


Abstract
Myrf is a key transcription factor for oligodendrocyte differentiation and central nervous system myelination. We and others have previously shown that Myrf is generated as a membrane protein in the endoplasmic reticulum (ER), and that it undergoes auto-processing to release its N-terminal fragment from the ER, which enters the nucleus to work as a transcription factor. These previous studies allow a glimpse into the unusual complexity behind the biogenesis and function of the transcription factor domain of Myrf. Here, we report that Myrf N-terminal fragments assemble into stable homo-trimers before ER release. Consequently, Myrf N-terminal fragments are released from the ER only as homo-trimers. Our re-analysis of a previous genetic screening result in Caenorhabditis elegans shows that homo-trimerization is essential for the biological functions of Myrf N-terminal fragment, and that the region adjacent to the DNA-binding domain is pivotal to its homo-trimerization. Further, our computational analysis uncovered a novel homo-trimeric DNA motif that mediates the homo-trimeric DNA binding of Myrf N-terminal fragments. Importantly, we found that homo-trimerization defines the DNA binding specificity of Myrf N-terminal fragments. In sum, our study elucidates the molecular mechanism governing the biogenesis and function of Myrf N-terminal fragments and its physiological significance.
INTRODUCTION
In the vertebrate central nervous system (CNS), nerve axons are tightly wrapped by myelin sheaths—the specialized plasma membrane domains of oligodendrocytes (OLs) (1–4). Myelin sheaths are critical to the rapid propagation of electrical signals along the axons. Myelin also provides the axons with essential trophic support (5,6). Myelin sheaths develop in the CNS as OLs undergo terminal differentiation, implicating OL differentiation as a crucial event for CNS myelination. In addition, OL differentiation represents a key rate-limiting step of remyelination in pathological conditions (7,8). Thus, insights into the regulatory mechanisms governing the differentiation of OLs are of profound importance for finding/improving cure for demyelinating disease as well as understanding normal CNS development.
Genetic studies have indicated that Myrf is a transcription factor indispensable for OL differentiation and CNS myelination (9,10). Consistently, recent studies have shown that signals that promote or inhibit myelination converge on Myrf, suggesting that Myrf serves as a rheostat for myelin growth and maintenance (11,12). Further, Richardson and colleagues have demonstrated that Myrf-governed myelin generation is pivotal to the acquisition of new motor skills in adult mice (13).
Unexpectedly, we and the Emery laboratory have found that Myrf is generated as a type-II membrane protein in the endoplasmic reticulum (ER) (14,15). Through a sophisticated sequence analysis, we discovered that Myrf harbors a domain distantly homologous to the intramolecular chaperone domain of bacteriophage tailspike proteins. In recognition of its well-characterized biochemical functions in viral proteins (16–18), we have proposed the name ICA (Intramolecular Chaperone Auto-cleavage) for this domain. During virus particle assembly, the ICA domain, which is part of phage tailspike proteins, induces the homo-trimerization of phage tailspike proteins by chaperoning the formation of a triple β-helix (the Intramolecular Chaperone function). Homo-trimeric phage tailspike proteins are then auto-cleaved by the ICA domain (the Auto-cleavage function). Strikingly, the ICA domain appears to carry out the same molecular feat for the proteolytic processing of Myrf (14,15). Namely, the ICA domain drives the homo-oligomerization of Myrf in the ER membrane. Homo-oligomeric Myrf is proteolyzed by the ICA domain, releasing its N-terminal fragments from the ER membrane. Freed Myrf N-terminal fragments enter the nucleus to work as a transcription factor. Functional assays showed that the auto-cleavage of Myrf by the ICA domain is essential for both its transcriptional activity and the differentiation of OLs in vitro (14,15).
These previous studies allow a glimpse into the unusual complexity behind the biogenesis and function of the Myrf transcription factor domain (i.e. its N-terminal fragment). However, many important mechanistic and functional issues remain to be elucidated. For example, how is the homo-trimeric complex of Myrf N-terminal fragments generated and maintained? Is homo-trimerization essential for the biological functions of Myrf N-terminal fragments? If so, why? Do Myrf N-terminal fragments bind to DNA as a homo-trimer? In this study, we have combined computational and experimental methodologies to address these issues. Our study elucidates the molecular mechanisms underlying the homo-trimerization of Myrf N-terminal fragments and the homo-trimeric DNA binding of Myrf N-terminal fragments. We further show that homo-trimerization is essential for the biological functions of Myrf N-terminal fragments, at least partly because it defines their DNA binding specificity.
MATERIALS AND METHODS
Constructs
Whenever a distinction needs to be made, the human and mouse proteins are referred to as hMyrf and mMyrf, respectively. Otherwise, we use Myrf as a generic term. The hMyrf cDNA (the 1111-amino-acid-long isoform [CCDS ID: 31579 and RefSeq ID: NP_037411]) was described previously (15). The mMyrf cDNA that encodes the 1139-amino-acid-long isoform was kindly provided by Dr Ben Emery. The hMyrf and mMyrf cDNAs were cloned into pcDNA3 and an in-house vector with an IRES-EGFP (internal ribosome entry site-enhanced green fluorescent protein) cassette. Luciferase reporters were generated by cloning rat genomic fragments into pGL3-promoter (Promega). All mutagenesis was carried out by a PCR-based method, and sequence information was verified by Sanger sequencing.
Animal procedures, tissue harvest and cell culture
Animal husbandry was carried out in accordance with Institutional Animal Care and Use Committee-approved protocols. Pregnant rats were purchased from Envigo. Oligodendrocyte precursor cells (OPCs) were purified from rat pups of P7–P9 by immunopanning (19). Primary OPCs and CG4 cells (20) were kept in a proliferative condition by supplementing the Sato media (19) with PDGF (10 μg/mL), NT3 (1 μg/mL), and CNTF (10 μg/mL). Primary OPCs and CG4 cells were maintained in a humidified 8% CO2 incubator at 37°C. HeLa and HEK293FT cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and maintained in a humidified 5% CO2 incubator at 37°C. Transient transfection for OPCs, CG4, HeLa and HEK293FT cells was performed using Lipofectamine 2000 as per the manufacturer's instructions.
Immuoprecipitation & immunoblotting
Cells were washed once with TBS and lysed with a lysis buffer (100 mM Tris pH 7.4, 300 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, protease inhibitor cocktail (Roche), 2 mM PMSF). Cell lysate was sonicated and spun down at 14 000g for 10 min at 4°C. Cleared lysate was mixed with antibody-coated beads (Sigma) and incubated for 1 h at 4°C on a rotating plate. The mixture was spun down at 6000g for 30 s, and the sup fraction was collected. The bead fraction was washed with a washing buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.25% Triton X-100). For the sequential immunoprecipitation experiment, proteins bound to the beads were eluted by peptides (0.8 mg/ml) with gentle agitation at 4°C for 30 min. The eluate was collected by centrifuge for 30 s at 6000g. The eluate was then mixed with an appropriate type of beads, together with equal volume of 5% skim milk that acts as a blocking reagent. The mixture was gently agitated at cold room for 1 h. If necessary, elution by peptide competition and incubation with another type of beads was repeated. Reagents used for immunoprecipitation are as follows: monoclonal mouse anti-FLAG® M2, HRP conjugated (Sigma #A8592, 1:5000), mouse anti-HA tag, 6E2, HRP conjugated (Cell signaling #2999, 1:10 000), mouse anti-Myc tag IgG, clone 9E11, HRP conjugated (Santa Cruz #sc-40, 1:5000), and mouse anti-V5 tag IgG (Abcam #ab27671, 1:5000). Samples were mixed with Laemmli sample buffer and boiled at 95°C for 5 min. Upon sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred to PVDF and probed with primary and HRP-conjugated secondary antibodies.
