


Abstract
Kink turns are widely occurring motifs in RNA, located in internal loops and associated with many biological functions including translation, regulation and splicing. The associated sequence pattern, a 3-nt bulge and G-A, A-G base-pairs, generates an angle of ∼50° along the helical axis due to A-minor interactions. The conserved sequence and distinct secondary structures of kink-turns (k-turn) suggest computational folding rules to predict k-turn-like topologies from sequence. Here, we annotate observed k-turn motifs within a non-redundant RNA dataset based on sequence signatures and geometrical features, analyze bending and torsion angles, and determine distinct knowledge-based potentials with and without k-turn motifs. We apply these scoring potentials to our RAGTOP (RNA-As-Graph-Topologies) graph sampling protocol to construct and sample coarse-grained graph representations of RNAs from a given secondary structure. We present graph-sampling results for 35 RNAs, including 12 k-turn and 23 non k-turn internal loops, and compare the results to solved structures and to RAGTOP results without special k-turn potentials. Significant improvements are observed with the updated scoring potentials compared to the k-turn-free potentials. Because k-turns represent a classic example of sequence/structure motif, our study suggests that other such motifs with sequence signatures and unique geometrical features can similarly be utilized for RNA structure prediction and design.
INTRODUCTION
The kink turn (k-turn) is a widespread structural motif found in double stranded RNA structures. Since it was first defined as a new structural element in the ribosome by Klein et al. (1), k-turns have been noted in many RNAs, including riboswitches and ribozymes, and associated with important regulatory and catalytic cellular roles. The Lilley group has recently explored k-turns extensively (2–10). The k-turn serves as a key architectural element that helps define specific ligand binding pockets by generating a kink between two helices with an angle of ∼50° (3). Hence, k-turns can offer important specialized components for the regulation of cellular functions, and can act as conformational switches upon binding to ligands (11–13). K-turns are also good elements for constructing nanostructures, as shown recently for 2–8 k-turns complexes (14).
K-turns occur in internal loops where two single strand regions are connected by two helices (Figure 1). The consensus k-turn sequence consists of a three-nucleotide single strand region and G•A and A•G base-pairs on the 3΄ side (9,15,16). The k-turn motif includes a tertiary interaction called A-minor motif where Adenine (A) close to a G–C base pair (17). Recent experimental studies by Lilley et al. have offered structural and functional insights into k-turn motifs (3,9. These researchers also collected a database of k-turn structures in which structures are grouped into classes based on base pair characteristics (2). They described a set of rules relating the sequence to the resulting structure and the folding process, emphasizing the important roles of k-turns in the folding of RNAs (9). More generally, such experimental structural information can provide folding rules to predict RNA structures from sequence, and thus assist in the understanding of RNA functional mechanisms. In recent studies, it was shown that an adjacent base pair to G–A, (3b, 3n), as illustrated in Figure 1, determines both the folding properties of the k-turn and the specific conformation class of the k-turn (7,10).
Figure 1.

The consensus sequence of simple kink-turns.
Experimental crystal structures have revealed that k-turn motifs have highly conserved sequence contents and secondary structures (1,15,16). In Figure 2, we illustrate the U4 snRNA k-turn as an example of a standard k-turn class. Yet, there are some cases where k-turns do not follow this rule. Lilley et al. divide the k-turns into simple and complex k-turns based on existence of consensus sequence signatures. In our study, we employ sequence signatures of simple k-turns in the context of structure prediction from sequence or secondary structure; complex k-turns do not follow the consensus sequence rule and are more difficult to identify. When such a target sequence contains a k-turn sequence signature, a modified potential is applied. We test this approach in our coarse-grained modeling protocol, RAG, where RNAs are represented as graphs, thereby simplifying atomic representations and allowing rapid sampling of the candidate conformations.
Figure 2.

The 2D and 3D structures of U4 snRNA k-turn, 1E7K. The k-turn comprising three-nucleotide bulge is colored magenta, and successive GA and AG base-pairs colored green.
In brief, coarse-grained graphs representing RNA secondary structures have been introduced in the 1970s by Waterman (18) and Nussinov (19) and developed later by others (20,21). See (22,23) for recent reviews.
Graphs are mathematical objects where nodes are connected by vertices to represent various connectivity networks, such as social, economic, and transportation networks. Though simplifications of the atomic structures are involved, the number of degrees of freedom is drastically reduced (from sequence space to node space), and this makes structure enumeration, analysis, and sampling much simpler with graphs.
We have introduced the RAG (RNA-As-Graphs) database in 2004 to aid the cataloging, analyzing, and designing RNA structures (24–30). RAG translates RNA 2D structures into tree graphs by representing helices as edges, and loops (hairpins, bulges, helix ends, junctions) as vertices. See Supplementary Figure S2. Recently, we have extended RAG to 3D representations to include both connectivity of 2D structure and helical orientations in 3D space (31). Specifically, in RAG-3D, we added vertices at helix ends and centers of junctions, as well as scaled edges to reflect helix lengths. See Supplementary Figure S2. This graph-based approach has proven effective for enumerating RNA motif space (25), suggesting RNA-like motifs for design (32), identifying modular units/recurring motifs in observed RNAs (28), and assembling RNAs from fragments (27). Here, we focus on our recent application of predicting tertiary topologies of RNAs from given or predicted secondary structures using RAGTOP.
