


Abstract
Whole genome sequencing (WGS) is a promising strategy to unravel variants or genes responsible for human diseases and traits. However, there is a lack of robust platforms for a comprehensive downstream analysis. In the present study, we first proposed three novel algorithms, sequence gap-filled gene feature annotation, bit-block encoded genotypes and sectional fast access to text lines to address three fundamental problems. The three algorithms then formed the infrastructure of a robust parallel computing framework, KGGSeq, for integrating downstream analysis functions for whole genome sequencing data. KGGSeq has been equipped with a comprehensive set of analysis functions for quality control, filtration, annotation, pathogenic prediction and statistical tests. In the tests with whole genome sequencing data from 1000 Genomes Project, KGGSeq annotated several thousand more reliable non-synonymous variants than other widely used tools (e.g. ANNOVAR and SNPEff). It took only around half an hour on a small server with 10 CPUs to access genotypes of ∼60 million variants of 2504 subjects, while a popular alternative tool required around one day. KGGSeq's bit-block genotype format used 1.5% or less space to flexibly represent phased or unphased genotypes with multiple alleles and achieved a speed of over 1000 times faster to calculate genotypic correlation.
INTRODUCTION
Whole genome sequencing (WGS) is becoming a mainstream strategy for characterizing DNA variants of human disease and trait variation (1–5). Given sequence variants called from WGS data by tools like GATK (6), a downstream analysis plays a key role in finding responsible DNA variants. However, currently two major issues are challenging the downstream analysis of whole-genome sequence for genetic mapping. First, the amount of sequence and genomic annotation data are often very huge. For example, the 1000 Genomes Project Phase 3 contains over 88 million variants (7) and has over 16 GB genotypes data in variant call format (VCF) even after compression by the GNU Zip Format. When processed by conventional algorithms, it often demands unfeasibly large computing time and space. Second, given tens of millions of sequence variants on the entire genome, it is also rather difficult to discriminate the causal variants of a disease or trait from so many neutral or irrelevant ones.
While adopting advanced general infrastructure (such as the cloud computing (8)) was a promising scheme, making use of characteristics of the sequencing data themselves to boost the capacity of existing systems is also a nontrivial strategy. For example, an algorithm was developed to compress sequencing text data by blocks so that one can quickly access part of sequencing data through an index file (Tabix) (9). Currently, sequence variants are usually stored in text formats, e.g. VCF, which are convenient for human read and pre-process. However, loading and parsing these data are often the bottleneck of computing speed. Although the compressed data by blocks provides a foundation for concurrent computing, the parallel algorithm to speed text processing for WGS study is not readily available. Recently, some algorithms were developed to represent genotypes of bi-allelic variants as compressed bitmap indices (10) or integer matrix by the positional Burrows–Wheeler transform (11), which were able to reduce the computational burden of variant queries and/or storage space. However, the compressed bitmap indices algorithm did not work for phased genotypes, and the positional Burrows–Wheeler transform approach was not designed for variants with more than two alleles, which will limit their applications to many downstream analyses in practice. Therefore, advanced algorithms for relieving the huge burden of computing space and time are in demand.
Recently a number of methods have been proposed for identifying risk alleles in sequencing studies. These include methods for annotation (12), pathogenic prediction (13) and statistical analysis (14,15). Meanwhile, it is reaching a consensus that an integrative analysis with multiple functions and resources will be more powerful than individual methods alone (16–18). ANNOVAR is a widely-used tool with various genomic resources to annotate sequence variants (19). SnpEff is a genetic variant annotation toolbox that supports multiple species and predicts coding effects (20). VEP also provides many unique resources to annotate sequence variants (21). PLINK/SEQ and RVTESTS (22) are toolboxes containing multiple methods for gene-based association analysis with rare variants. However, genetic mapping tools for a one-stop integrative downstream analysis from quality control, genomic annotation, pathogenic/functional prediction and statistics association is still limited although there have been some integrative analysis tools (21,23). Moreover, most of these tools are often very slow for large-scale WGS data due to lack of robust algorithms for the big data.
Finally, yet importantly, a problem often overlooked in a downstream analysis is the accurate annotation of gene features (e.g. stoploss or missense). Although high-quality reference sequence data have been published, there are still many gaps because of the repetitive regions or lying on the centromeres and short arms of acrocentric chromosomes that are difficult to clone or assemble (5,24). These intrinsic gaps in the human reference genome may result in many errors in the gene-feature annotation of variants. It has been noted that the gene-feature annotation of different tools is quite inconsistent for around 20% of exonic variants (25). More accurate gene feature annotation is fundamentally crucial for the following annotation and prioritization analysis of the sequence variants.
In the present study, we proposed three novel algorithms to solve three bottlenecks (gene feature annotation inaccuracy, space and speed burden) in a large-scale WGS data analysis. These three fundamental algorithms formed a robust infrastructure of a framework that flexibly integrates multiple analysis functions to prioritize sequence variants in WGS studies jointly. All algorithms and analysis methods have been implemented in a biological Knowledge-based mining platform for Genomic and Genetic studies using Sequence data (KGGSeq, http://grass.cgs.hku.hk/limx/kggseq/). These algorithms were tested with the whole sequencing data from the 1000 Genomes Project for performance comparison to existing tools. All of the source codes implementing the algorithms are deposited in the GitHub repository (https://github.com/limx54/KGGseq).
MATERIALS AND METHODS
A gap-filled gene-feature annotation algorithm
In some consensus coding sequence regions, the reference genome has short sequence deletions and/or insertions compared to the reference coding DNA that is updated periodically by the Bioinformatics Center of University of California, Santa Cruz (UCSC) (See an example in Figure 1). However, sequence reads produced by sequencing machine are often mapped onto a reference genome (e.g. Human Reference Genome HG19) and the coordinates of variants are given according to the reference genome. Therefore, the relative distance of a variant to the protein coding starting position has to be adjusted for the insertion or deletion gap on the reference genome. Otherwise, it will result in an incorrect calculation of involved amino acid residue in a transcript. Therefore, we developed a gap-filled gene-feature annotation algorithm to adjust the coordinate shifts in protein coding regions. The complete source codes are in the GeneFeatureAnnotTask.java file of our deposited source codes in the GitHub repository.
