–10 nM) with 50 pM of either 5΄ flap or HJ0 in the presence of EDTA to chelate the metal ions to prevent cleavage of the substrate. On the HJ0, two slower-migrating bands appear with increasing DmGen concentration, with the first shifted species occurring at concentrations of DmGen between 100 and 500 pM, followed by complete conversion to a supershifted species by 10 nM DmGen (Figure 3B), These results are consistent with DmGen binding to HJ0 with high affinity, similar to those seen with CtGEN1, which binds to HJs with a high affinity (∼10 nM). We determined the DmGen concentration at which half of the substrate is bound (K1/2,EMSA,HJ) by plotting the fraction of DNA bound (both shifted bands) as a function of DmGen concentration for four independent EMSAs and fitting them to the Hill equation (Supplementary Figure S9A). This analysis yields a K1/2,EMSA,HJ for binding of DmGen to the HJ0 equal to 0.19 ± 0.24 nM. In addition, at 10 nM DmGen, all of the DNA is in the supershifted band, consistent with 100% of the HJ0 being bound by a dimer or two monomers of DmGen (Figure 3B).
Interestingly, DmGen also forms two shifted species on a 5΄ flap substrate (Figure 3C), with the first shifted species occurring at concentrations of DmGen between 100 and 500 pM, followed by complete conversion to a supershifted species by 1 nM DmGen (Figure 3C). The K1/2,EMSA,5΄FLAP for the 5΄ flap substrate determined from three independent experiments is 0.18 ±.08 nM. By 1 nM DmGen, all of the DNA is in the supershifted band (Figure 4B and Supplementary Figure S9B). The supershifted (dimer) band is unexpected because for HsGEN1 a monomer is sufficient for 5΄ flap cleavage (9). No band shifts were observed when the same experiment was performed with linear dsDNA (Supplementary Figure S9C), indicating that the supershifted bands do not result from nonspecific binding of DmGen to the DNA.
Figure 4.

DmGen cleaves 5΄ flaps faster than HJs. Graphs show time courses of nuclease progression under conditions of excess (A) 5΄ flap or (B) HJ0. For each time course, aliquots were taken at various time points (note that the time scales differ in each panel). The intensity of each cleavage product was quantified by ImageQuant, and the data were normalized to the expected amount of detectable product (see Experimental Procedures). Each point represents the mean of three experiments, except in (A), which is the mean of two experiments. Error bars indicate standard error of the mean. The curves drawn though the data are best fits to single exponentials (C) The rate of cleavage as a function of DmGen concentration under conditions of excess DmGen. The data from each individual replicate in (A and B and Supplementary Figure S10) were fit to a single exponential curve given by the equation y = m1*x /(m2 + x), where m1 = maximum rate (kapp,max) at saturating protein concentrations and m2 = the apparent dissociation constant (K1/2). Note that the first point (3 nM protein) was performed with 5 nM 5΄ flap or HJ0, whereas the rest of the experiments (20, 60, 100 and 200 nM protein) were performed with 2 nM DNA. From these fits K1/2 and kapp,max are 62 ± 3 nM and 46 ± 9 s−1 for the flap and 660 ± 500 nM and 31 ± 19 s−1 for the 5΄ flap and HJ0, respectively.
DmGen cleaves 5΄ flaps faster than Holliday junctions
We investigated the kinetics of flap and HJ0 cleavage by DmGen as a function of both substrate and enzyme concentration in multiple-turnover assays (i.e. excess substrate relative to protein) (Figure 4A and B). 5 nM 5΄ flap is completely cleaved within 1–4 min using DmGen concentrations ranging from 0.5 to 3 nM (10- to 1.7-fold excess DNA), with the rate of cleavage increasing with increasing concentration. In contrast, the rates of HJ0 cleavage are ∼10-fold slower, with reactions taking 10–30 min to plateau (Figure 4B). The reaction containing 3 nM DmGen goes to completion, but reactions with the lower concentrations (0.5–1 nM) plateau at 20–60% of the substrate being cleaved, respectively (Figure 4B). Enzyme death, substrate inhibition and product inhibition can result in <100% of the substrate being cleaved. The observation that DmGen can completely cleave the 5΄ flap at the same concentrations (Figure 4A) and that the plateau levels decrease as the HJ0:DmGen ratio increases (i.e. increasing HJ0 concentrations exhibit decreasing product formation; Figure 4B) suggest that the excess HJ0 may be inhibiting the reaction. This suggestion is supported by our observation that the monomer of DmGen binds more tightly than the dimer. Consequently, as the HJ0 concentration increases above the concentration of DmGen, the excess HJ0 acts as a trap, binding monomers of DmGen and removing them from solution, thereby reducing the concentration of active substrate-dimer complexes. HsGEN1 shows similar inhibition of cleavage when the HJ substrate is in excess (9,43). We do not observe such substrate inhibition with the flap, likely because flap cleavage requires only a monomer of DmGen.
