

Abstract
Linker histone H1 regulates chromatin structure and gene expression. Investigating the dynamics and stoichiometry of binding of H1 to DNA and the nucleosome is crucial to elucidating its functions. Because of the abundant positive charges and the strong self-affinity of H1, quantitative in vitro studies of its binding to DNA and the nucleosome have generated results that vary widely and, therefore, should be interpreted in a system specific manner. We sought to overcome this limitation by developing a specially passivated microscope slide surface to monitor binding of H1 to DNA and the nucleosome at a single-molecule level. According to our measurements, the stoichiometry of binding of H1 to DNA and the nucleosome is very heterogeneous with a wide distribution whose averages are in reasonable agreement with previously published values. Our study also revealed that H1 does not dissociate from DNA or the nucleosome on a time scale of tens of minutes. We found that histone chaperone Nap1 readily dissociates H1 from DNA and superstoichiometrically bound H1 from the nucleosome, supporting a hypothesis whereby histone chaperones contribute to the regulation of the H1 profile in chromatin.
Graphical abstract
Linker histone H1 affects the higher-order chromatin structure in vivo and in vitro. Eukaryotic chromosomes are compacted in a hierarchical manner. The first level of compaction is to wrap DNA into nucleosomes around an octameric histone protein core containing two copies of four histones, H2A, H2B, H3, and H4.1,2 Each nucleosome core particle contains ~147 bp of DNA with ~1.7 left-handed superhelical turns.3 Two adjacent nucleosome core particles are connected by 20–90 bp of linker DNA. The second level of compaction is to stack nucleosomes into a fibril form, where linker histone H1 is indispensable.4,5 Linker histones bind to the nucleosome at the entry/exit sites where it occupies a region of 20 bp.6 As DNA compaction in chromatin imposes a physical barrier to factor binding,7 H1 and its post-translational modifications8–10 are important factors regulating various genome transactions.9,11,12
Binding of H1 to DNA and the nucleosome has been studied both in vivo and in vitro. With a thermal denaturing method, Mamoon et al. studied the binding of H1, its C-terminal domain (C-H1), and the globular domain (G-H1) to DNA.13 H1 and C-H1 bind strongly to DNA with binding constants of 108–109 M−1, while G-H1 binds much weaker to DNA with a binding constant of ~104 M−1. The size of an H1 binding site on DNA varies from 10 to 40 bp in the literature.13,14 On the basis of structural studies of linker histones H1 and H5, it has been suggested that the conserved globular domain plays a critical role in its interaction with chromatin.15 This hypothesis has been tested by a mutagenesis study in combination with structural modeling revealing at least two distinct DNA binding sites in the globular domain.16 H1 binds to the nucleosome at the dyad axis and the entry or exit site with a stoichiometry of approximately one H1 molecule per nucleosome17,18 that varies slightly depending on the nucleosome reconstitution conditions.18–20 It has also been shown that the C-terminal domain of H1 undergoes conformational changes upon binding to DNA or the nucleosome.18
Despite its tendency to form polydisperse oligomers free in solution and bound to DNA,21,22 H1 in a nucleus is dynamic, and only a small fraction is immobilized on DNA for a long period of time,23 suggesting that H1 binding may be regulated by histone chaperones. Only limited information about linker histone chaperones is currently available. Nuclear autoantigenic sperm protein (NASP),12,24–27 nucleosome assembly proein-1 (Nap1),28–31 prothymosin α,32 nucleophosmin,33 and template activating factor-134 have been suggested as linker histone chaperones. Revealing the interactions between linker histone chaperones and linker histones would further our understanding of their roles in gene regulation mechanisms.