Immunofluorescence
Cells were cultured on coverglass and fixed with 4% formaldehyde and permeabilized with 0.1% Triton X-100. Upon blocking with 1% BSA, they were incubated with primary antibodies diluted in blocking buffer at 4°C overnight, followed by incubation with fluorochrome-conjugated secondary antibodies. Nuclei were stained with Hoechst 33342 (Invitrogen). Fluorescence was visualized with a Leica DMi8 microscope with an ORCA-Flash4.0 sCMOS camera. Reagents used for immunofluorescence are as follows: monoclonal ANTI-FLAG® M2 antibody (Sigma #F1804, 1:1000), anti-Myc tag (Abcam #ab9106, 1:1000), anti-GFP (Abcam #ab290, 1:5000), anti-MBP (Millipore #MAB386, 1:500), goat anti-rabbit, Alexa Fluor® 594 conjugate (ThernoFisher #A11037, 1:10 000), donkey anti-mouse, Alexa Fluor® 488 conjugate (ThernoFisher # A21202, 1:10 000), goat anti-rat, Alexa Fluor® 594 conjugate (ThernoFisher #A21209, 1:10 000), and donkey anti-rabbit, Alexa Fluor® 488 conjugate (ThernoFisher # A21206, 1:10 000).
In vitro differentiation assay of primary OPCs
DNA plasmids that express Myrf cDNA or its variants fused to the IRES-EGFP cassette were transiently transfected into primary OPCs using Lipofectamine 2000 (Invitrogen). Transfected OPCs were kept in a proliferative condition for 2 days that deters spontaneous differentiation. Cells were immunostained for EGFP and myelin basic protein (MBP). For each cDNA construct, 100 random fields were counted in a blind fashion.
Luciferase assay
Luciferase assays were performed by using the Promega dual luciferase reporter assay kit as per the manufacturer's instructions. Cells were co-transfected with pGL3-promoter-based luciferase reporters, pRL-TK (an internal control), and Myrf-expressing plasmids. The rn4 genomic coordinates of the luciferase reporters are as follows: 126 (chr1:169920248–169920647), and 320 (chr10:71034166-71034749).
DNA pulldown assay
HEK293FT cells were transfected with hMyrf, mMyrf, or their variants. Cell lysate was cleared by centrifugation at 15 000g for 20 min at 4°C. Biotinylated duplex oligonucleotides were conjugated to Dynabeads (Invitrogen) in buffer A (5 mM Tris pH 8.0, 0.5 mM EDTA, 1 M NaCl). Oligonucleotide-conjugated beads were washed twice with 500 μl of buffer A and three times with buffer C (20 mM Tris pH 8.0, 1 mM EDTA, 10% glycerol, 1 mM DTT, 50 mM NaCl). 300 μg of cell lysate were incubated with oligonucleotide-conjugated beads in buffer C and sheared salmon sperm DNA (final concentration 0.2 mg/ml) for 20 min at room temperature with rotation. The mixture was spun down to separate into the bead and the sup fractions. The bead fraction was washed five times with 500 μl buffer C, and both fractions were analysed by immunoblotting. The DNA sequences used are as follows. The Myrf motif is underlined, and the mutated portions are shaded by grey.
RESULTS
Myrf N-terminal fragments form homo-trimers
Our previous study concluded that Myrf N-terminal fragments form homo-trimers based on the size exclusion chromatography of a truncated hMyrf protein that was expressed and purified in E. coli (15). This conclusion was corroborated by the recent electrophoretic mobility shift assay from Küspert et al. (21). To gain further support for the homo-trimerization of Myrf N-terminal fragments, we tried SDS-PAGE with a low percentage of SDS. Homo-trimerization of Myrf N-terminal fragments is predicted to be driven by a triple-β helix (see below). The triple β-helix is a structural motif where three polypeptide chains are intertwined around a 3-fold common axis, leading to strong homo-trimerization (18) (Figure 1A). When we carried out SDS-PAGE at 0.1% of SDS, which did not affect protein migration as judged by the separation of size markers, we were able to observe high-molecular-weight (HMW) species (Figure 1C). The electrophoretic mobility of the HMW species from the wild-type protein (mMyrf, Figure 1B) was the same as that of the mMyrf-Δ861-1139 or mMyrf-Δ921-1139 HMW species, but not the mMyrf-Δ1-116 HMW species. Further, when the mMyrf sample was boiled before loading, its HMW species disappeared. Thus, the HMW species are specific complexes of mMyrf N-terminal fragments. The electrophoretic mobility of mMyrf N-terminal fragments is abnormal in that we consistently detect them at ∼90 kDa, even though their theoretical molecular weight is 64 kDa. Owing to the uncertainty associated with the determination of molecular weights from electrophoretic mobility, the result in Figure 1C did not allow us to determine the exact homo-oligomerization status of Myrf N-terminal fragments.
Figure 1.

Myrf N-terminal fragments form homo-trimers. (A) A triple β-helix (derived from PDB ID: 3GW6). (B) Schematic diagram of the constructs. (C) The constructs in panel B were expressed in HEK293FT cells. Cell lysate was subject to SDS-PAGE with 0.1% of SDS in a cold room. (D) Sequential immunoprecipitation. Flag-, Myc-, HA-, and V5-mMyrf were co-expressed in HEK293FT cells. Cell lysate was subject to immunoprecipitation with Flag beads, generating the ‘1F’ sample. Bound proteins were eluted by Flag peptides. The eluate was subject to immunoprecipitation with Myc beads, along with 5% milk to block non-specific binding, generating the ‘2M’ sample. Bound proteins were eluted by Myc peptides. One half of the eluate was subject to immunoprecipitation with HA beads, along with 5% milk, generating the ‘3H’ sample. The other half was subject to immunoprecipitation with Flag beads, along with 5% milk, generating the ‘3F’ sample. (E) Control immunoprecipitation experiment to confirm that HA- and V5-mMyrf N-terminal fragments can be found in the same complexes. (F) Control immunoprecipitation experiments to confirm the specificity of each immunoprecipitation step of the sequential immunoprecipitation experiment. Of note, Group 1 of Control 4 is the sequential immunoprecipitation experiment shown in panel D, which is why the immunoblot result in panel D is replicated here. IB: immunoblotting.
We turned to an alternative approach, which is sequential immunoprecipitation. We co-expressed Flag-mMyrf, Myc-mMyrf, HA-mMyrf and V5-mMyrf in HEK293FT cells. The four tags were all attached to the N terminus for the detection of Myrf N-terminal fragments. Cell lysate was subject to immunoprecipitation with Flag beads (1F in Figure 1D), and bound proteins were eluted by Flag peptides. Subsequently, the eluate was subject to immunoprecipitation with Myc beads (2M in Figure 1D), and bound proteins were eluted by Myc peptides. Finally, one half of the eluate was subject to immunoprecipitation with HA beads (3H in Figure 1D) while the other half to immunoprecipitation with Flag beads (3F in Figure 1D). If mMyrf N-terminal fragments formed homo-trimers, the N-terminal fragments of Flag-mMyrf, Myc-mMyrf, HA-mMyrf and V5-mMyrf would not all be found in the same complexes. This means that V5 signals would be detected in the 3H-sup fraction, but not the 3H-bead fraction. Consistent with this prediction, the N-terminal fragments of V5-mMyrf were found exclusively in the 3H-sup fraction (Figure 1D). For this result to serve as proof that Myrf N-terminal fragments form homo-trimers, the homo-trimerization of Myrf N-terminal fragments should be irreversible in that once they form homo-trimers, they may be dissociated into monomers, but monomeric Myrf N-terminal fragments do not reassemble into homo-trimers. Our previous study showed that this is indeed the case (15). Further, this is consistent with the computational prediction that the homo-trimerization of Myrf N-terminal fragments is driven by the triple-β helix. The triple-β helix is such an elaborate structure that the intramolecular chaperone function of the ICA domain is essential for its formation. Once full-length Myrf protein undergoes auto-cleavage, the N-terminal fragment cannot benefit from the intramolecular chaperone function of the ICA domain any longer (Figure 1B). Consequently, the homo-trimers of Myrf N-terminal fragments may be dissociated, but dissociated monomers cannot reassemble into homo-trimers.