Classic steady-state enzyme kinetics (multiple turn-over assays) allow for the determination of the kcat and Km of an enzyme by plotting the rate of reactions as a function of substrate concentration. Because substrate inhibition occurs if the concentration of DmGen is below the concentration of HJ0 substrate, such assays could not be performed. To circumvent the problem of substrate inhibition, we measured rates of cleavage of the flap and HJ0 as a function of DmGen concentration, maintaining DmGen in excess over substrate (Figure 4C and Supplementary Figure S10). The rates measured in these reactions are the rate at which DmGen cleaves a single substrate molecule and therefore represent steps at or before cleavage. To determine the dependence of the cleavage rate on DmGen concentration, we first determined the pseudo-first-order rate constants, kapp, by fitting the rate curves for each DmGen concentration to single exponentials and then plotted kapp as a function of DmGen concentration. The rate of cleavage (or kapp) of both the flap and HJ0 as a function of DmGen concentration are fit well by simple hyperbolic binding curves, allowing us to determine the K1/2,KINETIC (DmGen concentration necessary to achieve half maximal cleavage rate or simply ‘apparent affinity’) and the apparent maximal cleavage rate (Figure 4C). The K1/2,KINETIC and apparent maximal cleavage rates are 62 ±3 nM and 46 ± 9 s−1 for the flap and 660 ± 500 nM and 31 ±19 s−1 for the HJ0, respectively. In all concentrations of DmGen tested, the rate of 5΄ flap cleavage is least ∼7-fold greater than the rate of HJ0 cleavage (Figure 4C). These results are in striking contrast to the published data on HsGEN1. Published studies on HsGEN1 using excess protein found that HsGEN1cleaves a HJ more rapidly than a 5΄ flap, suggesting that HJs are preferred substrates over 5΄ flaps for HsGEN1 (9). In contrast, our data suggest that 5΄ flaps may be the preferred substrates for DmGen.
HsGEN1 cleaves 5΄ flaps faster than HJs
We next determined whether the difference in substrate specificity of DmGen and HsGEN1 may reveal a bona fide difference in enzyme activity between the orthologs or may result from dissimilar assay conditions (buffer and DNA sequence of substrates). The published study on HsGEN1 (9) employed a low-salt phosphate buffer (referred to here as Rass buffer), whereas we used a medium-salt Tris buffer (Bellendir buffer); in addition, the sequences of the substrates were different in the two studies (Figure 5A). Consequently, we compared HsGEN1 with DmGen side-by-side. The human and Drosophila proteins cleave each substrate (5΄ flap and HJ created from Bellendir or Rass oligos) at the same site (Supplementary Figure S5). DmGen and HsGEN1 have very similar rates of cleavage on both HJ substrates in both buffers (Figure 5B, dark blue and light blue bars); however, rates of 5΄ flap cleavage vary with sequence and with buffer. In the Bellendir buffer, the two proteins have similar cleavage rates on both 5΄ flap substrates, and 5΄ flap cleavage is significantly faster than HJ cleavage (Figure 5B). In the Rass buffer, cleavage rates differ for the two sequences, with a 34-fold and a 6-fold difference between the highest and lowest rates of flap cleavage for HsGEN1 and DmGen, respectively (Figure 5B and Supplementary Table S3). In summary, DmGen cleaves 5΄ flaps faster than HJs in all conditions tested, and HsGEN1 cleaves 5΄ flaps faster in all but one of the conditions we tested (Figure 5B and Supplementary Table S3). Thus, varying the buffer conditions and oligo sequences used in kinetic assays reveals additional properties of HsGEN1 beyond those described previously. These results suggest that simpler branched substrates, such as flaps, are preferred by both DmGen and HsGEN1 and that both enzymes utilize similar mechanisms of recognition and cleavage.
The rate-limiting step of HJ0 cleavage is assembly of a productive dimer complex on the substrate
Examination of the DmGen kinetic data together with the EMSAs can shed light onto possible mechanisms of 5΄ flap and HJ cleavage. Given that the EMSAs show very tight binding of a DmGen monomer (K1/2,EMSA ∼ 0.2 nM) to both the flap and the HJ0 (Figure 3B–C and Supplementary Figure S9), it is likely that the concentration dependence of the cleavage rate that we observe (Figure 4C) results from a second monomer of DmGen binding to the substrate prior to cleavage. From our EMSAs, we estimate that the concentration for a second monomer binding to either the HJ0 or the 5΄ flap is 0.5–10 nM. The K1/2,KINETIC,5’FLAP for the flap from our kinetic data is slightly larger than would be predicted for dimerization on the 5΄ flap from the EMSAs, but this difference may be due to the different temperatures at which the two experiments were performed (EMSAs at 4°C and kinetic assays at 25°C). The K1/2,KINETIC,HJ for the HJ0 (660 nM), however, is significantly larger than apparent binding affinities for the dimer seen in the EMSAs, in which we observe 100% dimer at 10 nM DmGen. These differences indicate that the K1/2,KINETIC,HJ value for the HJ0, determined from kinetic measurements represents a step other than the binding seen in the EMSAs.