By providing an efficient means to gather information about subpopulation dynamics, single-molecule methods are valuable tools in chromatin dynamics studies.35–37 The dynamics and stoichiometry of binding of H1 to DNA and the nucleosome, despite their importance in chromatin structure and dynamics, have yet to be reported at single-molecule resolution. This deficiency is largely due to the highly concentrated electrostatic charges on H1 making it extremely prone to nonspecific binding on glass or quartz surfaces and nearly impossible to investigate on a typical single-molecule setup. Upon initial attempts to study the behavior of binding of H1 to DNA and the nucleosome, we found that the popular polyethylene glycol (PEG)-coated glass surface35 resulted in an overwhelming amount of H1 nonspecific binding on the surface, making this surface unsuitable for the intended measurements. To this end, we developed a new surface passivation technique to eliminate the nonspecific H1 binding on a quartz microscope slide surface to reliably investigate binding of H1 to these targets at the single-molecule level. In the work presented here, we utilized a supported lipid bilayer38 to block nonspecific binding of H1. We created a hybrid surface by combining a supported 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) bilayer and a biotin-PEG submonolayer to specifically immobilize fluorescently labeled H1 via its binding to DNA or a nucleosome without any interference from nonspecifically bound H1 on the surface.
With the newly prepared microscope slides free from nonspecifically bound H1, we were able to study the dynamics and stoichiometry of binding of H1 to DNA and nucleosomes in the absence and presence of Nap1 at the single-molecule level for the first time. Our study revealed that H1 binding is not as dynamic as previously thought. Instead, once bound to DNA or the nucleosome, H1 does not dissociate spontaneously within our observation time window of 25 min. We found that the stoichiometry of binding of H1 to DNA and the nucleosome is heterogeneous and that the heterogeneity is due to H1 self-association. We also revealed that Nap1 removes H1 almost completely from the DNA and to a lesser extent from the nucleosome. These results suggest a mechanism by which a linker histone chaperone directs H1 preferentially to a nucleosomal site and consequently contributes to the dynamic regulation of chromatin structure.
MATERIALS AND METHODS
DNA Substrate Preparation
Naked and nucleosomal DNA substrates were prepared by ligating several oligonucleotides as previously reported.35–37 All DNA oligomers were purified via high-performance liquid chromatography and purchased from IDT (Coralville, IA); notations for the sequences in this paper follow the convention used on the IDT Web site. A 46 bp biotin-tagged DNA was prepared by annealing an oligomer (sequence, 5′-/5BioSg/CAACGAAATCCTCCGAGAGGTCCAAATATCCACCTGCAGATTCTAC) with its complementary oligomer carrying a Cy3 label (sequence, 5′-GTAGAATCTGCAGGTGGATATTTGGACCTCTCGGAGGATTTCGTTG/3Cy3Sp/). A 147 bp biotin-tagged DNA substrate was prepared by annealing and ligating three forward segments, (1) 5′-/5Biosg/CAACGAAATCCTCCGAGAGGATCGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAG, (2) 5′-/5Phos/ACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTTCCCCCGCGTT, and (3) 5′-/5Phos/GTAATCCCCTTGGCGGTTAAAACGCGGGGGACAGCGCGTACGTGCGT, with three reverse segments, (1) 5′-ATCGGATGTATATATCTGACACGTGCCTGGAGACTAGGGA, (2) 5′-/5Phos/GTAATCCCCTTGGCGGTTAAAACGCGGGGGACAGCGCGTACGTGCGT, and (3) 5′-/5Phos/TTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTCTCGAT/3Cy3Sp/. The resulting DNA substrate carries a Cy3 label at the 3′-end of the reverse strand. A 217 bp biotin-tagged double-stranded DNA substrate, containing a 147 bp Widom-601 sequence and a 70 bp linker sequence, was prepared by annealing and ligating four forward segments, (1) 5′-/5Biosg/GCATGTAAGTGCATGTAAGTATCGAGAATCCCGGTGCCGAGGCCGCTCAATTGG, (2) 5′-/5Phos/TCGTAGA-CAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTAC, (3) 5′-/5Phos/TCCCTAGTCTCCAGGCACGTG/iCy3/TCAGATATATACATCCGAT, and (4) 5′-/5Phos/GCGGCCGCGTATAGGGTCCATCACATAAGGGATGAACTCGGTGTGAAGAATCATGCTTTCCTTGGTCATT, with three reverse segments, (1) 5′-AATGACCAAGGAAAGCATGATTCTTCACACCGAGTTCATCCCTTATGTGATGGACC, (2) 5′-/5Phos/CTATACGCGGCCGCATCGGATGTATATATCTGACACGTGCCTGGAGACTAGGGAGTAATCCCCTTGGCGGTTAAAACGCGGGG, and (3) 5′-/5Phos/GACAGCGCGTACGTGCGTTTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTCTCGAT. For each DNA substrate, equal amounts of segments were mixed and annealed in purified and deionized (DI) water (Nanopure, Barnstead) with a polymerase chain reaction machine (MJ Mini, Bio-Rad Laboratories Inc., Vallejo, CA). A mixture was heated to 95 °C for 3 min and subsequently cooled to 4 °C in 1.5 °C steps, each 1 min in duration. The nicks in the annealed DNA were sealed with T4 ligase (New England Biolabs, product no. M0202T) in the buffer recommended by the manufacturer (product no. M0202S) at 16 °C for 16 h. The ligated DNA substrate was characterized via native 5% polyacrylamide gel electrophoresis in 0.2× TBE buffer [1× TBE consists of 89.0 mM Tris (pH 8.0), 89.0 mM boric acid, and 20 mM EDTA].