The RAGTOP (RNA-As-Graphs-Topologies) hierarchical graph sampling approach has shown promise for characterizing 3D helical arrangements in RNAs and predicting riboswitch 3D structures from a given 2D structure represented by a graph (33,34). Starting with a given RNA sequence, we predict the 2D structure using programs such as Mfold (35), RNAfold (36) and MC-fold (37), or extract 2D information from known 3D structures using programs such as RNAView (38), FR3D (39), and MC-Annotate (40). A 2D RNA graph is then built using the rules shown in Supplementary Figure S2. Then, we use our data-mining prediction program called RNAJAG (RNA-Junction-As-Graphs) to predict junction topologies based on classifications of junction families for three-way and four-way junctions (30). The junction family is predicted based on a random forest data-mining approach that classifies a family type by base content, loop length and free energy estimates of base pairs. Then 3D graphs are built and sampled by knowledge-based statistical potentials using a Monte Carlo/Simulated Annealing (MC/SA) protocol (34) to find low scored graph states. These guiding potentials include terms for bending and torsion angles of internal loops and radii of gyration patterns, and are calculated based on known RNAs. This overall structure prediction combination has shown significant improvements over current approaches (NAST (41), FARNA (42,43) and MC-Sym (37)) for predicting 3D global helical arrangements in various RNAs computed as graphs (33,34). The final graph model was previously converted into an atomic model manually, but now this process is being automated using our fragment assembly approach based on the RAG-3D search tool for similar RNA motifs (S. Jain and T. Schlick, in preparation). Thus, RAGTOP predicts a 3D graph structure from a given secondary structure and then converts that graph candidate into an atomic model.
In this study, we focus on kink-turn structures, which are special cases of internal loop topologies. Our goal is to employ sequence signatures into our graph sampling approach by using k-turn statistical potentials. Thus, we calculate two different sets of knowledge-based potentials based on two separate datasets for potential calculations: (i) a dataset of internal loops including k-turn motifs, (ii) a dataset of internal loops without k-turn motifs. These datasets are determined by identifying the k-turn motifs using the DSSR (Dissecting the Spatial Structure of RNA) software, designed for RNA structure analysis (44) over a non-redundant RNA dataset, and dividing the whole dataset into these two subsets accordingly. The non-redundant dataset contains 1445 RNA structures and was taken from the available Nucleic Acids Database (NDB) by April 2016 (45,46). We then apply our scoring function and the RAGTOP graph sampling approach to predict global topologies of 12 known RNAs, from small RNAs including only k-turn motifs to large RNAs including k-turn motifs as well as other motifs. During the RAGTOP protocol, we identify the k-turn motifs based on the consensus k-turn sequence signatures and apply k-turn specific potentials if relevant. This approach of using sequence patterns could be generalized to other motifs, as there exist many recurrent modules in RNAs, such as described by Leontis et al. (47).
MATERIALS AND METHODS
Here we describe: (i) annotation of the k-turn motifs by DSSR for a priori potential calculations, (ii) resulting potentials for k-turn and other remaining internal loops and (iii) RAGTOP protocol with annotation of the k-turn motifs by conserved sequence and secondary structure analysis of internal loops.
Annotated k-turn motifs by DSSR
We collected 1445 non-redundant PDB structures for 3D geometry analysis from NDB (45,46) representing a total of 2742 internal loops. The internal loops are identified using the DSSR (Dissecting the Spatial Structure of RNA) tool (44). In RAG, a helix is defined only if at least two consecutive Watson–Crick base pairs are present. Hence, we filtered out internal loops having only one base pair in one of the helices among the internal loops annotated by DSSR (44) and continued with remaining 1401 internal loops. Because RAG defines an unpaired nucleotide as a bulge, we identified internal loops having only one unpaired nucleotide in the bulge by our code and added those loops into our dataset. The final dataset used in generation of statistical potentials includes 1835 internal loops. Among those, 112 loops are annotated as k-turn-like. For deriving the k-turn potential, we used 66 k-turn motifs defined as normal k-turns by the DSSR tool (44). The remaining 39 were classified as either ‘undecided’ or reverse kink-turns, and seven k-turn motifs have bend angles larger than 120° according to our bend angle definition and therefore filtered out from the dataset. See Supplementary Table S1 for the list of all k-turn motifs used to derive the k-turn statistical potential.
Within this final dataset, we group internal loops into 27 different families with respect to the sizes of their single stranded regions, L and R, where L ≤ R as (see Figure 3): 0/1, 0/2, 0/3, 0/4, 0/5, 0/6+, 1/1, 1/2, 1/3, 1/4, 1/5, 1/6+, 2/2, 2/3, 2/4, 2/5, 2/6+, 3/3, 3/4, 3/5, 3/6+, 4/4, 4/5, 4/6+, 5/5, 5/6+, 6+/6+, where 6+ means greater than or equal to six (34). Figure 3 provides examples of L/R families.
Figure 3.

Examples of L/R families of internal loops.
Statistical potentials
We define our knowledge-based statistical potential 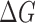 as
as
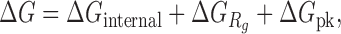 |
where 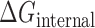 denotes the bending and torsion component,
denotes the bending and torsion component, 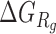 denotes radii of gyration term, and
denotes radii of gyration term, and 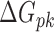 denotes the pseudoknot term.
denotes the pseudoknot term.
We calculate the bending and torsion angles of the internal loops and classify then by L and R classes to include 2D information for both k-turn-like and non k-turn-like datasets separately (34). See SI for definitions of internal loops, bend angles, and torsion angles.