For each fraction analysed, the same amount was loaded for the four immunoblots. Thus, the lack of V5 signals in the 3H-bead fraction was not due to insufficient sample loading because the other three immunoblots for the same fraction generated clear signals (Figure 1D). The lack of V5 signals in the 3H-bead fraction may be explained by the possibility that complexes of Myrf N-terminal fragments were significantly dissociated during the third immunoprecipitation. We rejected this possibility for two reasons. First, the N-terminal fragments of V5-mMyrf were abundantly detected in the 3F-bead fraction, indicating that complexes containing the N-terminal fragments of Flag-, Myc- and V5-mMyrf remained intact. Second, the N-terminal fragments of Flag- and Myc-mMyrf were found in the 3H-bead fraction, demonstrating that complexes containing the N-terminal fragments of Flag-, Myc- and HA-mMyrf were stable. Under the homo-tetramer hypothesis, the expected ratio of V5 signals between the 3H-bead and the 3H-sup fractions is 1:2 in our experimental condition, which does not agree with the observed ratio (Figure 1D). Finally, one may argue that the lack of V5 signals in the 3H-bead fraction is due to the mutual exclusion of the N-terminal fragments with HA and V5 tag from the same complexes. Our control experiment, however, revealed that the N-terminal fragments of HA- and V5-mMyrf can be found in the same complexes if the second Myc immunoprecipitation step is omitted (Figure 1E). Critical to this sequential immunoprecipitation experiment was to ensure that there is no non-specific binding in each immunoprecipitation step. We optimized several experimental variables to eliminate non-specific binding. Indeed, control experiments showed that the three immunoprecipitation steps were without significant non-specific contaminants (Figure 1F). Overall, our sequential immunoprecipitation experiment demonstrates that only up to three different types of Myrf N-terminal fragments (distinguished by different tags) can be found in the same complexes, indicating that Myrf N-terminal fragments form homo-trimers.
The auto-cleavage of Myrf proceeds in cis
Full-length Myrf proteins form homo-oligomers in the ER membrane (15), upon which the ICA domain carries out an auto-cleavage reaction to release the N-terminal fragments from the ER membrane. A catalytic dyad of the ICA domain consisting of a lysine and a serine residue performs the auto-cleavage reaction (14,15). For hMyrf (the human protein), K583 serves as a base that activates the hydroxyl group of S578, and the activated serine cleaves the peptide bond that connects it to P577 (15,18). It remains unknown whether the lysine residue of a Myrf polypeptide in the homo-oligomer pairs with the serine residue from the same polypeptide (cis configuration) or a different one (trans configuration) for the auto-cleavage reaction (Figure 2A). For a related intramolecular chaperone domain of bacteriophage ϕ29, the catalytic dyad is formed by residues that belong to different polypeptides, which are brought together by homo-trimerization (22). To determine whether this is also the case for Myrf, we examined the auto-cleavage pattern of Flag-hMyrf (Figure 2B) and Myc-hMyrf-K583A (a non-cleavable mutant, Figure 2B) in HeLa cells. When expressed alone, Flag-hMyrf underwent auto-cleavage while Myc-hMyrf-K583A did not (Figure 2C), as reported previously (14,15). If the auto-cleavage of Myrf proceeded in trans, Myc-Myrf-K583A could undergo proteolytic processing in the presence of Flag-hMyrf. We found that it was not the case (Figure 2C), indicating that the catalytic dyad consists of the serine and lysine residues that belong to the same Myrf polypeptide. Unexpectedly, however, we found that the post-translational modifications of Flag-hMyrf N-terminal fragments were altered in the presence of co-expressed Myc-hMyrf-K583A (see Discussion).
Figure 2.

The auto-cleavage of Myrf proceeds in cis. (A) The catalytic dyad of hMyrf consists of S578 and K583. In the cis configuration, both catalytic residues come from the same Myrf polypeptide in the homo-oligomer. In the trans configuration, they come from different polypeptides. (B) Schematic diagram of the constructs. (C) Flag-hMyrf and Myc-hMyrf-K583A were transfected into HeLa cells alone or together to distinguish between the cis or trans configuration. Cell lysate was subject to SDS-PAGE. IB: immunoblotting.
Myrf N-terminal fragments form stable homo-trimers before ER release
The above result demonstrates that each Myrf polypeptide in the homo-oligomer is cleaved by its own catalytic dyad. In light of our finding above that Myrf N-terminal fragments form homo-trimers, does this mean that cleaved Myrf N-terminal fragments are released from the ER individually and then assemble into homo-trimers in the cytoplasm or nucleus? Given our previous finding that Myrf N-terminal fragments, if expressed directly as a truncated construct, do not self-associate (15) and the sequence homology with viral proteins (see below), we prefer the alternative hypothesis that Myrf N-terminal fragments assemble into stable homo-trimers prior to ER release, with the implication that they are released from the ER only as homo-trimers. This hypothesis predicts that if there were a non-cleavable Myrf mutant in the homo-oligomer, even cleaved Myrf N-terminal fragments would not be released from the ER membrane. To test this prediction, we determined the subcellular localization of the N-terminal fragments of wild-type and a non-cleavable Myrf in CG4 cells (a widely used oligodendrocyte cell line) (20). When expressed alone, the N-terminal fragment of Flag-hMyrf was localized to the nucleus while that of Myc-hMyrf-K583A was excluded from it (Figure 3A), as previously reported (14,15). Remarkably, when co-expressed with Myc-hMyrf-K583A, the N-terminal fragment of Flag-hMyrf was frequently excluded from the nucleus (Figure 3A). Cell counts showed that the fraction of cells with extra-nuclear FLAG signals were significantly increased upon co-expression of the uncleavable mutant (P value < 5.72 × 10−119 by the binomial test, Figure 3B). Together with the data in Figure 2, these results indicate that, in the presence of non-cleavable Myrf mutants, wild-type Myrf is proteolyzed normally, yet fails to be released from the ER.
Figure 3.

Myrf N-terminal fragments form homo-trimers prior to ER release. (A) Flag-hMyrf and Myc-hMyrf-K583A were transfected into CG4 cells alone or together to determine the subcellular localization of their N terminal fragments. Scale bar, 10 μm. (B) The immunofluorescence result in panel A was quantified by cell counting. The binomial test showed that the N-terminal fragments of Flag-hMyrf were found outside the nucleus more frequently in the presence of co-expressed Myc-hMyrf-K583A (P value < 5.72 × 10−119). (C) Flag-mMyrf and Myc-mMyrf-K592A were co-transfected into HEK293FT cells. Cell lysate (‘L’) was subject to immunoprecipitation with Myc beads, leading to the bound (‘B’) and sup (‘S’) fractions. Flag-mMyrf-Δ750-1139 was used for a control immunoprecipitation experiment. (D) Two luciferase reporters (126 and 320), chosen from the published Myrf ChIP-seq data (14), responded well to transfected Myrf. The response of pGL3-promoter was set to 1, and the reported values are means and standard errors of 3 biological replicates. *P < 0.05 by two-tailed one sample Student's t test with Bonferroni correction for multiple hypothesis testing. (E) Luciferase assay determining how the co-expression of mMyrf-K592A affects the transcriptional activity of mMyrf or mMyrf-Δ750-1139. The transcriptional activity of mMyrf was set to 1, and the reported values are means and standard errors of 4 biological replicates. Two-way ANOVA showed that there was a significant interaction between the type of mMyrf (mMyrf or mMyrf-Δ750-1139) and the amount of co-expressed mMyrf-K592A. *P < 7.5 × 10−6 for 320, and P < 0.01 for 126. Post-hoc analysis indicated that mMyrf-K592A significantly suppressed the transcriptional activity of mMyrf, but not mMyrf-Δ750–1139. *P < 0.0003 for 320 and P < 0.02 for 126. IP: immunoprecipitation. IB: immunoblotting.