Purification and Labeling of Protein
Expression and purification of Xenopus laevis core histone H2A, H2B, H3, and H4 and subsequent assembly of a histone octamer were performed following published protocols.39 H1G101C, a mutant of X. laevis linker histone H1° (termed H1 in this paper), was expressed and purified according to a previously published method.40 After purification with a cation exchange column (Bio-Rex 70, 50–100 mesh, Bio-Rad Laboratories Inc., product no. 142-5832), H1G101C was labeled with the Cy5 maleimide monoreactive dye (GE Healthcare, product no. PA25031) according to the instructions recommended by the manufacturer. The labeling reaction mixture was purified with another cation exchange column (Bio-Rex 70, 100–200 mesh, Bio-Rad Laboratories Inc., product no. 142-5842) to remove any free dye. The labeling efficiency was close to 100% according to the absorbance values at 280 and 650 nm utilizing a correction factor of 0.05 for the dye at 280 nm. N-Terminally His6-tagged yeast Nap1 (termed Nap1 in this paper) was overexpressed and purified according to previously published protocols.41 Briefly, yNap1 was purified with a Ni-NTA column (HisPur Ni-NTA, Thermo Scientific, product no. 88222) and a Mono-Q anion exchange column (Mono Q 4.6/100 PE, GE Healthcare, product code 17-5179-01).
Reconstitution of Nucleosomes
Mononucleosomes were reconstituted with the 217 bp DNA substrate and the histone octamer core using a salt dialysis method.42 The DNA:octamer molar ratio was 1.0–1.2. The nucleosome was characterized via native 5% polyacrylamide gel electrophoresis in 0.2× TBE on ice.
Preparation of the Biotin-PEG-Silane Submonolayer
A submonolayer of biotin-PEG-silane (MW 3400, Laysan Bio, Arab, AL) was deposited on a thoroughly cleaned quartz surface as follows. A set of new quartz slides was sonicated in acetone, dichloromethane, and methanol sequentially for 15 min each, followed by thorough rinsing with deionized (DI) water. The slides were then flamed with a propane torch to remove residual organic adsorbents. The slides were subsequently immersed in a piranha solution (four parts of 98% H2SO4 mixed with one part of 30% H2O2) for 1 h, followed by thorough rinsing with DI water. After being blow-dried with 99.99% N2, the slides were treated with the following functionalization steps. Biotin-PEG-silane was dissolved in anhydrous acetonitrile (spectrophotometric grade, low water, OmniSolv, product no. AX0151-1) to a concentration of 20 pM with sonication. Cleaned quartz slides were immersed in the biotin-PEG-silane solution for 1 min with sonication, followed by additional sonication in acetonitrile twice for 5 min each to remove any unreacted biotin-PEG-silane. The slides were further sonicated twice for 5 min each in DI water for cleaning. The resulting slides were blown dry with 99.99% N2 and stored under vacuum.