The knowledge based potential for internal loops, 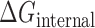 , is calculated for each L/R category based on Boltzmann statistics as
, is calculated for each L/R category based on Boltzmann statistics as 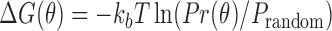 . The bend and torsion angle probabilities are calculated as number of occurrences in
. The bend and torsion angle probabilities are calculated as number of occurrences in 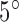 and
and 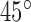 intervals, respectively, over the total number of internal loops, and the random probabilities are the uniform distribution probabilities proportional to the size of the angular intervals. See our previous work for full details (34).
intervals, respectively, over the total number of internal loops, and the random probabilities are the uniform distribution probabilities proportional to the size of the angular intervals. See our previous work for full details (34).
The total internal loop potential 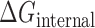 is determined by summing up the potentials for all internal loops of a structure:
is determined by summing up the potentials for all internal loops of a structure:
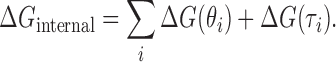 |
We calculate bend and torsion potentials for our k-turn-like dataset, non k-turn-like dataset, and all dataset separately. In Supplementary Figure S3, we plot bend and torsion angle potentials for k-turn-like, non k-turn-like, and all internal loops for categories L/R = 2/5, 2/6+ and 3/6+ since most of the k-turn-like motifs are found in these three categories. The bend angles of the kink-turns are calculated to be ∼50° as expected.
The statistical potentials also include terms for radii of gyration (as also used by Hofacker et al. (48)) and pseudoknot length. See SI for details.
RAGTOP
Our RAGTOP approach for predicting a graph candidate from a given 2D structure involves two main steps: (i) prediction of junction topologies using RNAJAG, (ii) MC/SA graph sampling guided by k-turn and non k-turn statistical potentials. We analyze the resulting ensemble using score, RMSD and clustering. The next step of RAGTOP is to generate atomic models based on the predicted graph structure (S. Jain and T. Schlick, in preparation).
Assessment of initial graphs and junction prediction
The initial tree graph is generated based on the 2D structure information given in BPSEQ format. To generate 2D structures, we use available tools such as RNAView (38), FR3D (39) and MC-Annotate (40) based on known 3D structures. We also tested our approach by predicting 2D structures by RNAfold (36) based on the dynamic programming algorithm proposed by Nussinov (49,50). We translate the given 2D structure into a graph topology. In our graph representation, helices are represented as edges and loops (hairpins, internal loops, helix ends, junctions) as vertices (31,33,34). Then, if the structure has junction(s), we use our RNAJAG tool to predict the coordinates of the junction vertices. RNAJAG predicts 3 three-way and 9 four-way junction families based on the random forests data-mining technique using three measures (adenine base content, loop length, and free energy estimates of base pairs) and determines junction families and coaxial stacking (30,34). Then, we generate the 3D graph topology from the 2D graph by adding vertices at the helical ends, and scaling edges to reflect helix lengths and base-pair content. We also add a vertex at the center of the junction to represent spatial properties of the junctions.
MC/SA graph sampling
After initial graph and junction predictions, we perform Monte Carlo/Simulated Annealing sampling until satisfactory convergence for a fixed number of steps (50,000 steps). For each move, we randomly select an internal loop, and then randomly select one of the helices of the internal loop for rotation (along with all linked vertices) along a randomly selected axis (x, y and z). We apply ‘random moves’ (see (34)) with an angle randomly selected between 0° and 360° at each MC step together with a simulated annealing cooling protocol (see below) to ensure convergence. The sample graphs are scored based on knowledge-based statistical potentials (bend and torsion angles of internal loops, radii of gyration and pseudoknot length) (33,34).
We use sequence signatures to identify all possible k-turn motifs. If the selected internal loop has a k-turn sequence signature, we use k-turn bend and torsion angle potentials and use non k-turn potentials otherwise. We search for simple k-turn sequence signatures having (i) ‘GAGG’ and ‘CC’, or (ii) ‘GAGC’ and ‘GC’, or, (iii) ‘GAGC’ and ‘GU’, or, (vi) ‘CAGC’ and ‘GC’, or (v) ‘UAGC’ and ‘GC’ sequences in their (1b, 2b, 3b, 4b) and (3n, 4n) positions (see Figure 1). We list the k-turn structure data with sequence patterns in Supplementary Table S2.
At each step, if the score is lower than that of previous conformation, the move is accepted. If it is higher, we calculate probability of acceptance at step  as
as
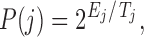 |
where 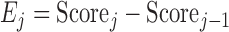 is the change in the score,
is the change in the score, 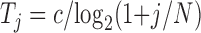 is the cooling temperature, and
is the cooling temperature, and 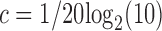 . Hence, the acceptance probability depends on the score and the effective temperature. We start with Ti = 521° K and slowly decrease the temperature until Tf = 0.015° K. In general, the number of accepted moves is higher at high temperatures and decreases at low temperatures. The move is accepted if the acceptance probability is higher than a randomly generated number between 0 and 1. We further extended the simulations to 105 steps to check if we reached a minimum (data not shown). The system is cooled until T = 0.009° K. The number of accepted moves is around 300–400 steps after the first 50,000 steps. We also apply a steric clash criteria based on the minimum distance between any two edges of a graph. If the minimum distance is <1 Å, the move is rejected.