To corroborate our conclusion, we determined whether cleaved Myrf N-terminal fragments bind to non-cleavable full-length Myrf proteins in the ER membrane. Myc-mMyrf-K592A (equivalent to hMyrf-K583A) was expressed in HEK293FT cells together with either Flag-mMyrf or Flag-mMyrf-Δ750-1139 (Figure 3C). Cell lysate was subject to immunoprecipitation with Myc beads. A significant fraction of Flag-mMyrf N-terminal fragments bound to Myc beads (Figure 3C), revealing their physical association with the non-cleavable mutants in the ER membrane. As a control experiment, we looked into the physical association between Myc-mMyrf-K592A and the N-terminal fragment of Flag-mMyrf-Δ750–1139 (a mutant mMyrf truncated before the transmembrane domain). Owing to this truncation, it is not integrated into the ER membrane, but still undergoes homo-oligomerization and auto-cleavage (15). The N-terminal fragments of Flag-mMyrf-Δ750-1139 were not associated with Myc-mMyrf-K592A in the ER membrane (Figure 3C). These results highlight the interaction specificity between Flag-mMyrf N-terminal fragments and Myc-mMyrf-K592A in the ER membrane. Overall, we conclude that Myrf N-terminal fragments assemble into stable homo-trimers before ER release.
Non-cleavable Myrf suppresses the transcriptional activity of wild-type Myrf
The foregoing finding predicts that non-cleavable Myrf mutants would suppress the transcriptional activity of wild-type Myrf because they prevent the ER release of wild-type Myrf N-terminal fragments, abrogating their transcriptional activity in the nucleus. To test this hypothesis, we performed luciferase assay in CG4 cells. Two Myrf-bound genomic regions were chosen from the published Myrf ChIP-seq (chromatin immunoprecipitation coupled with high-throughput sequencing) data (14) and cloned into pGL3-promoter to be used as Myrf luciferase reporters (reporters 126 and 320). They responded well to transfected mMyrf (Figure 3D). Using these reporters, we determined how mMyrf-K592A impacts the transcriptional activity of mMyrf. mMyrf-Δ750–1139 was used as a control because mMyrf-K592A did not bind to its N-terminal fragments (Figure 3C) and thus would not affect its transcriptional activity. For both reporters, two-way ANOVA (analysis of variance) revealed that there was a significant interaction between the type of mMyrf (mMyrf versus mMyrf-Δ750–1139) and the amount of co-expressed mMyrf-K592A (Figure 3E, *P < 7.5 × 10−6 and 0.01 for 320 and 126, respectively). Post-hoc analysis showed that mMyrf-K592A suppressed the transcriptional activity of mMyrf, but not mMyrf-Δ750-1139 (*P < 0.0003 and 0.02 for 320 and 126, respectively), supporting our hypothesis that non-cleavable Myrf mutants interfere with the transcriptional activity of wild-type Myrf.
The bridge region between the DNA-binding and ICA domains assembles Myrf N-terminal fragments into stable homo-trimers
What would drive the homo-trimerization of Myrf N-terminal fragments? Sequence homology with viral proteins predicts that the bridge region between the DNA-binding and ICA domains might be key to the homo-trimeric interaction (Figure 4A), most likely by adopting the triple β-helix conformation (15–18). We found strong evidence that supports our hypothesis by re-analyzing a previously published genetic screening result in C. elegans.
Figure 4.

Homo-trimerization is mediated by the bridge region between the DNA-binding and ICA domains and is essential for the functions of Myrf N-terminal fragments. (A) Multiple sequence alignment of the bridge region. (B) The G566R mutation did not interfere with the proteolytic processing of hMyrf. (C) An immunoprecipitation experiment showed that hMyrf N-terminal fragments self-associate, as reported before (14,15). (D) The self-association of hMyrf N-terminal fragments is not observed for the mutants. Same constructs with Flag and Myc tags were co-expressed in HEK293FT cells. Cell lysate (L) was subject to immunoprecipitation with Flag beads, leading to the bound (B) and sup (S) fractions. (E) The transcriptional activity of mMyrf-G575R is much weaker than that of mMyrf and is in fact as low as that of pcDNA3. The transcriptional activity of mMyrf was set to 1, and the reported values are means and standard errors of three biological replicates. *P < 0.001 by two-tailed one sample Student's t test with Bonferroni correction. (F and G) mMyrf-G575R failed to drive the in vitro differentiation of primary rat OPCs. EGFP, mMyrf or mMyrf-G575R was transfected into primary rat OPCs. After two days of culture in a proliferation condition that deters spontaneous differentiation, we determined the fraction of transfected cells (marked by EGFP) that had differentiated to express MBP. The reported values are means and standard errors of 5 biological replicates. *P < 0.05 by two-tailed unpaired Student's t test with Bonferroni correction. Scale bar, 10 μm. IP: immunoprecipitation. IB: immunoblotting.
Russel and colleagues performed a genome-wide mutagenesis screen to find genes important for molt regulation in C. elegans (23). From a screen of >105 mutagenized genomes, they identified PQN-47 (the C. elegans ortholog of Myrf) as a key gene regulating molt. Worms homozygous for the let-25 allele (a mutant PQN-47 allele) exhibited a fully penetrant L1 (larval stage 1) arrest and molting defective phenotype. This phenotype was reproduced in PQN-47 null worms, suggesting that the mutant PQN-47 protein encoded by the let-25 allele is functionally null. The let-25 allele specifies the substitution of an absolutely conserved glycine residue with arginine. However, the molecular basis for the striking phenotype of the let-25 allele remained unknown. We found that the glycine residue mutated in the let-25 allele corresponds to G566 for hMyrf (or G575 for mMyrf). G566 of hMyrf is located in the bridge region (Figure 4A), where a triple β-helix is thought to be formed for the homo-trimerization of Myrf N-terminal fragments. G566 is believed to be critical to the packing interaction that sustains the triple β-helix conformation. A replacement by arginine would put three bulky side chains as well as three positive charges in the packed space, greatly destabilizing the putative triple β-helix. Consequently, the N-terminal fragment of hMyrf-G566R would not be able to maintain its homo-trimerization status.
To test this exciting hypothesis, we expressed Flag-hMyrf-G566R in HeLa cells and found that it is normally processed (Figure 4B). However, there were changes in the post-translational modifications of the N-terminal fragment, similar to hMyrf co-expressed with hMyrf-K583A in Figure 2C. Strikingly, our immunoprecipitation experiment revealed that the N-terminal fragment of hMyrf-G566R exists as a monomer (Figure 4D), unlike the wild-type protein (Figure 4C). Consistently, the N-terminal fragments of mMyrf-G575R (equivalent to hMyrf-G566R) failed to form HMW species in our SDS-PAGE with 0.1% of SDS (Figure 1C). The C-terminal fragment of hMyrf-G566R formed homo-oligomers (data not shown), suggesting that G566R specifically disrupts the homo-trimerization of the N-terminal fragment. These results support the hypothesis that the bridge region is critical to the homo-trimerization of Myrf N-terminal fragments. To corroborate this conclusion, we made a mutant, hMyrf-Δ549–566, where the region spanning residues G549–G566 is deleted (Figure 4A). Since this is the bridge region postulated to be critical to the homo-trimerization of Myrf N-terminal fragments, the N-terminal fragment of hMyrf-Δ549-566 would exist as a monomer. We found that this is the case (Figure 4D). To elucidate the specific effect of the G566R mutation, we designed two further mutants: G566D to maintain the charge repulsion while minimizing the effect of steric hindrance, and G566W to maintain the steric hindrance while minimizing the effect of charge repulsion. The N-terminal fragments of both mutants were found to exist as monomers (Figure 4D), suggesting that both charge repulsion and steric hindrance contribute to the disruption created by the G566R mutation. Overall, these results demonstrate that the bridge region is pivotal to the homo-trimerization of Myrf N-terminal fragments.