Generate coding DNA sequence according to the reference genome. Extract the DNA sequences from the reference genome according to exon coordinates in the RefGene file from UCSC (e.g. http://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/refGene.txt.gz). For transcripts on the reverse strand, the extracted sequences are converted to reverse complementary sequences.
Mark the gaps in the extracted coding DNA. Align the generated coding DNA sequence with the latest reference coding DNA sequences from UCSC (e.g. http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/refMrna.fa.gz) by Smith–Waterman local alignment algorithm. In scoring profile, the mismatch and the match are given score −4 and 5, respectively. To produce fewer gaps, a deletion or insertion is given a high penalty score −50. The resulting deletion- and insertion-gaps in the coding regions of extracted coding DNA sequences are marked, compared to the coding DNA sequences.
Compute the positions of affected amino acid while considering the gaps. Check whether a variant is in a protein-coding region according to its coordinate and the boundaries of exons in a gene model database. If it is in a coding region, calculate its relative distance (l) to the protein coding start site. If there are n bp insertions and m bp deletions between the protein coding start site and the position of the variant in the coding DNA, the actual relative distance is equal to f = l+n−m. Compute the affected amino acid residue by the variant according to f and infer the exact mutation type, e.g. start loss, stop loss, stop gain, missense or synonymous.
Figure 1.

An example that the reference genome has a gap compared to the latest reference cDNA provided by the refGene database. Note: The alignment result was copied from a webpage of UCSC (https://genome.ucsc.edu/cgi-bin/hgc?hgsid=570008099_N9gTjX22Iv9RKMv0HA8tgBeJn4Oj&g=htcCdnaAli&i=NM_001146344&c=chr1&l=12884617&r=12891264&o=12884617&aliTable=refSeqAli&table=refGene). The rectangle in red denotes the amino acid codon used for gene feature annotation. For SNPEff, the UCSC reference genome hg19 was used.
Note that the steps 1 and 2 are only need to be done once. The step 3 has to be done for each variant but is very fast. We have implemented step 3 in a parallel computing framework so that millions of variants can be annotated in minutes through multiple CPUs.
Besides the RefGene model, KGGSeq also provides the compiled resource data for two other widely used gene models, UCSC Known Genes (26) and GENCODE (27). However, the gap-filled gene feature annotation algorithm is only necessary for the former two because their coding DNA sequences are updated even weekly. As the coding DNA sequences of GENCODE are directly extracted from the reference genome, there would be no relative gaps for the GENCODE model. Moreover, KGGSeq also allows for a customized genome model when the input gene model file with the same format is provided (See KGGSeq online manual, http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#GeneFeatureFiltering).
Genotype bit-block encoding algorithm
Encoding of phased and unphased genotypes into parsimonious bit-blocks
We developed a new encoding to flexibly represent and store genotypes in RAM and hard disk by minimal computer bits. For unphased genotypes, the number of possible genotypes is s = [n*(n−1)/2+n] given the allele number n. The minimal number of bits required to denote all possible genotypes (including missing genotype) is equal to the ceiling of 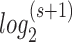 . The genotypes are then simply encoded into binary bits according to ascending order of alphabets. The missing genotype was in the last place. The encoding details can be seen in the online manual, http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#BinaryOutputs.
. The genotypes are then simply encoded into binary bits according to ascending order of alphabets. The missing genotype was in the last place. The encoding details can be seen in the online manual, http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#BinaryOutputs.
For phased genotypes, the number of possible genotypes is s = (n*n) given the allele number n. The minimal number of bits required to denote all possible genotypes (including missing genotype) is also equal to the ceiling of 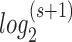 . The genotypes are then encoded into binary bits according to the ascending order of alphabets as well. The missing genotype was in the last place. The encoding details can be seen in the online manual, http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#BinaryOutputs.
. The genotypes are then encoded into binary bits according to the ascending order of alphabets as well. The missing genotype was in the last place. The encoding details can be seen in the online manual, http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#BinaryOutputs.
To speed up the analyses for genotypes, e.g. computing pairwise genotype correlation or counting homozygous genotypes in patients, we designed a unique structure, bit-block, to store the encoded bit genotypes. Assume a variant needs b bits to denote one genotype and it has m genotypes of different subjects. The first bit of each encoded genotype is extracted and put into a bit block of size m. The second bit of each encoded genotype forms the second bit block. This procedure is repeated until all of the b bits of a genotype are extracted. The b bit blocks are stored consecutively into t bytes, where t is equal to the ceiling of b*m/8 (Illustrated in Figure 2). The bits can be conveniently retrieved by blocks for further analyses. We demonstrate the usage of bit-block genotypes to enhance the speed of pairwise genotype correlation substantially. When the frequencies of all alternative allele are low (say <3%), the binary genotypes will be compressed by replacement of bytes recoding the position of ‘1’ bits. Each of such position is denoted by three bytes.
Figure 2.

Illustration of the bit-block encoding of genotypes in KGGSeq. Note: (A) The genotypes in a text file with the standard variant cell format (VCF) format; Genotypes of three subjects at three different variants are shown as an example. The first two variants have missing genotypes ‘./.’. (B) The genotypes are encoded into binary bits (http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#BinaryOutputs) and stored by blocks. The size of a block is equal to the number of subjects. The consecutive blocks are marked by different colors. (C) For rare variants, say, minor allele frequency <1%, only a tiny fraction of bits has the value ‘1’. The corresponding indices of bit ‘1's are stored in a hash set and the original space storing the genotype bits is cleaned. (D) The bit block variants can be used to speed up some time-consuming analyses. (E) The bit blocks can be stored in a hard disk in binary format by the unit of bytes. A byte contains 8 bits. The first two bytes are magic numbers for validating the KGGSeq bit-block format. The third byte tells whether genotypes are unphased (00000000) or phased (00000001). The genotypes start from the fourth byte. In this example, the fourth and fifth bytes denote genotypes of V1 and V2. The sixth and seventh bytes denote genotypes of V3.
The encoded genotypes (either compressed or not) are saved in a file with an extension name of ‘ked’. At the same time, the pedigree information and variant information are saved in a file with an extension name of ‘kam’ and a file with an extension name of ‘kim’, respectively. The encoded genotypes can be further compressed by standard compression algorithms, say, GNU Zip Format. The three files make up a KGGSeq binary genotype file set. The complete source codes are in the VCFParseTaskFast.java file of our deposited source codes in the GitHub deposit.