. Hence, the acceptance probability depends on the score and the effective temperature. We start with Ti = 521° K and slowly decrease the temperature until Tf = 0.015° K. In general, the number of accepted moves is higher at high temperatures and decreases at low temperatures. The move is accepted if the acceptance probability is higher than a randomly generated number between 0 and 1. We further extended the simulations to 105 steps to check if we reached a minimum (data not shown). The system is cooled until T = 0.009° K. The number of accepted moves is around 300–400 steps after the first 50,000 steps. We also apply a steric clash criteria based on the minimum distance between any two edges of a graph. If the minimum distance is <1 Å, the move is rejected.
Selection of candidate graph from MC graph ensemble
After generating the pool of accepted graphs, we calculate the root mean square deviations (RMSD) using VMD (51) for a candidate graph, as follows. P1 measures the best answer among all ensemble graphs with respect to the graph of the solved structure; this is a reference value only, since the known structure cannot be considered in general. When the reference structure is not known, for use in the selection of the candidate graph, we apply two different procedures, as follows. The lowest-scored graph in the pool is selected as one candidate (C1), and the last accepted graph is selected as another candidate (C2). The RMSDs of graphs C1 and C2 with respect to graph of the solved structure are given in Table 1.
Table 1. Graph results for RNAs including k-turn and non k-turn motifs using k-turn versus general potentials.
| K-turn potentials applied | General potentials applied | |||||||
|---|---|---|---|---|---|---|---|---|
| PDB | L | RNA class | P1 (best RMSD) | C1 (best score) | C2 (last graph) | P1 (best RMSD) | C1 (best score) | C2 (last graph) |
| K-turns | ||||||||
| 1E7K_D | 17 | snRNA | 2.64 | 2.70 | 2.76 | 2.63 | 3.16 | 3.15 |
| 3SIU_C | 28 | snRNA | 2.46 | 3.23a | 3.22 | 2.28 | 4.46 | 4.45 |
| 2XEB_AB | 33 | snRNA | 3.73 | 4.37 | 4.37 | 3.61 | 6.27 | 6.05 |
| 2OZB_C | 33 | snRNA | 3.49 | 6.10 | 5.68 | 3.25 | 6.90 | 6.90 |
| 2HW8_B | 36 | mRNA | 2.33 | 3.00 | 3.04 | 2.34 | 7.16 | 6.44 |
| 1ZHO_B | 38 | mRNA | 2.45 | 2.84a | 2.90 | 2.45 | 7.11 | 6.55 |
| 2VPL_B | 48 | mRNA | 2.78 | 3.91 | 3.77 | 2.67 | 7.59 | 7.05 |
| 1U63_B | 49 | mRNA | 2.99 | 4.66 | 4.62 | 2.99 | 9.34 | 6.86 |
| 1MZP_B | 55 | rRNA | 2.68 | 4.66 | 4.62 | 3.70 | 6.72 | 6.60 |
| 2GIS_A | 94 | SAM Ribosw. | 13.58 | 17.87 | 18.07 | 13.45 | 18.31 | 18.52 |
| 3V7E_D | 127 | SAM Ribosw. | 13.15 | 21.17 | 21.23 | 13.15 | 22.26 | 22.30 |
| Non K-turns | ||||||||
| 1RLG_D | 25 | Box C/D RNA | 2.43 | 3.81 | 3.97 | 2.49 | 3.80 | 3.58 |
| 1OOA_D | 29 | Aptamer | 2.65 | 3.76 | 3.15 | 2.65 | 3.76 | 3.15 |
| 2IPY_C | 30 | IRE RNA | 2.01 | 2.22 | 2.18 | 2.01 | 2.22 | 2.18 |
| 1MJI_C | 34 | 5S rRNA | 2.38 | 3.26 | 3.24 | 2.38 | 3.44 | 3.39 |
| 1I6U_D | 37 | rRNA fragment | 1.56 | 2.44 | 2.30 | 1.61 | 2.39 | 2.39 |
| 1F1T_A | 38 | Aptamer | 1.93 | 2.77 | 2.77 | 1.96 | 2.67 | 2.65 |
| 1S03_B | 47 | mRNA | 1.94 | 4.18 | 3.87 | 1.98 | 4.29 | 4.28 |
| 1XJR_A | 47 | Viral RNA | 3.99 | 6.26 | 6.32 | 4.23 | 6.43 | 6.30 |
| 2PXB_B | 49 | SRP | 1.99 | 3.88 | 2.72 | 1.99 | 3.88 | 2.72 |
| 2OIU_P | 51 | Ribozyme ligase | 4.51 | 6.61 | 6.87 | 4.51 | 6.61 | 6.87 |
| 2HGH_B | 55 | 5S rRNA | 4.24 | 6.15 | 6.40 | 4.24 | 6.43 | 6.24 |
| 1DK1_B | 57 | rRNA fragment | 4.42 | 6.76 | 8.67 | 4.42 | 6.73 | 10.43 |
| 1MMS_C | 58 | rRNA fragment | 4.64 | 9.19 | 9.26 | 4.64 | 9.19 | 9.26 |
| 1D4R_AB | 58 | SRP | 5.95 | 8.17 | 7.87 | 5.95 | 8.17 | 7.87 |
| 1KXK_A | 70 | Group II intron | 2.99 | 4.07 | 5.48 | 3.48 | 5.52 | 4.58 |
| 1SJ4_R | 73 | HDV ribozyme | 6.51 | 7.36 | 7.92 | 6.07 | 7.00 | 7.06 |
| 1P5O_A | 77 | HCV IRES | 5.58 | 11.21 | 11.75 | 5.49 | 10.40 | 10.33 |
| 3D2G_A | 77 | TPP ribosw. | 7.16 | 16.81 | 17.05 | 6.06 | 13.11 | 13.46 |
| 2HOJ_A | 79 | TPP ribosw. | 6.63 | 17.24 | 16.24 | 6.63 | 18.06 | 16.85 |
| 2GDI_X | 80 | TPP ribosw. | 7.14 | 18.60 | 19.57 | 7.03 | 17.56 | 17.80 |
| 1LNG_B | 97 | SRP | 5.53 | 14.79 | 17.51 | 5.61 | 15.01 | 13.85 |
| 2LKR_A | 111 | U2/U6 snRNA | 14.25 | 21.82 | 22.32 | 14.25 | 18.20 | 20.66 |
| 1MFQ_A | 128 | SRP | 15.41 | 27.24 | 27.44 | 16.48 | 30.22 | 26.97 |
| 1GID_A | 158 | Group I intron | 14.66 | 26.18 | 25.65 | 14.66 | 25.13 | 26.19 |
aAll-atom models built using our fragment assembly approach (S. Jain and T. Schlick, in preparation) yield RMSDs of 3.62 Å for 3SIU, and 1.89 Å for 1ZHO, with respect to the experimental structures, as sketched in Supplementary Figure S6.