Homo-trimerization is essential for the function of Myrf N-terminal fragments
The lethal phenotype of the let-25 allele in C. elegans strongly suggests that homo-trimerization is essential for the biological functions of Myrf N-terminal fragments, even a matter of life or death. To test this hypothesis, we performed luciferase assay in CG4 cells where the transcriptional activity of mMyrf was compared with that of mMyrf-G575R. For the two reporters 126 and 320, the transcriptional activity of mMyrf-G575R was significantly lower than that of mMyrf (Figure 4E). In fact, the transcriptional activity of mMyrf-G575R was as low as that of pcDNA3 (empty vector), indicating that monomeric Myrf N-terminal fragments completely failed to elicit transcription for the two reporters. Western blot analysis of the luciferase samples for the reporter 320 showed that the wild-type and mutant proteins were expressed at similar levels (Figure 4E). The same was true for the luciferase samples for the reporter 126 (data not shown). However, as noted above for the human protein, the post-translational modifications of mMyrf-G575R N-terminal fragments were altered compared to mMyrf N-terminal fragments (Figure 4E). We also confirmed that the N-terminal fragments of mMyrf-G575R were localized to the nucleus (data not shown). Taken together, these results demonstrate that homo-trimerization is indispensable for the transcriptional activity of Myrf N-terminal fragments.
To determine whether homo-trimerization is also essential for Myrf to drive the in vitro differentiation of primary OPCs, we performed a differentiation assay with primary OPCs purified from rat pups by immunopanning (19). Primary rat OPCs were transfected with EGFP, mMyrf, or mMyrf-G575R. Transfected OPCs were cultured for 2 days in a proliferative condition that deters spontaneous differentiation. We found that 13% of the cells transfected with mMyrf had differentiated to express MBP, a mature OL marker (Figure 4F and G). In contrast, about 1% of the cells transfected with mMyrf-G575R had differentiated to express MBP, a level similar to the transfection of EGFP. These results highlight that homo-trimerization is required for Myrf N-terminal fragments to drive OL differentiation in vitro.
A novel homo-trimeric DNA motif explains the homo-trimeric DNA binding of Myrf N-terminal fragments
Since we did not detect significant differences in the steady-state protein expression and nuclear localization of the N-terminal fragment between mMyrf and mMyrf-G575R, we wondered whether homo-trimerization is essential for the DNA binding of Myrf N-terminal fragments. To address this issue, we need to know the DNA motif recognized by the homo-trimer of Myrf N-terminal fragments. A previous study presented solid biochemical data indicating that the DNA motif CTGG[C/T]AC is critical to the DNA binding and transcriptional activity of Myrf N-terminal fragments (14). However, it is hard to imagine how this motif would mediate the homo-trimeric DNA binding of Myrf N-terminal fragments. Does this suggest that the homo-trimer of Myrf N-terminal fragments uses only one of its three DNA-binding domains to bind to DNA? To resolve this enigma, we re-analyzed the published Myrf ChIP-seq data (14). Surprisingly, our computational analysis uncovered a new DNA motif (Figure 5A, e-value: 1.9 × 10−148 by MEME (24)). The new motif comprises three degenerate copies of C[A/T]GGCA[C/G], with each consecutive pair arranged as an inverted repeat. The new motif is hereafter referred to as the Myrf motif. C[A/T]GGCA[C/G], the underlying unit of the Myrf motif, is similar to the previous motif CTGG[C/T]AC. The Myrf motif is highly attractive for two reasons. First, its trimeric architecture is consistent with our findings that Myrf N-terminal fragments exist as homo-trimers and function as such. Second, the Myrf motif seamlessly accounts for the biochemical data presented for the previous motif because the Myrf motif incorporates it. Of note, our MEME run also found two other motifs. But they are just simple runs of A's and found only in few sequences, and thus we did not pursue them further.
Figure 5.

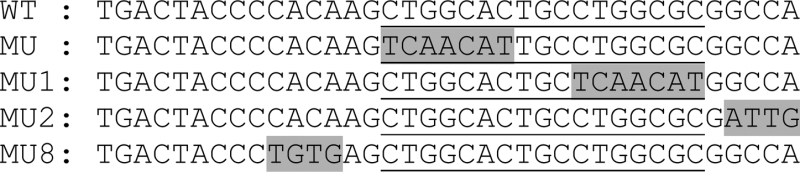
The Myrf motif mediates the homo-trimeric DNA binding of Myrf N-terminal fragments. (A) The Myrf motif identified by re-analysis of the published Myrf ChIP-seq data (14) by MEME (24). The three underlying units are marked to highlight the homo-trimeric architecture of the Myrf motif. (B) An incidence of the Myrf motif found near the gene Rffl (the underlined bases in WT). A rat genomic fragment (rn4 chr10:71034166–71034749) that includes this Myrf motif incidence was cloned into pGL3-promoter and used as a luciferase reporter to validate the Myrf motif. For a functional validation of the Myrf motif, the Myrf motif incidence was mutated as shown (MU through MU9). (C) The Myrf motif was validated by luciferase assay in CG4 cells. The ratio between firefly and renilla luciferase levels was determined to measure transcriptional output. The reported values are means and standard errors of 4 biological replicates. *P < 0.01 by two-tailed unpaired Student's t test with Bonferroni correction. (D) To extend the validity of the Myrf motif to endogenous Myrf, the motif validation experiment was repeated in rat OPCs without exogenous Myrf. Vector means pGL3-promoter. Transfected OPCs were cultured in a differentiation condition for 3 days to induce the expression of endogenous Myrf. The reported values are means and standard errors of 6 biological replicates. *P < 0.01 by two-tailed unpaired Student's t test with Bonferroni correction. (E) DNA pulldown assay using duplex oligonucleotides corresponding to the Myrf motif incidence shown in panel B. The homo-trimeric complex of hMyrf N-terminal fragments, while avidly bound to WT, MU2 and MU8, did not bind to MU and MU1. The sup fractions show that similar amounts of hMyrf N-terminal fragments were present in the binding reactions.