Rapid computing pairwise genotype correlation by bit-blocks
The genotypic correlation is often used to approximate the linkage disequilibrium of variants with unphased genotypes. Assume genotypes are denoted by the number of alternative alleles, xi, for a biallelic variant. The Pearson genotype correlation of two variants, Vi and Vj, can be calculated according to the following formula:
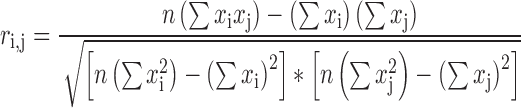 |
Assume the two bit blocks of a variant i are saved in two bit vectors, bi1 and bi2.
Produce an accessory bit vector,
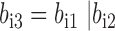 and non-missing genotype bit vector
and non-missing genotype bit vector 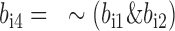 , where |, & and ∼ are bitwise inclusive OR AND and COMPLEMENT, respectively. The same can be done for variant j.
, where |, & and ∼ are bitwise inclusive OR AND and COMPLEMENT, respectively. The same can be done for variant j.- For variant i and variant j, the non-missing genotypes variant i and variant j at are denoted by
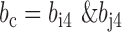 . We will have,
. We will have,
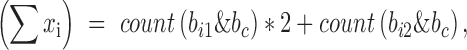
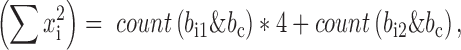
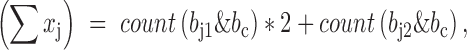
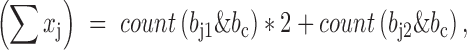
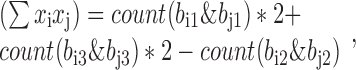
where count(x) is a function of counting the number of bits with value ‘1’, which is very fast on computers. Given these components, the genotype correlation can be calculated by the above formula straightforwardly. The implementation was in LDCalcTask.java of the GitHub deposit.
Fast accessing and parsing text algorithm
Sectional and random access of compressed text lines
The input variants data and annotation data in WGS analysis organized by lines are commonly compressed by GNU Zip format in blocks, which can save storage space in hard disks. We developed a new algorithm to read the compressed text lines in parallel. The algorithm made use of the features of blocked compression format (Blocked GNU Zip Format, or BGZF for short (9)) to read and parse the data by blocks. The following are the main steps of the algorithm. The specific source codes are in the BGZFInputStream.java file of our deposited source codes in the GitHub deposit.
Calculate initial positions in a compressed file for a given number, n, to partition the file into approximately equal parts.
Search valid start and end reading positions around the initial positions for each part by matching the block identifier code of BGZF, e.g. 00011111100010110000100000000100 (in bits).
Read and parse the compressed data from the valid start reading positions by lines after reserving data from the valid start reading position to the first new line delimiter (except for the first part).
Merge all reserved data across the parts to splice the truncated lines between blocks after all parts have been read out.
Moreover, to facilitate random access according to genomic coordinates, we also developed an algorithm to build an index file for each BGZF file. The index file recorded the chromosome name, starting position of a variant or region on the chromosome, and physical starting position of a block in the BGZF file. The index records were sorted according to the chromosome positions and file positions. Given a chromosome position, the conventional binary search algorithm was adopted to quickly locate the physical starting position of a block in the compressed file. This function allowed that only interesting blocks were loaded into memory and parsed.
Optimized text splitting and parsing
Attribute values of the same variants are commonly organized by lines in a text file (i.e. a variant-centred format), which is a space-efficient way to store information. For example, genotypes of multiple subjects of a variant are organized by lines in a VCF file and multiple populations’ minor allele frequencies of a variant are stored by lines in an annotation resource file as well. However, these attribute values must be parsed one by one for analysis through a text splitting function. According to our observations, the text splitting procedure took over 60% time when the text splitting functions (StringTokenizer or String.split) in Java library was employed. Therefore, we designed a new text splitting algorithm to speed up the text splitting. This new function made use of two important features to enhance the text splitting speed. First, it directly reads bytes (rather than the standard computer text) from hard disk and converts the bytes into a computer text only when necessary. The bytes are converted to numeric values much faster than text values. Second, in most text files, each row has fixed number of values to be split. So we use a shared array to temporally store the splitting results for every line in a text file, which saves the time for applying a temporary array to store each row. In our test with the genotype splitting of text lines on chromosome 1 of 2504 subjects from 1000 Genomes Project, the optimized splitting algorithm only needs ∼40% of the time, compared to the standard Java library functions (StringTokenizer and String.split). This algorithm has been combined with the above algorithm for sectional and random access of compressed text lines. See detailed implementation in the BZPartReader.java file of the GitHub deposit.
Compare gene feature annotation results of different tools
In the present paper, we compared gene feature annotation of different tools. It should be noted that an exact comparison is difficult because some variants can be mapped onto multiple transcripts or even multiple genes. To simplify the comparison, we encode the features into numbers according to http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html#GeneFeatureFiltering, which roughly suggests the potential effect of a variant on protein function. The top five features (startloss, stoploss, stopgain, splicing and missense) are considered in the comparison. If one variant was annotated with multiple features, the one with the highest priority was used for comparison. The unique feature of one tool refers to the feature annotated by this tool is different from others. The default parameters settings of each tool were used for the annotation. For each kind of feature, KGGSeq was compared with other annotation tools under both RefGene and GENCODE (v19) models. The unique and overlapped variants with the same annotation were counted. The unique variants were validated with the variants in dbSNP (b144_GRCh37p13).
Data analysis on KGGSeq
The phased genotype data (phase 3 version 5, 2504 subjects) in VCF format downloaded from 1000 Genomes Project (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20130502/) were used to test computing performance and analysis functions of KGGSeq V1.0 throughout of this paper. These data in the website had already been compressed in BGZ format, which can be efficiently processed by KGGSeq in parallel. The KGGSeq options for an integrative gene-based association analysis between Southern Asian and Eastern Asian with the 1000 Genomes Project sample are listed in Supplementary Table S1.
Compare the speed with PLINK/SEQ and vcftools
The above mentioned whole genome VCF data from 1000 Genomes Project were used for the comparison. All tools directly use the compressed data as input. We mainly compared the speed of loading and paring the text for simply counting allele frequency from VCF files. The default parameter setting was used for the tools. The key commands used in PLINK/SEQ and vcftools were ‘counts’ and ‘–freq’. KGGSeq was customized to not store intermediate data into hard disk in the comparison. CPU frequency of the computer was 3.457GHz. The version of Java running environment for KGGSeq was 1.7.