Best RMSDs when the reference structure is not known are indicated in bold in each row. After MC/SA sampling, the lowest graph RMSD with respect to the reference graphs from solved structures (P1), lowest scored graph (C1) and last accepted graph (C2) are shown.
RESULTS
In Table 1 and Supplementary Table S2, we show results of RAGTOP applications with the updated k-turn statistical potential to 30 RNAs from our previous work and five new RNAs. Our set includes 11 structures with a k-turn sequence signature (Supplementary Table S2) and 24 structures without a sequence signature (non k-turn). One of the structures (1OOA) has a reverse k-turn which is not modeled as a special pattern in this work. In reverse k-turns, the turn towards the major groove rather than the minor groove, so it bends in the opposite direction compared to canonical k-turns (52). We categorize the internal loops based on the number of nucleotides in their single stranded regions; L and R, where L ≤ R. (See Figure 3 for examples of L/R families and the Materials and Methods for details.) Our dataset includes four snRNAs of 2/5 family, four mRNAs and an rRNA of 2/6+ family and two riboswitches of 3/6+ family. In terms of identifying the k-turns, our approach works for all family classes, but these three are the most common families recognized by our sequence signature definition. Although our k-turn sequence signature definition includes most common k-turn sequences, there are some k-turns that our definition misses. This is because we only included the most common sequence signatures to avoid false k-turn predictions. See Supplementary Table S1 for the all k-turn structures and their corresponding sequences found in our dataset. In particular, snoRNA 1RLG, is missed because its sequence signature ‘GACC’, and ‘GG’ is not included in our k-turn definition. Although we have no cases where a non k-turn is classified as k-turn in this work, false positive predictions may occur in general.
RAGTOP's MC/SA procedure locates low energy configurations corresponding to the knowledge-based scoring function (see Materials and Methods). To select candidate graphs, we use two procedures: lowest scored graph (C1) and last graph in the MC/SA protocol (C2). These graphs are then compared to the graphs of the solved RNA to determine the graph RMSD for C1 and C2. In addition, we provide P1 for reference, the graph in the entire MC ensemble that has the lowest graph RMSD with respect to experimental reference graph. C1 and C2 are the predictions when the reference graph is unknown. Table 1 provides P1, C1 and C2 results based on k-turn specific potentials and general potentials for 35 structures in our set. Supplementary Figure S4 illustrates predicted graph models for all structures based on both the k-turn and non k-turn specific potentials.
We had already shown that comparing graph RMSDs is similar to comparing atomic RMSDs (30). The results in Table 1 show overall improvement compared to the general statistical potentials. Significantly, the prediction RNAs with k-turn sequence signatures improves for all k-turns. The graph RMSDs of six k-turns (2XEB, 2VPL, 1ZHO, 2HW8, 1U63 and 1MZP) decrease from 6–9 Å values to 3–4 Å. Although C1 and C2 values are higher than P1 values (when the reference is known), the predictions are good. We see that the predictions of four snRNA structures of the 2/5 family (1E7K, 2XEB, 3SIU and 2OZB) slightly improve, while the predictions of four mRNAs of the 2/6+ family (2VPL, 1ZHO, 2HW8 and 1U63) and one rRNA (1MZP) significantly improve. In Supplementary Figure S5, we present the score-RMSD landscapes as well as MC/SA score convergence for 35 structures. The improvement is also clear from score-RMSD landscapes where the landscapes indicate downhill shapes when k-turn specific potentials are used. The predictions of pseudoknot structures, two SAM riboswitches (2GIS and 3V7E) and one rRNA (1MZP), also improve with k-turn specific potentials.
To determine which RNA parts realize improvement, we show in Supplementary Table S3 the local RMSDs calculated as the RMSDs of only kink-turn parts (five vertices indicated by cyan in Supplementary Figure S4) with respect to native graph structure. The improvements are indeed in the kink-turn regions of the structures.