To validate the Myrf motif, we performed luciferase assay in CG4 cells. For the sake of inter-study comparability, we used the same luciferase reporter that was used for the validation of the previous motif (rn4 chr10:71034166–71034749) (14). This rat genomic fragment contains one incidence of the Myrf motif (the underlined bases in WT, Figure 5B). It also has a match of the previous motif (the first seven underlined bases in WT, Figure 5B) (14). The previous study showed that when the DNA sequence of WT is mutated to that shown for MU (Figure 5B), mMyrf neither elicits transcription from it nor binds to it (14). We successfully replicated these results (Figure 5C and E), setting the stage for further mutagenesis to thoroughly validate the Myrf motif. We now find that the DNA segment mutated in MU constitutes the first unit of the Myrf motif. Thus, this result may well be interpreted as validating the first unit of the Myrf motif. To validate the second and third units of the Myrf motif and thus its trimeric architecture, we mutated the second and third units as shown in Figure 5B (MU1). There was as little transcription from MU1 as from MU (Figure 5C), demonstrating that the second and third units of the Myrf motif are as critical to the transcriptional activity of Myrf as the first unit. Mutations outside of the Myrf motif had negligible effects on the transcriptional activity of Myrf (MU2 and MU8, Figure 5C), validating the Myrf motif. To probe the nature of interaction between Myrf N-terminal fragments and DNA, we mutated contiguous segments of the Myrf motif (MU3∼MU7 and MU9, Figure 5B). Most segments had significant effects on the transcriptional activity of mMyrf, suggesting that the homo-trimer of Myrf N-terminal fragments makes extensive contacts with DNA for transcriptional regulation. The above data were obtained by transfecting CG4 cells with mMyrf-expressing plasmids. To extend the validity of the Myrf motif to endogenous Myrf, we repeated the motif validation experiment with primary rat OPCs. They were transfected with reporter plasmids and cultured for 3 days in a condition that induces the expression of endogenous Myrf (withdrawal of PDGF and addition of T3) (19). We found the Myrf motif to respond to endogenous Myrf similarly to transfected Myrf (Figure 5D).
To directly show that the Myrf motif mediates the sequence-specific DNA binding of Myrf N-terminal fragments, we carried out a DNA binding assay. Flag-hMyrf was expressed in HEK293FT cells, and cell lysate was incubated with magnetic beads coated with WT DNA fragments shown in Figure 5B. hMyrf N-terminal fragments, which exist as homo-trimers, avidly bound to WT (Figure 5E). Consistent with the luciferase assay results, they also strongly bound to MU2 and MU8, but not MU and MU1. Immunoblotting of the sup fractions showed that similar amounts of Myrf N-terminal fragments were present in the binding reactions (Figure 5E). In sum, the luciferase and DNA binding assays demonstrate that the Myrf motif mediates the homo-trimeric DNA binding of Myrf N-terminal fragments.
Detailed analysis of the Myrf motif - prevalence
During its run, MEME randomly sampled 600 peaks from a total of 2052 Myrf ChIP-seq peaks and found 268 of them to have matches of the Myrf motif (Figure 6A), suggesting that the Myrf motif is found in about 45% of the Myrf peaks. As a further way of addressing the prevalence of the Myrf motif among the Myrf peaks, we used FIMO (25), scanning them to find motif matches. The matches were ranked by the FIMO score that reflects how good a match is. 72% of the 2052 Myrf ChIP-seq peaks were found to have at least one motif match with a FIMO score ≥6 (Figure 6A). When we restricted the analysis to those peaks that overlap with broad H3K27ac peaks (26), an activating histone modification mark, about 80% of them were found to have such matches. For experimental analysis, we randomly chose 5 Myrf peaks with FIMO scores of 6 ∼ 17. They were cloned into pGL3-promoter. We determined how they respond to transfected mMyrf, with pGL3-promoter serving as a negative control (Vector, Figure 6B). They all responded well to mMyrf. When the Myrf motif was mutated, four of them stopped responding to mMyrf (MUAll, Figure 6B), indicating that the Myrf motif mediates the transcriptional activity of Myrf N-terminal fragments at these four peaks. Their FIMO scores are 17.7, 12.9, 6.4 and 6.3, respectively, revealing that motif matches with a FIMO score of ∼6 can serve as bona fide Myrf recognition sites. Together with the computational analysis by MEME, we conclude that the Myrf motif mediates the transcriptional activity of Myrf at a majority of the Myrf-bound genomic regions.
Figure 6.

Detailed analysis of the Myrf motif. (A) MEME randomly chose 600 Myrf ChIP-seq peaks from a total of 2052 for its run and found 268 of them to have an incidence of the Myrf motif. As a further way of evaluating the prevalence of the Myrf motif, FIMO was used to scan the 2052 Myrf ChIP-seq peaks, searching for matches of the Myrf motif. Motif matches were ranked by the FIMO score, and a cumulative distribution was computed. (B) Luciferase assay was performed for an in-depth analysis of the Myrf motif. Four motif matches were cloned into pGL3-promoter. The rn4 genomic coordinates are as follows: 126 (chr1:169920248–169920647), 1463 (chr4:123025896–123026295), 1030 (chr2:28381432–28381831) and 1491 (chr4:147568152–147568551). The numbers in parentheses are FIMO scores. For each motif match, the first, second, and third unit was selectively mutated in MU1, MU2 and MU3, respectively. MUAll combines the mutations of MU1, MU2 and MU3. For all samples, mMyrf-expressing plasmids and pRL-TK were co-transfected. pGL3-promoter was used as a control (Vector). The transcriptional readout from the WT sequence was set to 1, and the reported values are means and standard errors of 4 biological replicates. *P < 0.05 by two-tailed one sample Student's t test with Bonferroni correction. (C) DNA pulldown assay using duplex oligonucleotides corresponding to the Myrf motif incidence shown in Figure 5B. The N-terminal fragments of Flag-hMyrf-G566R (which exist as monomers) bound to WT, but very weakly. Further, they failed to distinguish between WT and MU, unlike their homo-trimeric counterparts. IB: immunoblotting.
Detailed analysis of the Myrf motif - the contribution of each unit
Given the degenerate nature of the Myrf motif, it is of interest to dissect the contribution of each of the three units toward its overall activity. Bases at positions 7 and 11–13 of the Myrf motif belong to more than one unit (Figure 5A). Consistently, they are among the most information-rich bases of the Myrf motif. To delineate the contribution of each unit to the overall activity of the Myrf motif, we disrupted individual units of the Myrf motif (Figure 6B) by mutating the bases at positions other than 7 and 11–13. This mutagenesis scheme was applied to the four peaks for which we successfully identified the Myrf motif incidence mediating the transcriptional activity of Myrf N-terminal fragments. In all cases, the unit on the 5΄ side was designated as the first unit. Selective disruption of the first unit significantly suppressed the transcriptional activity of Myrf for the peaks 1463 and 1030 (MU1, Figure 6B). It was also the case for the reporter 320 (Figure 5C). The disruption of the second unit significantly impaired the transcriptional activity of Myrf for the peaks 1463, 1030, and 1491 (MU2, Figure 6B). Lastly, when the third unit was mutated, the transcriptional activity of Myrf was significantly decreased for the peaks 126, 1463, and 1030 (MU3, Figure 6B). It should be noted that our selective disruption of individual units is only partial because the bases at positions 7 and 11–13 still contribute to the binding of Myrf N-terminal fragments to the mutated units. Thus, for example, one cannot conclude that one unit instead of three is sufficient for the peaks 126 and 1491 (see also Discussion). Rather, a sound conclusion for these results would be that the contribution of individual units is not uniform across the Myrf-bound genomic regions, most likely reflecting context-specific interactions with other transcription factors for coordinated transcriptional regulation.