If the rate-limiting step to HJ0 cleavage is a conformational change after binding, the weak K1/2,KINETIC,HJ (660 nM) we observe for cleavage of the HJ0 could represent nonspecific of binding of the second monomer of DmGen prior to a conformational change that leads to the stable DmGen2:HJ0 complexes seen in the EMSAs. To test this possibility, instead of adding DNA, DmGen, and Mg2+ simultaneously as done above (Figure 4A–C), we pre-incubated DmGen with HJ0 in the absence of Mg2+, allowing time for the dimer to assemble on the HJ0, then initiated cleavage by the addition of Mg2+ (Figure 6). If the rate-limiting step is conformational change after the second monomer binds the HJ0 (i.e. productive assembly of the dimer on HJ0) that results in a stable DmGen2:HJ0 complex that is slow to dissociate, a burst of rapid cleavage with an amplitude equal to the concentration of pre-formed DmGen2:HJ0 complexes will be observed in the pre-incubation experiment (47). Using 3 nM DmGen with of 5 or 10 nM HJ0, we observe a burst of cleavage before the first time point (5 s), followed by a slow rate of cleavage similar to that seen in the simultaneous addition experiment (Figure 6A and B). This observation strongly suggests that given sufficient time, DmGen can cooperatively assemble into a productive complex on the HJ0. We used a DmGen concentration of 3 nM, corresponding to 1.5 nM dimer with 5 or 10 nM HJ0. If all the DmGen molecules were active and pre-bound as dimers to HJ0 and poised to undergo cleavage, we would expect burst heights of 30% for 5 nM HJ0 and 15% for 10 nM HJ0. We observed burst heights of 28% and 10%, respectively (Figure 6A), suggesting that the majority of DmGen is active and bound in a productive dimer complex prior to the addition of Mg2+. These results are consistent with our equilibrium binding EMSAs, which suggest that the binding affinity of a dimer of DmGen for the HJ0 is between ∼3 and 10 nM (Figure 3B). Taken together, our results strongly suggest that the rate-limiting step to HJ0 cleavage is a conformational change after binding of the second monomer of DmGen to the HJ. We conducted similar pre-incubation experiments with the flap (Figure 6C). In contrast to the results of pre-binding with the HJ0, no initial burst of cleavage is discernable, suggesting that the interaction of DmGen with the flap is dynamic and that binding of a monomer or dimer to the flap does not result in a complex that is slow to dissociate (Figure 6C and 6D).
Figure 6.

Production of an active complex is the rate-limiting step in HJ0 cleavage. (A) To determine whether the rate-limiting step of the HJ0 reaction is a slow conformational change that results in a stable Gen2:HJ0 complex that can rapidly cleave the DNA, 3 nM DmGen was pre-incubated with 5 or 10 nM HJ0 for several minutes before starting the time course experiment with MgCl2. Quantification and analyses were done as in (Figure 4A–C). (B) Data from the corresponding simultaneous addition experiments in Figure 4B (3 nM DmGen with 5nM HJ0, pink) is replotted on a 1-minute time scale for comparison to (A). (C) The same as in (A), except with the 5΄ flap. (D) Data from the corresponding simultaneous addition experiments in Figure 4A (3nM DmGen with 5nM 5΄ flap, pink) was replotted on 1-minute time scale for comparison to (C). Note that the data in this figure are not normalized, as in Figure 4.
DISCUSSION
In this work, we show that DmGen functions as a key structure-selective endonuclease (SSE) during repair of DNA damage. Our genetic data reveals a fundamental difference between DmGen and the well-characterized human and fungal orthologs. In Drosophila, Gen single mutants have extreme hypersensitivity to DNA damaging agents but mus81 single mutants do not (Figure 1); as in other organisms, mus81 Gen double mutants have even more severe hypersensitivities. These data are in stark contrast to genetic results seen in other species, where Mus81 is the predominant SSE and Gen orthologs play a secondary role in repair (17,19,20,35). Interphase protein localization does not appear to account for the reversed dominance observed in Drosophila, as DmGen, like Yen1 and HsGEN1, seems to be primarily or exclusively cytoplasmic (Supplementary Figure S2) (40,48,49); however, it is possible that the order of activation during mitosis is switched in Drosophila relative to yeast and human cells.
DmGen, like HsGEN1, cleaves a variety of branched structures including 5΄ flaps, replication forks, splayed arms and HJs (Figure 2 and Supplementary Figure S5). The mechanism of HJ cleavage appears to be similar between DmGen and HsGEN1, as the two proteins exhibit similar rates of cleavage across various conditions (Figures 5–7). Both DmGen and HsGEN1 are robust 5΄ flap endonucleases. HsGEN1's cleavage rate on flaps is more sensitive to reaction conditions than that of DmGen, though even this difference might be due to different physiological temperatures (Figure 5). This combination of genetic differences and biochemical similarities observed between DmGen and HsGEN1 highlights the unique platform that Drosophila provides to the DNA repair/structure selective endonuclease fields to further our understanding of the functions of these enzymes in vivo. Below, we discuss insights into the mechanism of cleavage of HJs and flaps by DmGen orthologs, structural considerations that may underlie novelties across DmGen orthologs, and the importance of HJs versus other substrates.