Liposome Preparation
The liposome for deposition of a supported lipid bilayer was prepared following the protocol suggested by the manufacturer of the lipid and a previously published protocol.38 A chloroform solution of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) (Avanti Polar Lipids, Alabaster, AL, product no. 233935) was dried under a mild N2 flow followed by vacuum-drying for 2 h to remove chloroform. The dried lipid was suspended in buffer I [10 mM Tris (pH 7.8) and 100 mM NaCl] to a lipid concentration of 2 mg/mL. The DOPC suspension was subjected to 2 min sonication/cooling cycles on ice to prevent overheating with an Ultrasonic cell disruptor (Sonifier 450, Branson Ultrasonics) equipped with a microtip (Branson Ultrasonics, part no. 101-148-062) until the lipid suspension became a clear solution. Finally, the solution was extruded through a synthetic filter membrane with an 80 nm pore size (Avanti Polar Lipids, product no. 610014) to remove metal particles from the ultrasonic tip and large lipid vesicles. The liposome solution was monodispersed with a particle diameter of 50 nm determined with a particle size analyzer (90 Plus, Brookhaven Instruments Corp., Holtsville, NY) compared against DI water. The liposome solution was aliquotted, flash-frozen with liquid N2, and kept at −80 °C for long-term storage.
Deposition of a Lipid Bilayer
A flow cell was fabricated by sandwiching a piece of Parafilm M (Bemis Inc., Oshkosh, WI) cut in the shape of a channel between a biotin-PEG-coated quartz slide and a thoroughly cleaned coverslip (following the slide cleaning procedure described above). The quartz slide and the coverslip were glued together by the Parafilm slightly melting when placed in a 110 °C oven for 90 s. We followed a published protocol38 to deposit a lipid bilayer on the bioton-PEG-coated quartz slide by incubating the flow cell chamber with a 1 mg/mL DOPC liposome solution for 45 min. Free and loosely attached liposomes were rinsed with buffer I. The resulting supported lipid bilayer is stable for several hours.43
Single-Molecule Measurements
Lipid bilayer-treated flow cell chambers were incubated with 10 nM neutravidin in buffer I for 2 min and then rinsed with 10 chamber volumes (~50 μL) of TE50S buffer [10 mM Tris (pH 8.0), 1 mM EDTA, and 50 mM NaCl]. For H1–DNA binding experiments, the chamber was incubated with a 100 pM solution of Cy3-labeled DNA in TE50S buffer for 2 min followed by rinsing with TE150S buffer [10 mM Tris (pH 8.0), 1.0 mM EDTA, and 150 mM NaCl]. Ten chamber volumes of H1-Cy5 solution, 50 nM in TE150S buffer, was passed through the chamber, and H1-Cy5 was allowed to interact with the immobilized DNA for 15 min. Free H1-Cy5 was rinsed with 10 chamber volumes of TE150S buffer. The chamber was rinsed with 10 chamber volumes of imaging buffer [10 mM Tris (pH 8.0), 1.0 mM EDTA, 150 mM NaCl, and 2 mM Trolox], which was degassed under vacuum for 5 min, and then the inlet and outlet holes were sealed with vacuum grease. A series of single-molecule fluorescence images were taken with a home-built, prism-coupled total internal reflection fluorescence (TIRF) microscope at a frame rate of 100 ms/frame until most of the Cy5 fluorophores on H1 were photobleached with a 635 nm laser beam (50 mW, CUBE, Coherent Inc.). Optical filters that pass through ≥650 nm wavelength light (650DCXR and HQ650LP, Chroma Technology) were used to image the fluorescence signal from the Cy5 fluorophores. At the end of a Cy5 fluorophore measurement, the Cy3 fluorophores on the DNA substrate were imaged with a 532 nm laser beam (150 mW, Crystalaser) through 550–650 nm filters (650DCXR and HQ550LP, Chroma Technology) to confirm co-localization of the Cy5-H1 on the Cy3-DNA substrate. We took at least eight measurements of 3 min each with minimal delay between measurements (~10 s) at different locations within the sample chamber (i.e., a fresh unbleached region on the microscope slide). Therefore, the total length of the measurement per sample was approximately 25 min. For measurements with the nucleosome, 2.5 nM nucleosome in TE50S was incubated with 50 nM H1-Cy5 for 15 min. The mixture was then introduced into a flow cell chamber and incubated for 2 min. For Nap1 incubation to remove H1 bound to DNA or the nucleosome, 10 chamber volumes of the 60 nM Nap1 solution in TE150S buffer was passed through the sample chamber whose surface contained H1 bound to DNA or the nucleosome. Another 10 chamber volumes of the imaging buffer was passed through the chamber to remove the free Nap1 and H1.