Homo-trimerization defines the DNA binding behaviour of Myrf N-terminal fragments
The discovery of the Myrf motif enabled us to determine whether homo-trimerization is essential for the DNA binding of Myrf N-terminal fragments. Flag-hMyrf-G566R was expressed in HEK293FT cells, and cell lysate was incubated with magnetic beads conjugated with various DNA fragments. For a comparative analysis, we carried out the same experiment for Flag-hMyrf simultaneously. The DNA binding pattern of Flag-hMyrf N-terminal fragments reported in Figure 5E was reproduced (Figure 6C). When we compared the DNA binding pattern of Flag-hMyrf N-terminal fragments with that of Flag-hMyrf-G566R N-terminal fragments, two interesting points emerged. First, the N-terminal fragments of hMyrf-G566R, which exist as monomers, bound to the WT DNA sequence, but very weakly compared to those of hMyrf, which exist as homo-trimers (Figure 6C). This weak DNA binding was reproducibly observed in repeat experiments. This result indicates that homo-trimerization is important for tight DNA binding. Second, and more importantly, the N-terminal fragments of hMyrf-G566R bound to MU as strongly as to WT, unlike their homo-trimeric counterparts (Figure 6C). This promiscuous DNA binding was also reproducibly observed in repeat experiments. This result reveals that homo-trimerization imparts transcriptional specificity to Myrf N-terminal fragments. As shown in Figure 5B, the first and second units of the Myrf motif are mutated in MU, yet the third unit remains intact. Thus, monomeric Myrf N-terminal fragments recognize MU as a cognate binding site and bind to it. However, the homo-trimeric complex of Myrf N-terminal fragments does not recognize it as such, underscoring the importance of homo-trimerization in distinguishing between genuine and spurious spots. Taken together, these results demonstrate that homo-trimerization is indispensable for the tight sequence-specific DNA binding of Myrf N-terminal fragments. This is likely to explain why homo-trimerization is essential for the biological functions of Myrf N-terminal fragments.
DISCUSSION
Mechanistic model for the biogenesis of the Myrf transcription factor domain
Myrf is a key transcription factor for OL differentiation and CNS myelination (9,10). Previous studies have shown that Myrf is generated as a type-II membrane protein in the ER, that Myrf undergoes auto-processing to release its N-terminal fragment from the ER membrane, and that the freed N-terminal fragment enters the nucleus to work as a transcription factor (14,15). Based on the data presented in this study, we extend this model in three ways. First, the auto-cleavage of full-length Myrf homo-oligomers in the ER membrane proceeds in cis. Second, Myrf N-terminal fragments form stable homo-trimers prior to ER release. Therefore, even though each Myrf polypeptide in the homo-oligomer is cleaved by its own catalytic dyad, Myrf N-terminal fragments are released from the ER in an all-or-none fashion. If all Myrf polypeptides in the homo-oligomer undergo auto-cleavage, Myrf N-terminal fragments are released from the ER as homo-trimers. Otherwise, nothing is released from the ER, even cleaved Myrf N-terminal fragments. This all-or-none release behavior allows non-cleavable Myrf mutants to interfere with the transcriptional activity of wild-type Myrf by blocking its ER release. Third, the bridge region between the DNA-binding and ICA domains is pivotal to the homo-trimerization of Myrf N-terminal fragments, most likely by adopting the triple β-helix conformation. When the bridge region is mutated (e.g., the G566R, Δ549-566, G566D, and G566W mutations), the N-terminal fragments fail to form homo-trimers, existing as monomers.
Functional significance of the homo-trimerization of Myrf N-terminal fragments
Our re-analysis of a genetic screening result in C. elegans (23) led us to find that homo-trimerization is essential for the biological functions of Myrf N-terminal fragments. Russel and colleagues isolated the let-25 allele, a mutant allele of PQN-47 (the worm ortholog of Myrf), in their genetic screen for genes important for molt regulation in C. elegans. Worms homozygous for the let-25 allele exhibited a fully penetrant lethal phenotype at the molt between L1 and L2. The lethal phenotype was reproduced in PQN-47 null worms, suggesting that the mutant PQN-47 protein encoded by the let-25 allele is functionally null. The let-25 allele mutates an absolutely conserved glycine residue into arginine, which corresponds to G566 of hMyrf or G575 of mMyrf. However, the molecular basis for the striking phenotype of the let-25 allele remained unknown. In this study, we demonstrate that the net effect of the let-25 allele is to disrupt the homo-trimerization of Myrf N-terminal fragments, implicating that homo-trimerization is indispensable for the biological functions of Myrf N-terminal fragments. Consistently, we found that monomeric Myrf N-terminal fragments are incapable of inducing transcription and driving the in vitro differentiation of primary rat OPCs.
Homo-trimeric DNA binding of Myrf N-terminal fragments
Our mechanistic and functional analyses clearly indicate that Myrf N-terminal fragments form homo-trimers and work as such, making it highly likely that they bind to DNA as a homo-trimer. However, a previous study demonstrated that the DNA motif CTGG[C/T]AC mediates the DNA binding and transcriptional activity of Myrf N-terminal fragments (14). It was difficult to imagine how this motif would support the homo-trimeric DNA binding of Myrf N-terminal fragments. A biochemical study on MrfA (the amoeba ortholog of Myrf) indicated that MrfA is most likely to bind to DNA as a homo-oligomer (27). We re-analyzed the published Myrf ChIP-seq data to elucidate the DNA binding mode of Myrf N-terminal fragments. To our surprise, our computational analysis uncovered a novel homo-trimeric DNA motif, which was subsequently validated by thorough luciferase and DNA binding assays. The new motif, which we termed the Myrf motif, strongly supports the notion that Myrf N-terminal fragments bind to DNA as a homo-trimer. It further suggests that the Myrf homo-trimer makes extensive contacts with DNA. Importantly, the Myrf motif seamlessly accounts for the solid biochemical data presented for the previous motif because the Myrf motif incorporates it.
Notably, the Myrf motif is quite degenerate, suggesting that the importance of each unit of the Myrf motif may vary for different Myrf-occupied genomic regions. Consistently, selective disruption of individual units did not have the same impact on the transcriptional activity of Myrf for the 4 different Myrf peaks (Figure 6B). Therefore, not all DNA contacts of the homo-trimer of Myrf N-terminal fragments would be equally critical, and dimeric matches (i.e. imperfect matches to the Myrf motif that consist of two units) may be functional at some genomic sites. In fact, we speculate that the degeneracy of the Myrf motif might have been exploited by nature to enhance the synergistic interaction between Myrf and other transcription factors for coordinated transcriptional regulation. On the other hand, it is interesting to contrast this loose requirement for trimericity on the DNA side with the absolute requirement of homo-trimerization for the Myrf protein side, as illustrated by mMyrf-G575R (Figure 4E and F).
Why is homo-trimerization critical to Myrf N-terminal fragments?
Homo-trimeric transcription factors are extremely rare. Heat shock factor 1 may be the only known example for eukaryotes (28). Why is homo-trimerization so important for the biological functions of Myrf N-terminal fragments? We did not observe significant differences in the steady-state protein expression and nuclear localization between monomeric and homo-trimeric Myrf N-terminal fragments. Thus, we turned to the possibility that homo-trimerization may affect the DNA binding of Myrf N-terminal fragments. The discovery of the Myrf motif enabled us to pursue this hypothesis. We found that homo-trimerization is critical to the DNA binding of Myrf N-terminal fragments for two reasons. First, homo-trimerization imparts transcriptional specificity to Myrf N-terminal fragments. The homo-trimer of Myrf N-terminal fragments distinguishes between genuine and spurious spots while the monomeric counterpart is not so good at it. Second, homo-trimerization enables Myrf N-terminal fragments to bind to target sites tightly.
We speculate that the unusual specificity conferred by homo-trimerization is what distinguishes Myrf from other OL transcription factors. The DNA motifs for Olig2 and Sox10, two other essential OL transcription factors, are much simpler compared to the Myrf motif. Olig2 and Sox10 have been reported to bind to more than 15 000 genomic regions (26,29,30). This number is in stark contrast to ∼2000 reported for Myrf (14). This small number of genomic binding sites may stem from the technical limitations of the Myrf ChIP-Seq dataset (14), which was derived by transfecting primary OPCs with tagged mMyrf. However, it is tempting to speculate that it may be due to the increased interaction specificity between Myrf N-terminal fragments and DNA. In this vein, Myrf may work as a ‘sieve’. Under this hypothesis, Olig2 and/or Sox10, which are already expressed in OPCs, bind to genomic sites superfluously in OPCs, priming them for later activation. As OPCs exit the cell cycle and undergo differentiation, Myrf starts to be expressed and selectively activates only those sites that are necessary for OL differentiation and CNS myelination. This selective activation may derive from the highly specific DNA binding of Myrf N-terminal fragments. Our model is consistent with the observation that Myrf is not required for the genomic localization of Olig2 and Sox10 (29). Our computational analysis shows that ∼50% of the Myrf ChIP-seq peaks are pre-bound by Olig2 in OPCs and/or immature OLs (26), and that Olig2 leaves these sites as Myrf begins to occupy them (unpublished observation).