RESULTS
A sequence gap-filled gene feature annotation suggested more functionally important variants
As accurately annotating non-synonymous and synonymous variants is essential for most downstream analyses, we first investigated how our proposed sequence gap-filled gene feature annotation algorithm (detailed in the Materials and Methods section) worked among variants released by 1000 Genomes Project. Here the gap refers to the sequence gap in the reference genome relative to coding DNA in RefGene model. Among around 400 000 exonic variants in the African (AFR) panel, which has the largest number of variants among the five ancestrally different panels, the algorithm considering sequence gap reported 549 unique missense, 30 unique stopgain and 2 unique stoploss variants (Table 1, see details in the Supplementary Excel Table S1), compared to the algorithm only not considering sequence gap. More importantly, these unique annotations are highly consistent with the annotations from an independent resource, dbSNP, a widely-used third-part database in which more information (flanking sequence) was used to compute the gene features (https://www.ncbi.nlm.nih.gov/books/NBK174586/). For example, the consistency rate of missense annotation is as high as 95% among the 443 variants available in dbSNP. In contrast, when the sequence gap was not considered, less than 2% annotations were consistent with those in dbSNP although up to 1409 variants also were reported to have unique non-synonymous and synonymous annotations. This comparison clearly shows that considering the genomic gap in reference genome substantially improved the gene feature annotation of variants.
Table 1. Number of exonic variants uniquely annotated by KGGSeq using gap-filled gene-feature annotation algorithm.
| Unique without considering gap (dbSNP#a, %b) | overlapped | Unique with considering gap (dbSNP#a, %b) | |
|---|---|---|---|
| startloss | 0 | 492 | 0 |
| Stoploss | 0 | 223 | 2(1, 100%) |
| Stopgain | 87(66, 1.52%) | 3905 | 30(22, 86.36%) |
| Splicing | 0 | 2287 | 0 |
| Missense | 829(651, 2%) | 225 145 | 549(443, 95%) |
| Synonymous | 493(396, 0.25%) | 166 857 | 858(672, 93.75%) |
| Total | 1409 | 398 910 | 1439 |
Note: The RefGene model and reference coding DNA sequences were provided by UCSC on Oct 10, 2015 for KGGSeq. The splicing variants were the variants within 2 bp sites of both ends of an intron. a: the number of variant available in dbSNP; b: proportion of variants with consistent annotations between the dbSNP (b144_GRCh37p13) and KGGSeq among the dbSNP available variants.
It may also be interesting to know how the gap-filled gene feature annotation algorithm on KGGSeq works, compared with other popular tools (ANNOVAR (19), SNPEff (20) and VEP (21)). Again, among the ∼400 000 exonic variants, the gap filled gene-feature annotation algorithm uniquely annotated up to 4000 or 5000 more non-synonymous variants in total than ANNOVAR and SNPEff with RefSeq model (Table 2). Compared to KGGSeq, ANNOVAR only annotated 122 unique non-synonymous variants while KGGSeq annotated 4200 unique non-synonymous variants against ANNOVAR. Around 97% (3853 out of 3973) variants with KGGSeq's unique annotation have no exact annotation in ANNOVAR's output (see details in Supplementary Excel Table S2). ANNOVAR seemed to not annotate a variant at which there is a sequence gap or inconsistency between the reference genome and coding DNA in RefGene model. With the gap-filled annotation algorithm, KGGSeq correctly predicted the gene feature after adjusting the sequence gaps. This may explain why KGGSeq produced more non-synonymous annotations. Moreover, the proportions of consistent annotations with the dbSNP for the KGGSeq unique annotations were also generally much higher than that of the unique annotations of the ANNOVAR and SNPEff as well.
Table 2. Compare non-synonymous gene feature annotation of three popular tools and two gene models for variants discovered in 1000 Genomes Project AFR panel.
| RefGene | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| KGGSeq versus ANNOVAR (2015 Jun 17) | KGGSeq versus SNPEff(v4_1k) | KGGSeq versus VEP(v81) | |||||||
| KGGSeq Unique (dbSNP#a, %b) | overlapped | ANNOVAR Unique (dbSNP#a, %b) | KGGSeq Unique (dbSNP#a, %b) | overlapped | SNPEff Unique (dbSNP#a, %b) | KGGSeq Unique (dbSNP#a, %b) | overlapped | VEP Unique (dbSNP#a, %b) | |
| startloss c | —c, d | — c | — c, d | 56 | 436 | 1 | 35 | 457 | 227 |
| stoploss | 7(5, 80%) | 218 | 2(2, 100%) | 21(15, 100%) | 204 | 81(72, 2.78%) | 15(10, 90%) | 210 | 141(126, 62.7%) |
| stopgain | 124(92, 71.74%) | 3811 | 4(4, 50%) | 171(139, 89.93%) | 3764 | 128(118, 1.69%) | 180(134, 83.58%) | 3755 | 597(530, 83.58%) |
| splicing | 96(90, 92.22%) | 2191 | 3(2, 100%) | 2286(2093, 98.09%) | 1 | 1902(1764, 0.4%) | 105(84, 73.81%) | 2182 | 43 322(40 572, 3.18%) |
| missense | 3965(87.08%) | 222 213 | 113(94, 55.32%) | 6422(5868, 96.1%) | 219 272 | 1197(1027, 11.88%) | 13 021(10 719, 94.88%) | 212 673 | 12 978(11 885, 94.26%) |
| total | 4192 | 228 433 | 122 | 8956 | 223 677 | 3309 | 13 356 | 219 277 | 18 275 |
| GENCODE(v19) | |||||||||
| startloss c | —c, d | — c | — c, d | 73 | 580 | 94 | 0 | 653 | 44 |
| stoploss | 3(2, 100%) | 337 | 7(5, 20%) | 29(23, 52.17%) | 311 | 129(115, 7.83%) | 2(1, 100%) | 338 | 40(25, 4%) |
| stopgain | 30(23, 56.52%) | 4222 | 156(52, 25%) | 178(152, 84.21%) | 4074 | 556(415, 13.01%) | 10(7, 100%) | 4242 | 163(58, 31.03%) |
| splicing | 157(146, 73.29%) | 2460 | 76(48, 31.25%) | 2614(2360, 88.18%) | 3 | 4283(3859, 0.34%) | 18(14, 64.29%) | 2599 | 49 203(41541, 1.56%) |
| missense | 680(509, 75.64%) | 231 245 | 5163(2121, 51.67%) | 6773(6091, 94.07%) | 225152 | 10 948(7239, 22.82%) | 6873(6314, 94.04%) | 225052 | 4679(1582, 40.39%) |
| total | 870 | 238 264 | 5402 | 9667 | 230 120 | 16 010 | 6903 | 232 884 | 54 129 |
Note: The RefGene model was provided by UCSC on Oct 10, 2015 for KGGSeq; and for ANNOVAR and the latest RefGene model (by Oct 10, 2015) from their websites was used. a: the number of variant available in dbSNP; b: the proportion of variants (out of the dbSNP available variants) with consistent annotations between the dbSNP (b144_GRCh37p13) and the tool. c: Because ANNOVAR has no startloss category but annotates startloss as missense, no comparison was made for this category. d: Because dbSNP has no startloss category, no consistency was calculated for this category.