RESULTS
Surface Passivation of a Microscope Slide for H1 Binding Studies
Xenopus linker histone H1° (termed H1 in this paper) contains 58 lysine residues and 7 arginine residues44 contributing to an isoelectric point of 11.3 and 55 positive charges at pH 7.5. The oxygen atoms on a water-covered glass surface at neutral pH attract positively charged molecules, which makes H1 extremely prone to binding nonspecifically to the surface. It is not trivial to passivate a glass surface to prevent nonspecific binding of positively charged molecules. A quartz surface, because of its lower background fluorescence, is preferred for single-molecule fluorescence measurements and is even less reactive than a glass surface. Polyethylene glycol (PEG) coating is the most popular method used to produce a biologically compatible quartz surface for single-molecule fluorescence measurements. PEGylation is usually implemented in two steps: (1) grafting primary amine groups to a quartz surface through a silanization reaction with (3-aminopropyl)-triethoxysilane (APTES) and (2) covalently attaching PEG/biotin-PEG chains on the surface through an amide bond between a surface amine group and the N-hydroxysuccinimide group of functionalized PEG (commercially available in several different forms). Despite numerous successful examples of a PEG surface in single-molecule research, H1 could not be studied on a PEG surface because of its overwhelming nonspecific binding (Figure 1A). After brief contact with a PEGylated surface (2–15 min), a 50 nM H1-Cy5 solution deposited a densely packed layer of H1-Cy5 on the surface, as shown in Figure 1A. This high level of nonspecific binding was the major obstacle in our attempts to employ a single-molecule method to investigate the behavior of binding of H1 to DNA and the nucleosome. Because PEG is neutral and hydrophilic, it is unlikely to form a stable complex with H1. Therefore, we concluded that the overwhelming binding of H1 on a PEGylated surface must be due to the incomplete coverage of the surface by PEG. Driven by this hypothesis, we sought to completely cover a quartz surface with a neutral and biologically compatible membrane.
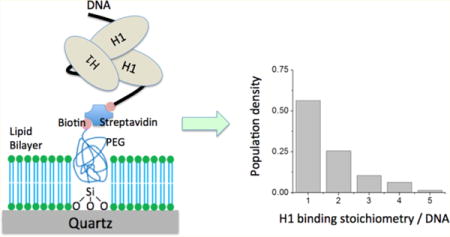
Figure 1.
Quartz microscope slide surface containing a supported lipid bilayer and a biotin-PEG submonolayer that prevents nonspecific binding of linker histone H1. (A) Nonspecific H1 binding to a polyethylene glycol (PEG)-coated quartz surface. The flow cell chamber was incubated with 50 nM H1-Cy5 in TE buffer supplemented with 150 mM NaCl for 15 min and then thoroughly rinsed with the same buffer. The sample was illuminated with a 635 nm laser on a prism-coupled total internal reflection fluorescence (TIRF) microscope setup. The image was taken at a rate of 100 ms/frame. The false color represents the fluorescence intensity as shown in panel B. The color scale shown in panel B applies to panels A, B, D, and E. The image contains two spectral channels, with the Cy5 channel on the right and the Cy3 channel on the left. The Cy5 fluorescence was so strong that it penetrated through the filter into the Cy3 channel, indicating overwhelming nonspecific binding of H1. (B) On a supported DOPC lipid bilayer-coated surface, nonspecific binding of H1 to the slide surface is negligible. The few dim spots could be contaminants or due to surface defects. All the experimental conditions were kept the same as in panel A. (C) Schematic depiction of the procedure to prepare the hybrid surface (see Materials and Methods for details). The red dot represents biotin, and the blue chains with green dots represent DOPC molecules. (D) On the hybrid surface of the lipid bilayer and biotin-PEG submonolayer, nonspecific H1 binding is negligible. The same experimental conditions were used as in panel A. (E) Fluorescence image of Cy3-labeled DNA (left, illuminated with a 532 nm laser) immobilized on the hybrid surface through biotin-neutravidin-biotin conjugation and the fluorescence image of Cy5-labeled H1 (right, illuminated with a 635 nm laser) specifically bound to DNA. The two images were taken from the same spot illuminated with two different lasers at two different time points.