The results for the 24 non k-turn structures do not exhibit substantial changes. Six of them (1OOA, 2IPY, 2PXB, 2OIU, 1MMS and 1D4R) remain unchanged since the potentials remain the same for those structures. For some structures, predictions are slightly better with general potentials compared to those with k-turn and non k-turn specific potentials. The score-RMSD landscapes of non k-turn structures also remain similar (Supplementary Figure S5). Three TPP riboswitches (3D2G, 2HOJ and 2GDI) have k-junction motifs, i.e., a kink-turn motif in a three-way junction (8). In this work, we only consider the kink-turn motifs in internal loops, because our potential accounts for motifs in internal loops only.
To demonstrate that the k-turn potential is effective on structures not included in the k-turn dataset, we also performed a 10-fold cross validation procedure for our kink-turn dataset. The entire dataset (Supplementary Table S1) is divided into 10 subsets randomly, then nine subsets are used to obtain training parameters, and one is used for testing. The procedure is repeated 10 times for each tenth of the dataset. The results (Supplementary Table S4) are in excellent agreement with the overall results presented in Table 1. That is, the difference is <0.5 Å.
In this work, we have used 2D structures derived from 3D information using available tools, namely RNAView (38), FR3D (39) and MC-Annotate (40), since our goal is to evaluate the k-turn potential's performance. However, we also experimented with using predicted 2D structures from sequence using RNAfold (36). The results are given in Supplementary Table S5. The predicted 2D structure is different from the true structure for some cases, resulting in RNA graphs with different number of vertices compared to the known graph. In such cases, we used the align function of PyMOL (The PyMOL Molecular Graphics System, Shrödinger LLC, http://www.pymol.org) to calculate graph RMSDs. The align function performs a sequence alignment followed by a structural superposition, and repeats refinement cycles to reject outliers. We set the number of cycles to zero to avoid outlier rejection and considered all vertices in graph RMSD calculations. From the comparisons in Supplementary Table S5, we see that when the predicted 2D structure is the same as the known structure (∼10 out of 35 total), the results are the same (1E7K, 3SIU, 2OZB, 2HW8, 1ZHO, 1OOA, 2IPY, 1MJI, 1F1T, 1XJR). When the predicted structure is different but close to the known structure (about 15 out of 35 total), we obtain somewhat higher RMSD values. However, using the kink-turn potential still improves the predictions. When the predicted 2D structure is incorrect, as in ∼10 of the total structures, the predictions are also poor.
To illustrate our automated fragment assembly modeling approach to generate all-atom models from candidate graphs (S. Jain and T. Schlick, in preparation), we use two examples, an mRNA and a snRNA (1ZHO and 3SIU, respectively). In Supplementary Figure S6, we show the steps taken for the RNA graph candidates C1 given in Table 1: graph partitioning by RAG-3D (31); assembly using 10 best matching graphs and their corresponding all-atom models using common loops; adjustment of bases, helix and loop lengths; geometry optimization by PHENIX (53); and ranking of final model with respect to C1. The all-atom RMSDs are 1.89 Å for 1ZHO, and 3.62 Å for 3SIU.
DISCUSSION
K-turn consensus sequence signatures are classic examples of sequence/structure motifs in RNAs. We have presented a first attempt to use sequence signatures for generating motif specific statistical potentials. We developed a priori knowledge-based potentials from known RNA structures and generated k-turn (and non k-turn) specific potentials. First, we predict initial graph geometries based on 2D topologies and junction predictions. Second, we sample graph topologies using MC/SA simulations, score them based on sequence signatures, using derived statistical potentials. When compared, results with k-turn specific potentials yield superior results to those with general potentials. More generally, other RNA sequence/motif patterns could be used to improve statistical potentials and predictions of RNA structures similarly.
The graph RMSDs are positively correlated to all-atom RMSDs as shown previously (30). Furthermore, the scores and graph RMSDs are positively correlated as shown in Supplementary Figure S5. Hence, the lowest scored graphs (C1) and the last accepted graphs (C2) in the MC/SA simulations are good candidates for all-atom models.
Although the junction topologies are mostly predicted well by RNAJAG at the beginning of RAGTOP, some predictions are imperfect due to different helical orientations. For example, for riboswitches 2GIS and 3V7E, the distances between helical arms are not predicted well, although the junction family and helical stacking are predicted correctly. Thus, the MC/SA procedure optimizes the internal loops while the junction prediction remains unchanged. In the future, extended moves to junctions and hairpins could be introduced to model different angular orientations of junction topologies. Furthermore, RNA junction families could also be extended by considering more families to represent the flexible helical orientations.
One of the difficulties in using sequence signatures is that it is not always possible to find an exact conserved sequence pattern, because many cases exist where the sequence breaks the general rules. One example is complex kink-turns (4). Some k-turns have unusual sequence variants: examples include Kt-23 from the 16S rRNA of T. thermophiles, and Kt-7 from the 23S rRNA of the H. marismortui ribosome (2). In such cases, the G–A pair of the (2b, 2n) positions is different.
Many RNA 3D motifs have been identified so far (54). The RNA 3D Motif Atlas developed by Leontis et al. (47,54) is one resource that is continuously updated. The motifs in the Motif Atlas are identified based on conserved interactions and overall geometry of the structure. Other resources that classify or extract RNA motifs also exist (55–58). However, the lack of integrated resources for annotating RNA motifs automatically remains a challenge in RNA modeling and design (59).
Besides kink-turns, motifs such as C-loops, sarcin-ricin motifs, RNA/protein or RNA/ligand binding sites also could be included in knowledge-based potentials in combination with structure prediction. RNA sequence and 2D structure properties are important in the identification of binding partners. Hence, including these properties could improve RNA structure prediction. Our results support the notion that employing motif specific parameters based on sequence/structure features could improve RNA structure prediction.