Figure 7.

Mechanism of DmGen cleavage of HJs and 5΄ flaps. (A) Mechanism of Gen function on HJs. DmGen monomer binds the HJ with a KD of ∼0.2 nM (K1/2,EMSA,HJ from EMSAs), followed by weak, nonspecific binding of a second monomer with a KD of ∼660 nM (K1/2,KINETIC,HJ from Kinetics). Formation of a productive dimer complex, exhibiting the correct DNA conformation required to position opposite DNA strands in the DmGen active sites, is slow. Once a productive dimer–HJ0 complex is formed, the dimer cooperatively nicks across the junction. (bottom left box) It is unlikely that a pre-formed dimer will encounter a HJ exhibiting the proper conformation required for cleavage. If the dimer–DNA complex is not productive, one monomer likely dissociates from the HJ, allowing other DmGen proteins access to the junction. (top right box) It is also possible that a DNA conformational change occurs prior to the second monomer binding; however, given our observation that production of a productive dimer–DNA complex is the rate-limiting step, this is unlikely to represent a main pathway. See discussion for additional details (B) Mechanism of DmGen function on 5΄ flaps. (top) DmGen monomer binds the 5΄ flap, triggering a DNA conformational change and rapid cleavage of the flap strand one nt 3΄ of the junction branch point. (bottom) A Drosophila-specific pathway is depicted in the box. (i) A pre-formed dimer can bind the 5΄ flap, (ii) Alternatively, two monomers can sequentially bind the 5΄ flap, with the first monomer having a very high affinity (∼0.2 nM from EMSAs) and the second monomer having a weaker affinity of ∼ 60 nM (based on the K1/2,KINETIC, 5’ flap determined from kinetic data). The additional DNA contacts provided by the second monomer may constrain the 5΄ flap conformation to facilitate cleavage.
The mechanism of 5΄ flap and HJ cleavage by DmGen
Comparison of our biochemical data with DmGen to those with the yeast and human orthologs suggests that they follow similar mechanisms of cleavage of 5΄ flaps and HJs. Taking both our kinetic and EMSA data into account allows us to elucidate important features of the mechanism of HJ and 5΄ flap cleavage. HJ cleavage requires the assembly of a dimer of DmGen (or orthologs) on the HJ prior to cutting (9,43,45,46). Our prebinding experiments (Figure 6A), which reveal a burst of cleavage followed by a slow turnover, indicate that after the second monomer binds the complex undergoes a conformational change that stabilizes the DmGen2:HJ0 complex and leads to rapid cleavage. These data support previous models for the activity of orthologs on HJs, which suggest that a rate-limiting conformational change occurs after binding of a second monomer (9,43,45,46). Our EMSAs show tight binding of both the monomer and dimer of DmGen to the HJ (Figure 4A and Supplementary Figure S9). Our observation of HJ0 substrate inhibition further supports tight monomer binding, and the near-stoichiometric burst amplitudes in prebinding experiments support tight dimer binding; however, in experiments in which DmGen, DNA, and Mg2+ were mixed simultaneously, we observe a very weak concentration dependence of DmGen for HJ cleavage, with a K1/2,KINETIC consistent with nonspecific DNA binding (Figures 4B and 6A). Accordingly, we propose that the first monomer binds tightly to the HJ (K1/2,EMSA ∼ 0.2 nM), followed by a second monomer binding with a weak affinity (K1/2,KINETIC ∼ 700 nM) to form a nonspecific dimer on the HJ (Figure 7A). Subsequently, this complex undergoes a rate-limiting conformational change that positions opposing DNA strands in the DmGen active sites to form a stable productive dimer-HJ0 complex that is slow to convert back to the nonspecifically bound form. After formation of a productive dimer–HJ complex, the dimer rapidly nicks the two opposing strands of the junction in a cooperative and symmetric manner, yielding two nicked duplexes. It is also possible that a DmGen dimer can form in solution prior to binding the HJ (Figure 7A, bottom left box); however, our results indicate that the HJ must be in the proper conformation for the second DmGen to bind tightly. The importance of the HJ conformation is also supported by the crystal structure of CtGEN1 bound to an HJ after cleavage. In the presence of cation, unbound HJs are found in the stacked X conformation (50). In the HJ model based on the crystal structure of CtGEN1 in the product complex, the HJ is bent into a non-planar conformation (46). Given the affinity of the monomer for DNA, binding of preformed dimers to the HJ may promote dissociation of one monomer. It is also possible that a DNA conformational change occurs prior to a second DmGen monomer binding the HJ (Figure 7A, top right box).