A supported lipid bilayer has been utilized in single-molecule fluorescence studies.45 Lipid 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) contains a headgroup of zero net charge and two unsaturated hydrocarbon bonds. A DOPC bilayer is a two-dimensional liquid that can form a continuous membrane over a large area on a flat surface. Its exceptional mechanical stability makes it an ideal candidate for inhibiting the electrostatic interaction between H1 and a quartz surface. Indeed, almost no H1 binding was observed on a quartz surface coated with a DOPC bilayer, as shown in Figure 1B. The small number of fluorescent spots may be attributed to contaminants or a small number of defect sites in the bilayer due to defects on the quartz surface.43
To immobilize DNA or nucleosomes on the surface through biotin-neutravidin conjugation, we grafted a submonolayer of biotin-tagged PEG through a silanization reaction with biotin-PEG-silane, prior to DOPC bilayer deposition (Figure 1C). The resulting hybrid surface contains biotin-PEG accessible to neutravidin. Via the variation of the concentration of the biotin-PEG-silane in the silanization reaction, the surface density of biotin-PEG can be controlled. The upper limit of biotin-PEG-silane concentration under our reaction condition (see Materials and Methods) is 100 nM, above which DOPC bilayer deposition is inhibited. The optimized biotin-PEG-silane concentration for our conditions and measurements was 20 pM. This concentration results in ~500 biotin-PEG molecules per 100 μm × 100 μm surface area.
It is possible that the biotin-PEG submonolayer may still slightly disrupt the lipid bilayer even at a low surface density, resulting in some nonspecific H1 binding to the surface. To examine this possibility, we monitored binding of H1 to the hybrid surface. No obvious H1 binding was observed (Figure 1D), except for a small number of dim spots not more than what was observed with the DOPC bilayer only. In contrast, many bright H1 spots were observed when a short 46 bp double-stranded DNA (ds-DNA) was immobilized through biotin-neutravidin-biotin conjugation prior to H1 injection (Figure 1E). These results indicate that the hybrid surface of a biotin-PEG submonolayer and a DOPC bilayer inhibits nonspecific binding of H1 almost completely while allowing specific binding of a biotinylated DNA substrate, thereby permitting investigations of binding of H1 to DNA and the nucleosome at single-molecule resolution.
Binding of H1 to DNA
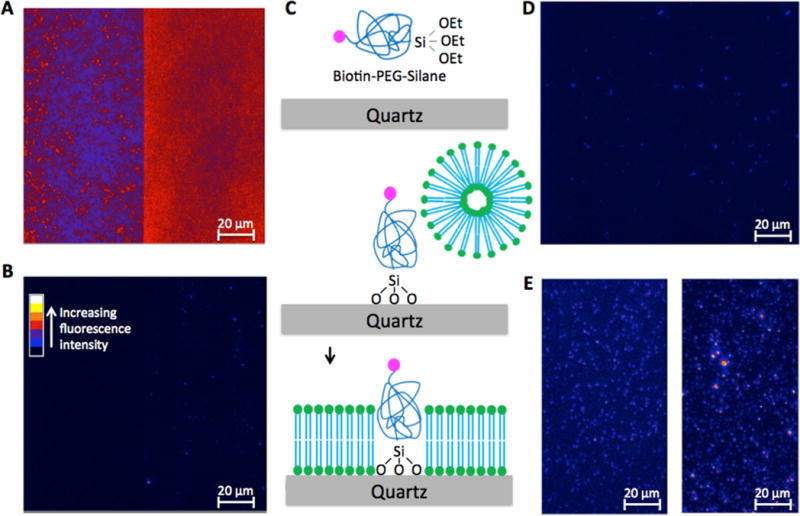
Binding of H1 to DNA is dynamic on a time scale of a few seconds to a few tens of seconds in vivo.46 On the other hand, it takes more than 1 h to completely remove H1 from DNA and nucleosomes isolated from a tissue sample.47 To reconcile these observations, we hypothesized that H1 binding is very stable with a negligibly low rate of dissociation and that removal of H1 from DNA requires a histone chaperone or a factor with chaperone function. To test this hypothesis, we first attempted to monitor H1 dissociation dynamics. We immobilized the 46 bp Cy3-labeled DNA on the hybrid surface and then allowed for Cy5-labeled H1 to bind the DNA for 15 min before rinsing away free H1. A 635 nm beam was illuminated on the surface until the Cy5 fluorophores in the DNA–H1 complex were photobleached, followed by illumination with a 532 nm beam to photobleach the Cy3 fluorophores on the DNA. Single-step