The importance of RNA motif prediction in RNA structure prediction was also emphasized in RNA-Puzzles II, a collective and blind experiment in 3D RNA structure prediction (60). That is, it was found that predictions improve using various motif search tools, such as a sequence-based motif prediction tool RMDetect (55) that can search for G-bulge loop, kink-turn, C-loop and tandem-GA loop; or JAR3D (61) that searches RNA motifs. However, these tools are not currently integrated with structure prediction tools for RNA. Nonetheless, as the RNA motif world becomes more annotated, sequence signatures for various motifs could define a strong foundation for structure prediction from sequence.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Swati Jain for providing data of all-atom models for our candidate graphs based on ongoing work (S. Jain and T. Schlick, in preparation).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of General Medical Sciences; National Institutes of Health (NIH) [GM100469, GM081410 to T.S.]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Klein D., Schmeing T., Moore P., Steitz T.. The kink‐turn: a new RNA secondary structure motif. EMBO J. 2001; 20:4214–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schroeder K.T., McPhee S.A., Ouellet J., Lilley D.M.. A structural database for k-turn motifs in RNA. RNA. 2010; 16:1463–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Daldrop P., Lilley D.M.. The plasticity of a structural motif in RNA: structural polymorphism of a kink turn as a function of its environment. RNA. 2013; 19:357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lilley D.M. The K-turn motif in riboswitches and other RNA species. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2014; 1839:995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang L., Lilley D.M.. The molecular recognition of kink-turn structure by the L7Ae class of proteins. RNA. 2013; 19:1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang L., Lilley D.M.J.. Structure of a rare non-standard sequence k-turn bound by L7Ae protein. Nucleic Acids Res. 2014; 42:4734–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McPhee S.A., Huang L., Lilley D.M.J.. A critical base pair in k-turns that confers folding characteristics and correlates with biological function. Nat. Commun. 2014; 5:5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J., Daldrop P., Huang L., Lilley D.M.. The k-junction motif in RNA structure. Nucleic Acids Res. 2014; 42:5322–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang L., Lilley D.M.J.. The kink turn, a key architectural element in RNA structure. J. Mol. Biol. 2016; 428:790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang L., Wang J., Lilley D.M.J.. A critical base pair in k-turns determines the conformational class adopted, and correlates with biological function. Nucleic Acids Res. 2016; 44:5390–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Breaker R.R. Riboswitches and the RNA world. Cold Spring Harbor Perspect. Biol. 2012; 4:a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barrick J.E., Breaker R.R.. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007; 8:R239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Serganov A., Patel D.J.. Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat. Rev. Genet. 2007; 8:776–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang L., Lilley D.M.J.. A quasi-cyclic RNA nano-scale molecular object constructed using kink turns. Nanoscale. 2016; 8:15189–15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Winkler W.C., Grundy F.J., Murphy B.A., Henkin T.M.. The GA motif: an RNA element common to bacterial antitermination systems, rRNA, and eukaryotic RNAs. RNA. 2001; 7:1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsumura S., Ikawa Y., Inoue T.. Biochemical characterization of the kink‐turn RNA motif. Nucleic Acids Res. 2003; 31:5544–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nissen P., Ippolito J.A., Ban N., Moore P.B., Steitz T.A.. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:4899–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waterman M.S. Secondary structure of single-stranded nucleic acids. Adv. Math. Suppl. Stud. 1978; 1:167–212. [Google Scholar]
- 19. Le S.-Y., Nussinov R., Maizel J.V.. Tree graphs of RNA secondary structures and their comparisons. Comput. Biomed. Res. 1989; 22:461–473. [DOI] [PubMed] [Google Scholar]
- 20. Shapiro B.A., Zhang K.. Comparing multiple RNA secondary structures using tree comparisons. Comput. Applic. Biosci.: CABIOS. 1990; 6:309–318. [DOI] [PubMed] [Google Scholar]
- 21. Benedetti G., Morosetti S.. A graph-topological approach to recognition of pattern and similarity in RNA secondary structures. Biophys. Chem. 1996; 59:179–184. [DOI] [PubMed] [Google Scholar]
- 22. Kim N., Petingi L., Schlick T.. Network theory tools for RNA modeling. WSEAS Trans. Math. 2013; 9:941. [PMC free article] [PubMed] [Google Scholar]
- 23. Kim N., Fuhr K.N., Schlick T.. Biophysics of RNA Folding. 2013; Springer; 23–51. [Google Scholar]
- 24. Fera D., Kim N., Shiffeldrim N., Zorn J., Laserson U., Gan H.H., Schlick T.. RAG: RNA-As-Graphs web resource. BMC Bioinformatics. 2004; 5:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gan H.H., Fera D., Zorn J., Shiffeldrim N., Tang M., Laserson U., Kim N., Schlick T.. RAG: RNA-As-Graphs database—concepts, analysis, and features. Bioinformatics. 2004; 20:1285–1291. [DOI] [PubMed] [Google Scholar]