Consistent with data on HsGEN1 (9,43), our EMSA data taken together with our kinetic data on the 5΄ flap under excess substrate conditions suggest that a monomer of DmGen can cleave a 5΄ flap. This result is not unexpected, as flap cleavage requires only a single nick. Our kinetic data using excess protein further suggest that a second monomer of DmGen can bind to the flap (with an apparent affinity/K1/2,KINETIC,5’FLAP of 60 nM) and increase the catalytic activity (Figure 4). This latter observation suggests that the rate-limiting step for flap cleavage is a conformational change that places the 5΄ flap in the proper orientation for cleavage and that the dimer facilitates this change. Based on these observations we propose the mechanism shown in Figure 7B. A DmGen monomer binds the 5΄ flap with high affinity (K1/2,EMSA ∼0.2 nM, based on EMSAs), followed by a conformational change that positions the DNA in the active site and the subsequent rapid cleavage of the 5΄ flap one nucleotide 3΄ of the branch point (Figure 7B, top). At low DmGen concentrations or in excess substrate, this path dominates; however as the concentration of DmGen increases, a second monomer or a preformed dimer of DmGen can bind the 5΄ flap and increase the rate of cleavage ∼ 5–10 fold. This proposed mechanism is supported by our observation that DmGen can form dimers in solution and that the rate of flap cleavage increases with DmGen concentration, with a K1/2,KINETIC = 60 nM (Figures 3, 4, 5 and Supplementary Figures S7-10). We suggest that this K1/2,KINETIC represents the binding affinity of the second monomer of DmGen to the flap (Figure 7B, (ii)). The additional DmGen–DNA binding interactions provided by the second monomer may help to constrain the DNA, facilitating the DNA conformational change that positions the flap strand in the active site of the other DmGen monomer. This second monomer, while not necessary for cleavage, can be equated to the role of the scaffolding protein SLX4. Although SLX4 does not harbor nuclease activity, it not only increases the cleavage rates of SAs by XPF-ERCC1, but also increases preference of XPF-ERCC1 for SAs over stem–loops or bubbles (51). Interestingly, examination of published EMSA data on Yen1 (Figure 6E from (40)) binding to 5΄ flaps reveals that dephosphorylated Yen1 exhibits two shifted bands similar to our results with DmGen. These results suggest that Yen1 can also dimerize on 5΄ flaps as well as HJs. It remains to be investigated whether other orthologs can also dimerize on flaps.
Protein structural comparisons across Gen orthologs highlight differences leading to substrate specificity and dimerization
Studies of DmGen orthologs across species show cleavage of a variety of branched DNA structures and different propensities for dimerization. Insight into possible structural reasons for these differences in biochemistry can be gleaned from analysis of the protein primary sequences and crystal structures of the DmGen orthologs and the 5΄ flap cutter FEN1, which is a monomeric family member. Sequence alignment of the DmGen and FEN1 orthologs reveals significant differences among the orthologs in the region of DmGen 76–125 (Supplementary Figure S11). This region is adjacent to the active site and forms part of the dimer interface in the crystal structure of CtGEN1and the proposed dimer interface of HsGEN1 (45,46). In FEN1, this region forms a helical arch with a cap through which the single-stranded 5΄ flap is threaded. It has been suggested that this cap would prevent DNA structures without free ends from threading into the catalytic site, thereby limiting the activity of FEN1 to substrates containing a single-stranded 5΄ end (46,52). In CtGEN1, both the arch and cap regions are absent, and the protein is predominately a monomer in solution and cuts HJs but not flaps, replication forks or splayed arms. In HsGEN1, only the cap region is absent; the protein is a monomer in solution, but it cleaves 5΄ flaps, replication forks, HJs, and splayed arms (Supplementary Figure S5). Flap cleavage by HsGEN1 shows greater sensitivity than DmGen to the buffer and to the sequence at the junction (8,45,53). DmGen has both the helical arch and cap (although the cap sequence is not conserved with FEN1); this protein can dimerize on substrates and can cut 5΄ flaps, replication forks, HJs, and splayed arms. Interestingly, Yen1 also contains both the helical arch and cap region, and it cuts 5΄ flaps, replication forks, and HJs, but not splayed arms, and appears to dimerize on flaps (Figure 6E from (40)). Taken together, these observations suggest that the presence and/or the sequence of helical arch and cap govern not only substrate specificity but also the propensity to DmGen and its orthologs to dimerize. The ability of DmGen to dimerize on substrates as well as its broad substrate specificity may contribute to its preferential usage over Mus81 in vivo.