Interestingly, both KGGSeq and VEP produced around 13 000 unique missense annotations and both tools’ unique missense annotations also have as high as 94% consistency with the dbSNP annotation. After checking the detailed annotation (as shown in the Supplementary Excel Table S2), it turned out most of the VEP unique annotations occur at transcripts from predicted mRNA model (i.e. the ID starting with XM_). Because of the uncertainty in the predicted mRNAs, KGGSeq (as well as ANNOVAR and SNPEff) does not use them for annotation. For example, the variant rs377389746 is a missense variant of XM_005271074 while it is a synonymous variant of NM_020888 and NM_001198972. Therefore, VEP gave a missense and a synonymous annotation but KGGSeq only had a synonymous annotation at this variant. Notably, VEP and SNPEff annotated many splicing variants but the consistency rates to dbSNP's annotation are only 0.4% and 3.18%, respectively. Probably, the two tools have different definitions on a splicing variant from the dbSNP. Under GENCODE model, KGGSeq produced less unique non-synonymous than ANNOVAR and SNPEff. This is because KGGSeq ignored all incomplete transcripts, e.g. ENST00000390243 that is incomplete at 3΄. This may explain why KGGSeq had much higher consistency with the dbSNP than all of the three tools under GENCODE model (see details of unique annotations the Supplementary Excel Table S2). To summarize, the higher consistency of KGGSeq's annotations with dbSNP suggests that KGGSeq can provide more accurate gene feature annotations than the other popular tools.
A genotype bit-block encoding algorithm saves a lot of space and time
Because genotypes in a large-scale WGS data set lead to a huge storage and computing burden in downstream analysis, we designed a new bit-block encoding algorithm to more efficiently store genotypes (illustrated in Figure 2). Compared to several existing genotype encoding algorithms (10,28), the proposed algorithm is able to flexibly encode both phased and unphased genotypes of variants with two or more alleles while most of the former ones can only process unphased genotypes of bi-allelic variants. As shown in Table 3, the bit-block encoding algorithm only needs ∼1.5% space to store the phased genotypes of 2504 subjects of 6 468 094 variants in chromosome 1, compared to the most popular format for sequence variants, VCF and the conventional linkage format. For the unphased genotypes, the space is even reduced to be ∼1.2%. Compared to another algorithm, BGT which used a sophisticated transform to compress the binary genotypes (29), the KGGSeq's binary genotype set is about 2 to 3 times larger. However, it should be noted BGT does not work for variants with over two alleles.
Table 3. Compare the size of genotypes in chromosome 1 of 1000 Genomes Project.
| VCF format | Plink linkage format file set | Plink binary genotype format file set | BGT format | KGGSeq binary genotype format file set | |
|---|---|---|---|---|---|
| Unphased | 64.234 GB | 66.093 GB | 3.92 GB | 342 MB | 793 MB |
| Phased | 64.234 GB | 66.093 GB | —a | 268 MB | 990 MB |
Note: Each file contained phased genotypes of 2504 subjects at 6 468 094 bi-allice variants in chromosome 1. a) The plink-binary genotype format cannot encode phased genotypes.
More importantly, another appealing advantage of the bit block genotypes is that it conveniently facilitates fast computation of genotype-related analysis. The bit-block uses bit vectors to represent genotypes of all subjects at each variant. A lot of statistical analysis at genotypes, say, calculating genetic correlation and counting alleles and genotypes, can be carried out straightforwardly by these bit vectors. As a vector contains genotypes of multiple subjects, a single bit-wise logical operation at the bit-vectors conducts analysis of multiple subjects in parallel. Therefore, these bit vectors can dramatically improve the analysis speed of many genetic analyses at genotypes. Table 4 shows the analysis speed for calculating genetic correlation on chromosome 22 of 2504 subjects from the 1000 Genomes Project. The algorithm based on the bit-block (see details in the Materials and Methods section) is over 1300 times faster than the conventional algorithm (directly calculating the Pearson correlation with coded genotypes 0, 1 and 2) when there are 16 000 variants. That is, a 2-week job by a conventional approach is done in half an hour by the new approach.
Table 4. Time used by a bit-block based algorithm and the conventional algorithm for computing Pearson correlation of genotypes.
| Number of variants | Bit-block | Conventional | Ratio |
|---|---|---|---|
| 4000 | 0:0:2.59 | 50:01.36 | 1:1159 |
| 7000 | 0:0:7.59 | 2:31:56.48 | 1:1201 |
| 10 000 | 0:0:14.99 | 5:08:54.14 | 1:1236 |
| 13 000 | 0:0:23.60 | 8:43:17.78 | 1:1330 |
| 16 000 | 0:0:35.18 | 13:18:33.96 | 1:1362 |
Note: The variants were randomly drawn from chromosome 22 and genotypes of 2504 subjects were used to calculate the correlation. The time format a:b:c.d stands for a hour(s), b minute(s) and c.d second(s), respectively. The version of Java running environment was 1.7. CPU frequency was 3.457 GHz. a: For the conventional approach, the genotypes were encoded according to the number of alternative alleles, i.e. 0, 1 and 2 first. The Pearson correlation coefficients were calculated for each pair of variants by a fast Java package Colt (https://dst.lbl.gov/ACSSoftware/colt/).