- 26. Izzo J.A., Kim N., Elmetwaly S., Schlick T.. RAG: an update to the RNA-As-Graphs resource. BMC Bioinformatics. 2011; 12:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koessler D.R., Knisley D.J., Knisley J., Haynes T.. A predictive model for secondary RNA structure using graph theory and a neural network. BMC Bioinformatics. 2010; 11:S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim N., Izzo J.A., Elmetwaly S., Gan H.H., Schlick T.. Computational generation and screening of RNA motifs in large nucleotide sequence pools. Nucleic Acids Res. 2010; 38:e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gopal A., Zhou Z.H., Knobler C.M., Gelbart W.M.. Visualizing large RNA molecules in solution. RNA. 2012; 18:284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laing C., Jung S., Kim N., Elmetwaly S., Zahran M., Schlick T.. Predicting helical topologies in RNA junctions as tree graphs. PLoS ONE. 2013; 8:e71947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zahran M., Bayrak C.S., Elmetwaly S., Schlick T.. RAG-3D: a search tool for RNA 3D substructures. Nucleic Acids Res. 2015; 43:9474–9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim N., Shiffeldrim N., Gan H.H., Schlick T.. Candidates for novel RNA topologies. J. Mol. Biol. 2004; 341:1129–1144. [DOI] [PubMed] [Google Scholar]
- 33. Kim N., Zahran M., Schlick T.. Shi-Jie C, Donald HB-A. Methods in Enzymology. 2015; 553:Academic Press; 115–135. [DOI] [PubMed] [Google Scholar]
- 34. Kim N., Laing C., Elmetwaly S., Jung S., Curuksu J., Schlick T.. Graph-based sampling for approximating global helical topologies of RNA. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:4079–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hofacker I.L., Stadler P.F.. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics. 2006; 22:1172–1176. [DOI] [PubMed] [Google Scholar]
- 37. Parisien M., Major F.. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008; 452:51–55. [DOI] [PubMed] [Google Scholar]
- 38. Yang H., Jossinet F., Leontis N., Chen L., Westbrook J., Berman H., Westhof E.. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003; 31:3450–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Petrov A.I., Zirbel C.L., Leontis N.B.. WebFR3D—a server for finding, aligning and analyzing recurrent RNA 3D motifs. Nucleic Acids Res. 2011; 39:W50–W55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gendron P., Lemieux S., Major F.. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001; 308:919–936. [DOI] [PubMed] [Google Scholar]
- 41. Jonikas M.A., Radmer R.J., Laederach A., Das R., Pearlman S., Herschlag D., Altman R.B.. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009; 15:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Das R., Baker D.. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:14664–14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Das R., Karanicolas J., Baker D.. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods. 2010; 7:291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu X.-J., Bussemaker H.J., Olson W.K.. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015; 43:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berman H.M., Olson W.K., Beveridge D.L., Westbrook J., Gelbin A., Demeny T., Hsieh S.H., Srinivasan A.R., Schneider B.. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992; 63:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coimbatore Narayanan B., Westbrook J., Ghosh S., Petrov A.I., Sweeney B., Zirbel C.L., Leontis N.B., Berman H.M.. The Nucleic Acid Database: new features and capabilities. Nucleic Acids Res. 2014; 42:D114–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Petrov A.I., Zirbel C.L., Leontis N.B.. Automated classification of RNA 3D motifs and the RNA 3D motif atlas. RNA. 2013; 19:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kerpedjiev P., Höner zu Siederdissen C., Hofacker I.L.. Predicting RNA 3D structure using a coarse-grain helix-centered model. RNA. 2015; 21:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nussinov R., Pieczenik G., Griggs J.R., Kleitman D.J.. Algorithms for loop matchings. SIAM J. Appl. Math. 1978; 35:68–82. [Google Scholar]
- 50. Nussinov R., Jacobson A.B.. Fast algorithm for predicting the secondary structure of single-stranded RNA. Proc. Natl. Acad. Sci. U.S.A. 1980; 77:6309–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 52. Strobel S.A., Adams P.L., Stahley M.R., Wang J.. RNA kink turns to the left and to the right. RNA. 2004; 10:1852–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. et al. . PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parlea L.G., Sweeney B.A., Hosseini-Asanjan M., Zirbel C.L., Leontis N.B.. The RNA 3D Motif Atlas: computational methods for extraction, organization and evaluation of RNA motifs. Methods. 2016; 103:99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cruz J.A., Westhof E.. Sequence-based identification of 3D structural modules in RNA with RMDetect. Nat. Methods. 2011; 8:513–519. [DOI] [PubMed] [Google Scholar]
- 56. Gardner P.P., Eldai H.. Annotating RNA motifs in sequences and alignments. Nucleic Acids Res. 2015; 43:691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chojnowski G., Waleń T., Bujnicki J.M.. RNA Bricks—a database of RNA 3D motifs and their interactions. Nucleic Acids Res. 2014; 42:D123–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roll J., Zirbel C.L., Sweeney B., Petrov A.I., Leontis N.. JAR3D Webserver: Scoring and aligning RNA loop sequences to known 3D motifs. Nucleic Acids Res. 2016; 44:W320–W327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pyle A.M., Schlick T.. Opportunities and Challenges in RNA Structural Modeling and Design. Biophys. J. 2017; doi:10.1016/j.bpj.2016.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miao Z., Adamiak R.W., Blanchet M.-F., Boniecki M., Bujnicki J.M., Chen S.-J., Cheng C., Chojnowski G., Chou F.-C., Cordero P. et al. . RNA-Puzzles Round II: assessment of RNA structure prediction programs applied to three large RNA structures. RNA. 2015; 21:1066–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zirbel C.L., Roll J., Sweeney B.A., Petrov A.I., Pirrung M., Leontis N.B.. Identifying novel sequence variants of RNA 3D motifs. Nucleic Acids Res. 2015; 43:7504–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.