Substrate specificities of DmGen and HsGEN1 suggest that HJs are uncommon repair intermediates
Although it may seem unusual for the roles of Gen and Mus81 to have switched in Drosophila relative to other species, comparison of the in vitro and in vivo activities of DmGen in flies and Mus81 in other species sheds light on the commonalities of these two enzymes that allow them to be interchangeable within cells. The predominant enzymes in flies (DmGen) and in yeast and human cells (Mus81) cut a variety of branched structures, with substantially higher activities on simpler structures, such as flaps, over the more physically restrained HJ. While DmGen and its orthologs cut 5΄ to a branch point and Mus81 cuts 3΄, this polarity difference may be less relevant in vivo and may simply dictate the orientation of binding of the endonuclease to the substrate. This suggestion is supported by in vitro data that indicate that both Mus81 and DmGen orthologs cleave nicked HJs and replication forks to form nick duplexed products (9,54,55). In addition, in vivo studies show that overexpression of Yen1 can rescue mus81 mutants in budding yeast (17), and expression of HsGEN1 or DmGen can rescue mus81 mutants in fission yeast, which lacks a Yen1 ortholog ((40) and Supplementary Figure S1). We propose that these simpler structures may be more important in vivo, perhaps because they are more frequently encountered. Consistent with this hypothesis, recent studies show that replicative stress in yeast results in nicked and gapped HJs, while another study highlights non-HJ intermediates of replication repair (56,57).
In repair, DNA intermediates arising from strand exchange that generates a displacement-loop (D-loop) resemble flaps or nicked HJs, and these intermediates have many potential advantages over a canonical HJ as substrates for SSEs. First, flaps or nHJs would presumably be an earlier intermediate in repair, and further synthesis and ligation would require additional energy. Second, flaps or nHJs only require one nuclease domain for cleavage and could explain why eukaryotic ‘resolvases’ are predominantly monomers or heterodimers with only a single catalytic subunit. These points suggest that there may be mechanisms to avoid formation of HJs in favor of less complex intermediates. Nonetheless, intact HJs may accrue if repair intermediates are not processed fast enough such that further synthesis and ligation occurs, or if blocked replication forks are regressed (58). Even so, the biological presentation of these intact HJ may be very different from the synthetic in vitro HJs used for testing cleavage activity. In vivo, intact HJs may be opened by helicases or ssDNA binding proteins, and consequently resemble the splayed arms or bubbles studied in vitro. Such intermediates have been proposed to explain the requirement for the Blm helicase to make meiotic crossovers in C. elegans meiosis (59). Accordingly, we propose that during damage repair, canonical resolvase activity of Mus81 and DmGen orthologs on intact HJs may be a backup or failsafe to resolve intact HJs if they accumulate, but the primary role of these endonucleases is to cleave other structures that arise.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Steve West for the kind gift of HsGEN1 protein. We thank Steve West and Anton Gartner for suggesting we move the tag to the C-terminus of DmGen; Gerry Smith for providing S. pombe strains and plasmids; Wolf-Dietrich Heyer for comments on an early manuscript; Ashutosh Tripathy and the UNC MacInFac core facility for performing SEC-MALS; and Tony Perdue and the UNC Microscopy core facility for assistance.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of General Medical Sciences (NIGMS) [5RO1GM099890, 1R35GM118127 to J.S., GM080294, GM109832 to J.S., D.A.E., 5RO1GM099890-03S1 to M.R.R.]; National Cancer Institute (NCI) [R01 CA084442 to D.A.R]; NIGMS [5T32GM007092 to S.P.B.] (in part); National Science Foundation Graduate Research Fellowship and The Graduate School at The University of North Carolina at Chapel Hill (to D.J.R.). Funding for open access charge: National Institute of General Medical Sciences [1R35GM118127].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lloyd R.G., Sharples G.J.. Genetic analysis of recombination in prokaryotes. Curr. Opin. Genet. Dev. 1992; 2:683–690. [DOI] [PubMed] [Google Scholar]
- 2. Holliday R. A mechanism for gene conversion in fungi. Genet.Res. 1964; 78:282–304. [DOI] [PubMed] [Google Scholar]
- 3. Valenzuela M.S., Inman R.B.. Visualization of a novel junction in bacteriophage lambda DNA. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:3024–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mizuuchi K., Kemper B., Hays J., Weisberg R.A.. T4 endonuclease VII cleaves holliday structures. Cell. 1982; 29:357–365. [DOI] [PubMed] [Google Scholar]
- 5. Bennett R.J., Dunderdale H.J., West S.C.. Resolution of Holliday junctions by RuvC resolvase: cleavage specificity and DNA distortion. Cell. 1993; 74:1021–1031. [DOI] [PubMed] [Google Scholar]
- 6. Sharples G.J., Benson F.E., Illing G.T., Lloyd R.G.. Molecular and functional analysis of the ruv region of Escherichia coli K-12 reveals three genes involved in DNA repair and recombination. Mol. Gen. Genet. 1990; 221:219–226. [DOI] [PubMed] [Google Scholar]