photobleaching of Cy3 from one surface site verifies that it contains a single DNA–H1 complex. More than 75% of the sites containing Cy3 showed single-step photobleaching under the experimental conditions (DNA concentration, buffer, and surface incubation time), indicating a single DNA molecule at a majority of the sites. We selected fluorescence time traces from these sites and analyzed the number of H1 molecules bound to a DNA substrate by counting the Cy5 photobleaching steps (Figure 2). Given the near unity labeling efficiency, the number of Cy5 photobleaching steps should be equal to the number of H1 molecules bound to a DNA substrate. Photobleaching of most of the Cy5 in the illuminated area took place mostly within 3 min under the experimental condition, after which the area of illumination was moved to a fresh region. A fresh area of illumination during the 25 min total measurement time always reveals a constant level of intensity and the number of fluorescence spots, verifying that the photobleaching steps are not due to dissociation of H1 from the DNA. This result validates the first point of our hypothesis postulating that binding of H1 to DNA is very stable and H1 does not spontaneously dissociate from DNA on a time scale of tens of minutes.
Figure 2.
Number of Cy5 photobleaching steps reveals the stoichiometry of binding of H1 to a substrate. Each panel shows time trajectories of Cy3 (green line) and Cy5 (red line) fluorescence intensities from a single site of DNA or nucleosome on a microscope slide. These sites were identified on the basis of a non-zero Cy3 intensity level recorded at the end of each measurement, which indicates the presence of a DNA or nucleosome labeled with Cy3. To identify Cy3 spots, a 532 nm laser beam was briefly turned on at the end of each measurement. Excited by a 635 nm laser beam, these sites would emit Cy5 fluorescence only when H1-Cy5 is bound to the DNA or nucleosome. The Cy5 intensity time trajectories at these sites show single or multiple stepwise intensity decrease(s). Each of these steps corresponds to a single Cy5 photobleaching event, and each of these photobleaching events (idealized by the straight black lines) should correspond to one H1 molecule bound to the substrate. Therefore, the number of these stepwise intensity decrease events at a single substrate site is the number of H1 molecules bound to the substrate. These numbers were counted by visual inspection for all substrate sites. Panels show examples of typical fluorescence intensity time trajectories of Cy3 and Cy5 in the cases of one to four H1 molecules bound to a substrate.
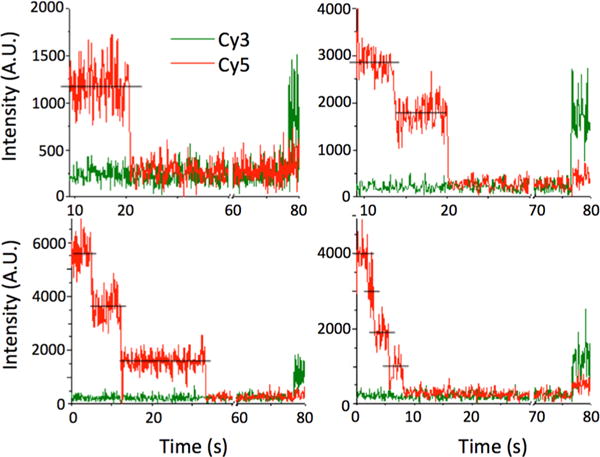
Next, we determined the stoichiometry of binding of H1 to DNA. From the fluorescence intensity time traces selected as described above, the number of photobleaching steps was manually counted (Figure 2). Interestingly, the number of H1 molecules bound to individual 46 bp double-stranded DNA fragments is heterogeneous with a wide distribution from one to five as shown in Figure 3A. The average stoichiometry is 1.71 ± 0.12 H1 molecules/DNA or 26.9 ± 1.9 bp/H1. The errors are from the Poissonian variance of the event counts assuming that our observation represents the average behavior of the system. Given the size of the DNA (46 bp), only one or two H1 molecules may directly bind to one substrate. Therefore, this wide distribution indicates self-association of H1 on the DNA.
Figure 3.