A sectional accessing and optimized parsing text algorithm substantially enhances data loading
Finally, WGS data analysis often involves thousands of gigabytes data (including the variants’ genotypes, quality scores and genomic annotations) saved in the hard disk in text format. Because so huge amount of data have to be loaded and parsed in RAM, the Input/Output speed is a challenging issue. Based on the block-wise compressed data (9), we developed an algorithm for sectional and random access to compressed text lines, which allows KGGSeq to load the compressed data into RAM in parallel directly. Besides, we also optimized the parsing in a text line by directly accessing a primitive data type, byte, rather than the senior data type, the Java text String. When using this algorithm to load and parse genotypes data in compressed VCF file from hard disk, KGGSeq was around 7 and 5 times faster than two popular alternative downstream analysis tools, PLINK/SEQ (with version 0.10, https://atgu.mgh.harvard.edu/plinkseq) and vcftools (with version 0.1.14). It only used around half an hour to process whole genome variants from the 1000 Genomes Project with 10 CPUs while the other two alternative tools (which cannot run jobs in parallel) need ∼24 and ∼18 h to produce the same results (Table 5).
Table 5. Compare the speed with PLINK/SEQ and vcftools for counting allele frequency of 1000 Genomes Project.
| Time | Maximal RAM used | |
|---|---|---|
| kggseq (v1.0) 10 CPUs | 35 min | 60 MB* |
| kggseq (v1.0) 1 CPU | 3 h 24 min | 36 MB* |
| PLINK/SEQ (v0.10)∧ | 24 h 25 min | 12 MB |
| vcftools(v0.1.14)∧ | 17 h 46 min | 6 MB |
Note: ∧: The PLINK/SEQ and vcftools do not support multiple CPUs. *:KGGSeq was customized to not reserve the intermediate data in the comparison. The version of Java running environment is 1.7. CPU frequency is 3.457 GHz.
A comprehensive framework for an integrative downstream analysis of WGS data
Based on the three key fundamental algorithms, we built a robust parallel-computing framework for flexibly integrating multiple analysis functions of WGS data. Currently, it has had five major modules: quality control, filtration, annotation, pathogenic prediction and statistic tests (summarized in Figure 3). There are both shared and unique functions for different types of diseases (e.g. Complex disease, Cancers and Mendelian diseases). All of the available methods and their usage are described in KGGSeq's user manual (http://grass.cgs.hku.hk/limx/kggseq/doc10/UserManual.html). Most of these individual methods have been published elsewhere. As these methods have been evaluated in their original papers, e.g. filtration of exonic variants for Mendelian diseases (18), regulatory variant prediction (13), gene-based association test for rare variants (30) and pathogenic prediction at genes (31), we would not re-evaluate them in the present paper except for a simple compare of pathogenic prediction with two latest approaches REVEL (32) and M-CAP (33). As shown in Figure 4, the combined pathogenic prediction model of KGGSeq (13) had a similar area under the curve of receiver operating characteristic with REVEL (0.938 vs.0.939) and higher area under curve than M-CAP(0.938 versus 0.815) when discriminating pathogenic variants from benign variants in ClinVar data set. More functions and methods would be added to this framework in the future. To demonstrate effectiveness of this integrative framework, we show two examples here.
Figure 3.

Main modules in KGGSeq for three types of diseases. Note: KGGSeq integrates a series of functions for quality control, filtration, annotation, pathogenic prediction and statistical tests for three types of human diseases: Mendelian diseases, complex diseases and cancers. Please see more description of the pipelines in the Supplementary text.
Figure 4.

The performance comparison of KGGSeq's pathogenic prediction with REVEL and M-CAP. Note: (A) area under the curve of receiver operating characteristic, (B) area under the curve of precision-recall. The pathogenic variants (within the clinical significance category 5) and benign variants (within the clinical significance category 2) in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) were extracted as a benchmark data set. After excluded overlapped variants with KGGSeq's training data set, 1898 non-synonymous pathogenic variants and 2180 benign variants were retained for the evaluation. A cut-off was used to produce the performance measures, including true positive, false positive, precision and recall rates. The curves were generated by varying the cut-off from the minimal score and maximal score of each prediction tool.
In the first example, KGGSeq was used to identify causal mutation in an exome-sequencing trio in which the offspring was affected with a Mendelian disease, neonatal-onset Crohn's disease. This is a small data set. The purpose of this example is to demonstrate the usefulness of KGGSeq for Mendelian diseases. The original study suggested two compound heterozygous mutations in IL10RA caused the disease (34). After the integrative analysis of KGGSeq, those two causal mutations chr11: 117860219 and chr11: 117860269 were pinpointed accurately with multiple annotations supporting their causality to the disease (see detailed output in the Supplementary Excel Table S3). For the purpose of comparison, we also used another tool, GEMINI, to analyze the same exome sequencing data. The variants’ information saved in VCF file was decomposed and normalized by a tool named VT (35) first and then annotated by SNPEff. The result VCF file was loaded to GEMINI and built an inside database. The ‘comp_hets’ function was called to identify potential compound heterozygotes and 17 genes including 60 variants were found in the data set of variants with ‘impact = MED or HIHG’. However, the two candidate variants were not in the final prioritized shortlist. Moreover, for the whole analysis, KGGSeq just took 4 min and 59 s, but GEMINI spent more than 1 h to run the analysis on the same machine. The detailed comparisons were listed in the Supplementary Tables S1 and S2.
In the second example, we show how an integrative gene-based association analysis in the WGS samples of 1000 Genomes Project can be conveniently and efficiently carried out on this framework. The analysis goal is to investigate the degree of population stratification between southern Asian and eastern Asian by the gene-based association at rare and functionally important variants on autosomes. Subjects from southern Asia and eastern Asia were assigned into two groups for the association analysis.
We used KGGSeq to filter out common variants, annotate variants with gene features by RefGene and GENCODE models, predict functional variants in both coding and non-coding regions of autosomes, and perform gene-based association analysis by statistical tests. The KGGSeq options are listed in the Supplementary Table S3. The analysis was run on 10 CPUs with 3.07 GHz in parallel. KGGSeq only took 7 min to load and parse the 5.4 GB compressed VCF data, which was 164.3 GB un-compressed text of ∼40 million variants actually. After eliminating variants with minor allele frequency >0.01 and on sex chromosomes, ∼29 million variants are left. In total, 18 629 256 variants were mapped onto genes according to the two gene models in which variants within 5000 base pairs of transcription start and end sites were included as upstream and downstream variants. The pathogenic and functional prediction at coding variants and non-coding variants ranked variants quantitatively (Supplementary Figure S1). As expected, the stopgain variants tend to have higher posterior probability than the other three types of non-synonymous variants. Among the non-coding variant categories, the upstream variants have the largest functional scores. After removing variants with negative predictions, only 6 683 904 variants are retained for gene-based association analysis.