- 7. Bell L., Byers B.. Occurrence of crossed strand-exchange forms in yeast DNA during meiosis. Proc. Natl. Acad. Sci. U.S.A. 1979; 76:3445–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ip S.C., Rass U., Blanco M.G., Flynn H.R., Skehel J.M., West S.C.. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008; 456:357–361. [DOI] [PubMed] [Google Scholar]
- 9. Rass U., Compton S.A., Matos J., Singleton M.R., Ip S.C., Blanco M.G., Griffith J.D., West S.C.. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010; 24:1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Castor D., Nair N., Declais A.C., Lachaud C., Toth R., Macartney T.J., Lilley D.M., Arthur J.S., Rouse J.. Cooperative control of Holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell. 2013; 52:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garner E., Kim Y., Lach F.P., Kottemann M.C., Smogorzewska A.. Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep. 2013; 5:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wyatt H.D., Sarbajna S., Matos J., West S.C.. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol. Cell. 2013; 52:234–247. [DOI] [PubMed] [Google Scholar]
- 13. Boddy M.N., Lopez-Girona A., Shanahan P., Interthal H., Heyer W.D., Russell P.. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol. Cell. Biol. 2000; 20:8758–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Interthal H., Heyer W.D.. MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000; 263:812–827. [DOI] [PubMed] [Google Scholar]
- 15. Dendouga N., Gao H., Moechars D., Janicot M., Vialard J., McGowan C.H.. Disruption of murine mus81 increases genomic instability and DNA damage sensitivity but does not promote tumorigenesis. Mol. Cell. Biol. 2005; 25:7569–7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fricke W.M., Brill S.J.. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003; 17:1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blanco M.G., Matos J., Rass U., Ip S.C., West S.C.. Functional overlap between the structure-specific nucleases Yen1 and Mus81-Mms4 for DNA-damage repair in S. cerevisiae. DNA Repair (Amst.). 2010; 9:394–402. [DOI] [PubMed] [Google Scholar]
- 18. Ho C.K., Mazon G., Lam A.F., Symington L.S.. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol. Cell. 2010; 40:988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tay Y.D., Wu L.. Overlapping roles for Yen1 and Mus81 in cellular Holliday junction processing. J. Biol. Chem. 2010; 285:11427–11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muñoz-Galvan S., Tous C., Blanco M.G., Schwartz E.K., Ehmsen K.T., West S.C., Heyer W.D., Aguilera A.. Distinct roles of Mus81, Yen1, Slx1-Slx4, and Rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol. Cell. Biol. 2012; 32:1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trowbridge K., McKim K.S., Brill S., Sekelsky J.. Synthetic lethality in the absence of the Drosophila MUS81 endonuclease and the DmBlm helicase is associated with elevated apoptosis. Genetics. 2007; 176:1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andersen S.L., Kuo H.K., Savukoski D., Brodsky M.H., Sekelsky J.. Three structure-selective endonucleases are essential in the absence of BLM helicase in Drosophila. PLoS Genet. 2011; 7:e1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yıldız Ö., Majumder S., Kramer B.C., Sekelsky J.. Drosophila MUS312 interacts with the nucleotide excision repair endonuclease MEI-9 to generate meiotic crossovers. Mol. Cell. 2002; 10:1503–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanai Y., Ishikawa G., Takeuchi R., Ruike T., Nakamura R., Ihara A., Ohashi T., Takata K., Kimura S., Sakaguchi K.. DmGEN shows a flap endonuclease activity, cleaving the blocked-flap structure and model replication fork. FEBS J. 2007; 274:3914–3927. [DOI] [PubMed] [Google Scholar]
- 25. Kaliraman V., Mullen J.R., Fricke W.M., Bastin-Shanower S.A., Brill S.J.. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 2001; 15:2730–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Benson F.E., West S.C.. Substrate specificity of the Escherichia coli RuvC protein. Resolution of three- and four-stranded recombination intermediates. J. Biol. Chem. 1994; 269:5195–5201. [PubMed] [Google Scholar]
- 27. Wright W.D., Ehmsen K.T., Heyer W.D.. Assays for structure-selective DNA endonucleases. Methods Mol. Biol. 2011; 745:345–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bennett R.J., West S.C.. RuvC protein resolves Holliday junctions via cleavage of the continuous (noncrossover) strands. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:5635–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okazaki K., Okazaki N., Kume K., Jinno S., Tanaka K., Okayama H.. High-frequency transformation method and library transducing vectors for cloning mammalian cDNAs by trans-complementation of Schizosaccharomyces pombe. Nucleic Acids Res. 1990; 18:6485–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lake C.M., Holsclaw J.K., Bellendir S.P., Sekelsky J., Hawley R.S.. The development of a monoclonal antibody recognizing the Drosophila melanogaster phosphorylated histone H2A variant (gamma-H2AV). G3 (Bethesda). 2013; 3:1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ratcliff G.C., Erie D.A.. A novel single-molecule study to determine protein–protein association constants. J. Am. Chem. Soc. 2001; 123:5632–5635. [DOI] [PubMed] [Google Scholar]
- 32. Yang Y., Wang H., Erie D.A.. Quantitative characterization of biomolecular assemblies and interactions using atomic force microscopy. Methods. 2003; 29:175–187. [DOI] [PubMed] [Google Scholar]