Unexpectedly, we found that Myrf N-terminal fragments undergo post-translational modifications in a homo-trimerization-dependent manner in the nucleus. When the ER release of Myrf N-terminal fragments was blocked, their pattern of post-translational modifications was altered (Figure 2C). The same phenomenon was observed for monomeric Myrf N-terminal fragments (Figure 4B). It remains unknown what sort of post-translational modifications Myrf N-terminal fragment undergoes and their functional significance. Our future study will elucidate the post-translational modifications of Myrf N-terminal fragments, their role in OL differentiation and CNS myelination, and how homo-trimerization impacts them. Given the apparent effect of homo-trimerization on the post-translational modifications of Myrf N-terminal fragments, homo-trimerization-dependent post-translational modifications may be another reason why homo-trimerization is indispensable for the biological functions of Myrf N-terminal fragments.
ACKNOWLEDGEMENTS
We are grateful to Dr Ben Emery for providing us with the mMyrf cDNA and teaching us how to purify OPCs from rat pups by immunopanning. We thank Drs Lawrence Wrabetz, Laura Feltri and Fraser Sim for their helpful comments on the manuscript. We are also grateful to the anonymous reviewers for their critical comments.
Footnotes
Present address: Patrick Busch, Weill Medical College of Cornell University, New York, NY 10021, USA.
FUNDING
University at Buffalo [startup fund to Y.P.]; National Institutes of Health [R01NS094181 to Y.P.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bercury K.K., Macklin W.B.. Dynamics and mechanisms of CNS myelination. Dev. Cell. 2015; 32:447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nave K.-A., Werner H.B.. Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014; 30:503–533. [DOI] [PubMed] [Google Scholar]
- 3. Aggarwal S., Yurlova L., Simons M.. Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol. 2011; 21:585–593. [DOI] [PubMed] [Google Scholar]
- 4. Miller R.H. Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol. 2002; 67:451–467. [DOI] [PubMed] [Google Scholar]
- 5. Lee Y.J., Morrison B.M., Li Y., Lengacher S., Farah M.H., Hoffman P.N., Liu Y.T., Tsingalia A., Jin L., Zhang P.W. et al. . Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012; 487:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Funfschilling U., Supplie L.M., Mahad D., Boretius S., Saab A.S., Edgar J., Brinkmann B.G., Kassmann C.M., Tzvetanova I.D., Mobius W. et al. . Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012; 485:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Franklin R.J.M., ffrench-Constant C.. Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 2008; 9:839–855. [DOI] [PubMed] [Google Scholar]
- 8. Back S.A., Rosenberg P.A.. Pathophysiology of glia in perinatal white matter injury. Glia. 2014; 62:1790–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Emery B., Agalliu D., Cahoy J.D., Watkins T.A., Dugas J.C., Mulinyawe S.B., Ibrahim A., Ligon K.L., Rowitch D.H., Barres B.A.. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009; 138:172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koenning M., Jackson S., Hay C.M., Faux C., Kilpatrick T.J., Willingham M., Emery B.. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. J. Neurosci. 2012; 32:12528–12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ishii A., Furusho M., Dupree J.L., Bansal R.. Role of ERK1/2 MAPK signaling in the maintenance of myelin and axonal integrity in the adult CNS. J. Neurosci. 2014; 34:16031–16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang H.-J., Vainshtein A., Maik-Rachline G., Peles E.. G protein-coupled receptor 37 is a negative regulator of oligodendrocyte differentiation and myelination. Nat. Commun. 2016; 7:10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKenzie I.A., Ohayon D., Li H., de Faria J.P., Emery B., Tohyama K., Richardson W.D.. Motor skill learning requires active central myelination. Science. 2014; 346:318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bujalka H., Koenning M., Jackson S., Perreau V.M., Pope B., Hay C.M., Mitew S., Hill A.F., Lu Q.R., Wegner M. et al. . MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS Biol. 2013; 11:e1001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Z.H., Park Y., Marcotte E.M.. A bacteriophage tailspike domain promotes self-cleavage of a human membrane-bound transcription factor, the myelin regulatory factor MYRF. PLoS Biol. 2013; 11:e1001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muhlenhoff M., Stummeyer K., Grove M., Sauerborn M., Gerardy-Schahn R.. Proteolytic processing and oligomerization of bacteriophage-derived endosialidases. J. Biol. Chem. 2003; 278:12634–12644. [DOI] [PubMed] [Google Scholar]
- 17. Schwarzer D., Stummeyer K., Gerardy-Schahn R., Muhlenhoff M.. Characterization of a novel intramolecular chaperone domain conserved in endosialidases and other bacteriophage tail spike and fiber proteins. J. Biol. Chem. 2007; 282:2821–2831. [DOI] [PubMed] [Google Scholar]
- 18. Schulz E.C., Dickmanns A., Urlaub H., Schmitt A., Muhlenhoff M., Stummeyer K., Schwarzer D., Gerardy-Schahn R., Ficner R.. Crystal structure of an intramolecular chaperone mediating triple-β-helix folding. Nat. Struct. Mol. Biol. 2010; 17:210–215. [DOI] [PubMed] [Google Scholar]
- 19. Dugas J.C., Emery B.. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harbor Protocols. 2013; 2013:745–758. [DOI] [PubMed] [Google Scholar]
- 20. Louis J.C., Magal E., Muir D., Manthorpe M., Varon S.. CG-4, a new bipotential glial cell line from rat brain, is capable of differentiating in vitro into either mature oligodendrocytes or type-2 astrocytes. J. Neurosci. Res. 1992; 31:193–204. [DOI] [PubMed] [Google Scholar]
- 21. Muth K.N., Piefke S., Weider M., Sock E., Hermans-Borgmeyer I., Wegner M., Küspert M.. The dual-specificity phosphatase Dusp15 is regulated by Sox10 and Myrf in myelinating oligodendrocytes. Glia. 2016; 64:2120–2132. [DOI] [PubMed] [Google Scholar]
- 22. Xiang Y., Leiman P.G., Li L., Grimes S., Anderson D.L., Rossmann M.G.. Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol. Cell. 2009; 34:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Russel S., Frand A.R., Ruvkun G.. Regulation of the C. elegans molt by pqn-47. Dev. Biol. 2011; 360:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu Y., Chen Y., Kim B., Wang H., Zhao C., He X., Liu L., Liu W., Wu L.M.N., Mao M. et al. . Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013; 152:248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Senoo H., Wang H.Y., Araki T., Williams J.G., Fukuzawa M.. An orthologue of the Myelin-gene Regulatory Transcription Factor regulates Dictyostelium prestalk differentiation. Int. J. Dev. Biol. 2012; 56:325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anckar J., Sistonen L.. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 2011; 80:1089–1115. [DOI] [PubMed] [Google Scholar]
- 29. Lopez-Anido C., Sun G., Koenning M., Srinivasan R., Hung H.A., Emery B., Keles S., Svaren J.. Differential Sox10 genomic occupancy in myelinating glia. Glia. 2015; 63:1897–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao C., Deng Y., Liu L., Yu K., Zhang L., Wang H., He X., Wang J., Lu C., Wu L.N. et al. . Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nat. Commun. 2016; 7:10883. [DOI] [PMC free article] [PubMed] [Google Scholar]