It turned out that most genes have different allele frequency at the rare variants between the southern Asian and eastern Asian, which also suggest a substantial population stratification between the two sub populations in Asian (Supplementary Figure S2A). According to a burden test (CMC (15)), 14 252 genes (out of 55 676 genes) have significant P-values (P ≤ 8.98 × 10−7, Bonferroni correction for family error rate 0.05). The variance-component test, SKAT (14), reported even more significant genes (41 611) than CMC. The inflation disappeared when the population membership was permuted among the subjects (Supplementary Figure S2B). This result suggests that population structure would be strictly controlled for gene-based association tests event at rare variants. Otherwise, it would introduce many false positives.
DISCUSSION
In the present study, we proposed three novel fundamental algorithms to improve gene feature annotation accuracy and to substantially relieve computing space and time burden in downstream analysis of large-scale whole genome sequence data. Moreover, these algorithms constitute the infrastructure of a robust analysis parallel-computing framework, KGGSeq, for flexibly integrating diverse functions to more effectively isolate genes or variants responsible for human traits or diseases. When tested with the WGS data from 1000 Genomes Project, KGGSeq annotated several thousand more non-synonymous variants than other popular tools. Moreover, it was over 20 times or much faster to load and parse sequence variants, and used ∼1.5% or less space to flexibly represent phased or unphased genotypes with multiple alleles, and achieved a speed of over 1000 times faster to calculate genotypic correlation. These advanced algorithms enable a rapid process of large-scale WGS data on computers that are affordable for most core facilities and laboratories. Besides its substantial capacity for large-scale sequencing data, KGGSeq is also a very comprehensive platform for integrative downstream analysis on the whole genome of monogenic diseases or complex diseases.
The bit-block algorithm provides an efficient way to store and represent genotypes in both hard disk and RAM. Compared with existing algorithms of binary encoding of genotypes (e.g. PLINK (28) GQT (10) and BGT (11) formats), our proposed algorithm has two unique features. First, it can flexibly encode both the phased and unphased genotypes with more than two alleles. In contrast, the existing algorithms either ignore multiallelic variants or collapse different heterozygous genotypes as an over-simplified heterozygous genotype, which will lead to loss of genotype information. It has been noted that multi-allelic genotypes are also important (36), which is common for Indels. Besides, PLINK (28) and GQT (10) do not support analyses of haplotypes. Second, it directly supports bitwise operation with bit-vectors, which can substantially speed up some time-consuming analyses on genotypes as we demonstrated in this paper. This is a powerful design to save both space and time without sacrifice of any other side. As the algorithm itself is generic, this genotype bit-block format will also be very useful for other WGS tools.
Among the four widely used tools, KGGSeq annotated the greatest number of unique non-synonymous variants and achieved consistency with dbSNP's annotations in most scenarios. Besides the gap-filled algorithm, the accuracy and completeness of reference gene models are also important for an accurate gene feature annotation. The differences in background reference gene model databases substantially attribute to the several thousand different annotations of SNPEff and VEP, compared to KGGSeq. Anyhow, a consideration of the sequence gap between reference genome and coding DNA helped rescue ∼600 non-synonymous variants and correct ∼900 false non-synonymous annotations in 645 African genomes from the 1000 Genomes Project. KGGSeq also covered the most majority of ANNOVAR's annotations under RefGene model. However, KGGSeq's algorithm only worked for the known gaps. There may be many unknown sequence gaps. Hopefully, the influence of reference databases on the annotation will decrease as they become more and more accurate and complete in the future. It is almost impossible to expect a single algorithm and/or database to achieve 100% annotation accuracy for all non-synonymous variants currently. According to the comparison, it seems a combination of KGGSeq and VEP under both the RefGene and Gencode models will lead to a higher coverage of gene-feature annotation with a relatively high accuracy in practice.
The fast text loading and parsing algorithm provides an efficient solution for processing biological data. To ease human reading and management, most bioinformatics data are stored in covenantal text files and organized by lines, e.g. the VCF format and the Browser Extensible Data (BED, https://genome.ucsc.edu/FAQ/FAQformat.html#format1) format. However, processing text needs more computing cost than other data types, e.g. integers. The text data can be compressed into blocks (9) to facilitate random access. Note that the text lines are often truncated between blocks, which complicates data parsing. The proposed algorithm enhanced the computing speed of the compressed data in two ways. First, it allows parallel loading of the compressed blocks while automatically joining the truncated text lines. Second, it makes use of a primitive data type, byte, to circumvent unnecessary conversions and operations of text data. Compared to alternative tools (PLINK/SEQ and vcftools which are implemented in C++), this algorithm substantially improved the speed of loading and parsing text data. We believe this algorithm will also be useful for other bioinformatics analysis tools.
KGGSeq was designed to integrate multiple methods and resources for WGS downstream analysis. Although it has integrated abundant functions for a comprehensive downstream analysis into five main functional modules (qualities control, filtration, annotation, pathogenic prediction and statistical tests), it is hard to conclude that these functions have been sufficient for all scenarios. For example, some methods using the tissue or cell-type specific gene expression or expression quantitative trait loci (37) may be added after careful evaluations for genetic mapping. In any case, a joint analysis by multiple powerful approaches tends to produce results that are more reliable. However, as there are so many individual methods available and even emerging, a systematic evaluation of the best method combination is unfeasibly difficult. Currently, we just integrated some representative methods and resources in the five major modules according to our own and some other colleagues’ experiences. The successful applications of KGGSeq to real data (17,38) have conceptually proved the effectiveness of this integrative strategy. In the present paper, we focus on the robust infrastructure to facilitate method integration for downstream analysis of large-scale WGS data.