- 33. Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. et al. . Clustal W and Clustal X version 2.0. Bioinformatics. 2007; 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 34. Nicholas K.B., Nicholas H.B. Jr, Deerfield D.W.I.. GeneDoc: analysis and visualization of genetic variation. EMBNEW.NEWS. 1997; 4:14 [Google Scholar]
- 35. Sarbajna S., Davies D., West S.C.. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014; 28:1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu L.F., Desai S.D., Li T.K., Mao Y., Sun M., Sim S.P.. Mechanism of action of camptothecin. Ann. N.Y. Acad. Sci. 2000; 922:1–10. [DOI] [PubMed] [Google Scholar]
- 37. Groth P., Auslander S., Majumder M.M., Schultz N., Johansson F., Petermann E., Helleday T.. Methylated DNA causes a physical block to replication forks independently of damage signalling, O(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J. Mol. Biol. 2010; 402:70–82. [DOI] [PubMed] [Google Scholar]
- 38. Alvino G.M., Collingwood D., Murphy J.M., Delrow J., Brewer B.J., Raghuraman M.K.. Replication in hydroxyurea: it's a matter of time. Mol. Cell. Biol. 2007; 27:6396–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lorenz A., West S.C., Whitby M.C.. The human Holliday junction resolvase GEN1 rescues the meiotic phenotype of a Schizosaccharomyces pombe mus81 mutant. Nucleic Acids Res. 2010; 38:1866–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blanco M.G., Matos J., West S.C.. Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell. 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan Y.W., West S.C.. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat. Commun. 2014; 5:4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eissler C.L., Mazon G., Powers B.L., Savinov S.N., Symington L.S., Hall M.C.. The Cdk/Cdc14 module controls activation of the Yen1 Holliday junction resolvase to promote genome stability. Mol. Cell. 2014; 54:80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan Y.W., West S.C.. GEN1 promotes Holliday junction resolution by a coordinated nick and counter-nick mechanism. Nucleic Acids Res. 2015; 43:10882–10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Freeman A.D., Liu Y., Déclais A.C., Gartner A., Lilley D.M.. GEN1 from a thermophilic fungus is functionally closely similar to non-eukaryotic junction-resolving enzymes. J. Mol. Biol. 2014; 426:3946–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee S.H., Princz L.N., Klugel M.F., Habermann B., Pfander B., Biertumpfel C.. Human Holliday junction resolvase GEN1 uses a chromodomain for efficient DNA recognition and cleavage. eLife. 2015; 4:e12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y., Freeman A.D., Declais A.C., Wilson T.J., Gartner A., Lilley D.M.. Crystal structure of a eukaryotic GEN1 resolving enzyme bound to DNA. Cell Rep. 2015; 13:2565–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Patel S.S., Wong I., Johnson K.A.. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991; 30:511–525. [DOI] [PubMed] [Google Scholar]
- 48. Matos J., Blanco M.G., Maslen S., Skehel J.M., West S.C.. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011; 147:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matos J., Blanco M.G., West S.C.. Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep. 2013; 4:76–86. [DOI] [PubMed] [Google Scholar]
- 50. Lilley D.M. Structures of helical junctions in nucleic acids. Q. Rev. Biophysi. 2000; 33:109–159. [DOI] [PubMed] [Google Scholar]
- 51. Hodskinson M.R., Silhan J., Crossan G.P., Garaycoechea J.I., Mukherjee S., Johnson C.M., Scharer O.D., Patel K.J.. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol. Cell. 2014; 54:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsutakawa S.E., Classen S., Chapados B.R., Arvai A.S., Finger L.D., Guenther G., Tomlinson C.G., Thompson P., Sarker A.H., Shen B. et al. . Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily. Cell. 2011; 145:198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shah Punatar R., Martin M.J., Wyatt H.D., Chan Y.W., West S.C.. Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ehmsen K.T., Heyer W.D.. Saccharomyces cerevisiae Mus81-Mms4 is a catalytic, DNA structure-selective endonuclease. Nucleic Acids Res. 2008; 36:2182–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matos J., West S.C.. Holliday junction resolution: regulation in space and time. DNA Repair (Amst.). 2014; 19:176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Garcıa-Luis J., Machin F.. Mus81-Mms4 and Yen1 resolve a novel anaphase bridge formed by noncanonical Holliday junctions. Nat. Commun. 2014; 5:5652. [DOI] [PubMed] [Google Scholar]
- 57. Olmezer G., Levikova M., Klein D., Falquet B., Fontana G.A., Cejka P., Rass U.. Replication intermediates that escape Dna2 activity are processed by Holliday junction resolvase Yen1. Nat. Commun. 2016; 7:13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Neelsen K.J., Lopes M.. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015; 16:207–220. [DOI] [PubMed] [Google Scholar]
- 59. Agostinho A., Meier B., Sonneville R., Jagut M., Woglar A., Blow J., Jantsch V., Gartner A.. Combinatorial regulation of meiotic Holliday junction resolution in C. elegans by HIM-6 (BLM) helicase, SLX-4, and the SLX-1, MUS-81 and XPF-1 nucleases. PLoS Genet. 2013; 9:e1003591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.