AVAILABILITY
KGGSeq has a simple command-line interface to run on most operating systems with Java running environment. It accepts multiple input formats of sequence variants with genomic coordinates of hg18, hg19 or GRCh38 (hg38) and output results for visualization and further analysis flexibly. KGGSeq is free for academic and non-profit usage, http://grass.cgs.hku.hk/limx/kggseq/. Built on a combination of abundant bioinformatics resources, bioinformatics/statistical methods and advanced computing algorithms, this software will play a useful role in genetic mapping studies of both Mendelian diseases and complex diseases (including cancer) with either small or large-scale of WGS data.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge a number of institutes and projects for their free data sets used in this study: 1000 Genomes Project, Online Mendelian Inheritance in Man, dbNSFP, etc. The authors also thank the anonymous reviewers for their useful comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Hong Kong Research Grants Council [GRF 17128515, HKU 776412M, N_HKU736/14]; Hong Kong Research Grants Council Theme-Based Research Scheme [T12-705/11]; National Natural Science Foundation of China [NSFC 91229105]; European Community Seventh Framework Programme Grant on European Network of National Schizophrenia Networks Studying Gene-Environment Interactions (EU-GEI); HKU Seed Funding Programme for Basic Research [201411159172, 201302159006, 201311159090]; Health and Medical Research Fund [01121436, 01121726]; National Institute of Health [R01HG008983, R21DA040177].
Conflict of interest statement. None declared.
REFERENCES
- 1. Cirulli E.T., Goldstein D.B.. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010; 11:415–425. [DOI] [PubMed] [Google Scholar]
- 2. Willig L.K., Petrikin J.E., Smith L.D., Saunders C.J., Thiffault I., Miller N.A., Soden S.E., Cakici J.A., Herd S.M., Twist G. et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 2015; 3:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Directors A.B.o. Points to consider in the clinical application of genomic sequencing. Genet. Med. 2012; 14:759–761. [DOI] [PubMed] [Google Scholar]
- 4. Genome of the Netherlands, C. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014; 46:818–825. [DOI] [PubMed] [Google Scholar]
- 5. Lander E.S. Initial impact of the sequencing of the human genome. Nature. 2011; 470:187–197. [DOI] [PubMed] [Google Scholar]
- 6. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao S., Prenger K., Smith L., Messina T., Fan H., Jaeger E., Stephens S.. Rainbow: a tool for large-scale whole-genome sequencing data analysis using cloud computing. BMC Genomics. 2013; 14:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011; 27:718–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Layer R.M., Kindlon N., Karczewski K.J., Exome Aggregation C., Quinlan A.R.. Efficient genotype compression and analysis of large genetic-variation data sets. Nat. Methods. 2016; 13:63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H. BGT: efficient and flexible genotype query across many samples. Bioinformatics. 2016; 32:590–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu X., Wu C., Li C., Boerwinkle E.. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 2016; 37:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li M.X., Kwan J.S., Bao S.Y., Yang W., Ho S.L., Song Y.Q., Sham P.C.. Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013; 9:e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X.. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011; 89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li B., Leal S.M.. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008; 83:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pabinger S., Dander A., Fischer M., Snajder R., Sperk M., Efremova M., Krabichler B., Speicher M.R., Zschocke J., Trajanoski Z.. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014; 15:256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li M.J., Deng J., Wang P., Yang W., Ho S.L., Sham P.C., Wang J., Li M.. wKGGSeq: a comprehensive strategy-based and disease-targeted online framework to facilitate exome sequencing studies of inherited disorders. Hum. Mutat. 2015; 36:496–503. [DOI] [PubMed] [Google Scholar]
- 18. Li M.X., Gui H.S., Kwan J.S., Bao S.Y., Sham P.C.. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012; 40:e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M.. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012; 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F.. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010; 26:2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhan X., Hu Y., Li B., Abecasis G.R., Liu D.J.. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 2016; 32:1423–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paila U., Chapman B.A., Kirchner R., Quinlan A.R.. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 2013; 9:e1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. International Human Genome Sequencing, C. Finishing the euchromatic sequence of the human genome. Nature. 2004; 431:931–945. [DOI] [PubMed] [Google Scholar]
- 25. McCarthy D.J., Humburg P., Kanapin A., Rivas M.A., Gaulton K., Cazier J.B., Donnelly P.. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsu F., Kent W.J., Clawson H., Kuhn R.M., Diekhans M., Haussler D.. The UCSC known genes. Bioinformatics. 2006; 22:1036–1046. [DOI] [PubMed] [Google Scholar]
- 27. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007; 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li M.J., Pan Z., Liu Z., Wu J., Wang P., Zhu Y., Xu F., Xia Z., Sham P.C., Kocher J.P. et al. Predicting regulatory variants with composite statistic. Bioinformatics. 2016; 32:2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ionita-Laza I., Lee S., Makarov V., Buxbaum J.D., Lin X.. Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet. 2013; 92:841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu J.S., Kwan J.S., Pan Z., Garcia-Barcelo M.M., Sham P.C., Li M.. Inheritance-mode specific pathogenicity prioritization (ISPP) for human protein coding genes. Bioinformatics. 2016; 32:3065–3071. [DOI] [PubMed] [Google Scholar]
- 32. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016; 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jagadeesh K.A., Wenger A.M., Berger M.J., Guturu H., Stenson P.D., Cooper D.N., Bernstein J.A., Bejerano G.. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016; 48:1581–1586. [DOI] [PubMed] [Google Scholar]
- 34. Mao H., Yang W., Lee P.P., Ho M.H., Yang J., Zeng S., Chong C.Y., Lee T.L., Tu W., Lau Y.L.. Exome sequencing identifies novel compound heterozygous mutations of IL-10 receptor 1 in neonatal-onset Crohn's disease. Genes Immun. 2012; 13:437–442. [DOI] [PubMed] [Google Scholar]
- 35. Tan A., Abecasis G.R., Kang H.M.. Unified representation of genetic variants. Bioinformatics. 2015; 31:2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campbell I.M., Gambin T., Jhangiani S.N., Grove M.L., Veeraraghavan N., Muzny D.M., Shaw C.A., Gibbs R.A., Boerwinkle E., Yu F. et al. Multiallelic positions in the human genome: challenges for genetic analyses. Hum. Mutat. 2016; 37:231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Consortium G.T. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heinen C.A., Jongejan A., Watson P.J., Redeker B., Boelen A., Boudzovitch-Surovtseva O., Forzano F., Hordijk R., Kelley R., Olney A.H. et al. A specific mutation in TBL1XR1 causes Pierpont syndrome. J. Med. Genet. 2016; 37:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.