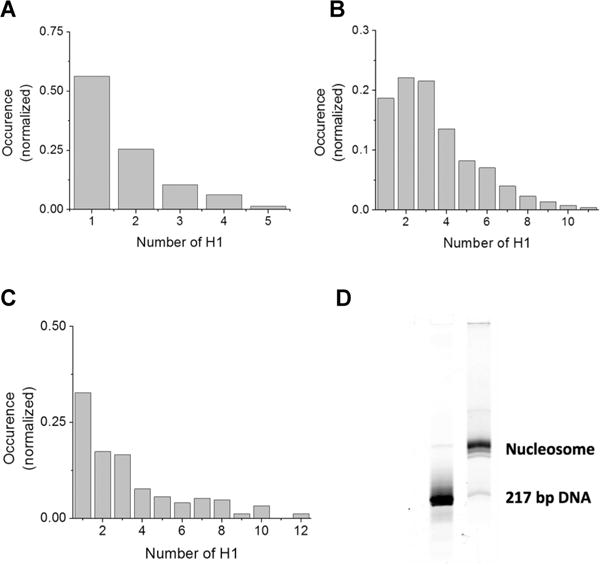
Analysis of the H1 binding stoichiometry reveals heterogeneity. (A) Histogram of the stoichiometry of binding of H1 to the 46 bp ds-DNA. The total number of complexes analyzed is 286. The total number of H1 molecules in these complexes is 489. (B) Histogram of the stoichiometry of binding of H1 to the 147 bp ds-DNA. The total number of complexes analyzed is 524. The total number of H1 molecules in these complexes is 1758. (C) Histogram of the stoichiometry of binding of H1 to the nucleosome. The total number of complexes analyzed is 247. The total number of H1 molecules in these complexes is 826. (D) A 5% polyacrylamide native gel of the Cy3-labeled 217 bp nucleosomal DNA and the reconstituted nucleosome visualized by Cy3 fluorescence reveals high nucleosome sample homogeneity.
We repeated the measurements with a longer DNA (147 bp) substrate and analyzed H1 binding stoichiometry for individual substrates. The distribution reveals a wide heterogeneity in the binding stoichiometry as shown in Figure 3B. The more abundant DNA–(H1)2 complex formation compared to DNA–H1 complex formation suggests that more than one H1 can directly bind DNA. The apparent average stoichiometry of H1 to DNA calculated from the histogram is 3.35 ± 0.17 H1 molecules/DNA or 43.9 ± 2.2 bp/H1.
These results reveal that binding of H1 to DNA is very much heterogeneous at the single-molecule level because of self-association among H1 molecules. It is worth noting that this binding mode is different from that previously observed in “tramtrack” structures because each site contains only a single DNA fragment that is nominally too short to fold back on itself.18,48
Binding of H1 to the Nucleosome
A 217 bp nucleosomal DNA construct containing the 147 bp Widom-601 strong nucleosome formation sequence with a 70 bp linker was assembled into a nucleosome with recombinant X. laevis core histones.42 The native gel image of the nucleosome shows reasonably high sample homogeneity (Figure 3D). To prevent nucleosome disassembly during the measurements, we incubated the nucleosome in TE buffer containing 50 mM NaCl with a 10-fold excess of H1 for 15 min and then loaded the mixture into a flow cell chamber for surface immobilization. To ensure identical conditions of the measurements, we rinsed the flow cell chamber with another TE buffer supplemented with 150 mM NaCl. The result reveals a wide distribution of the number of H1 molecules bound to a single nucleosome (Figure 3C). Even in repeated measurements with a second nucleosome containing a 147 bp DNA fragment, without any linker (Figure S1 of the Supporting Information), a wide distribution of stoichiometry was observed, confirming that the presence or length of linker DNA is not a dominant factor in determining this heterogeneity.
Effect of Nap1 on Binding of H1 to DNA
We investigated how the well-defined histone chaperone Nap1 affects binding of H1 to DNA in our system. Nap1 contains a negatively charged patch and competes for binding to histones with DNA.37 We first immobilized DNA on a sample chamber and formed the H1–DNA complex as described above. Then we flushed the chamber with imaging buffer containing 60 nM Nap1. Subsequent fluorescence imaging and analysis revealed a dramatic decrease in the number of H1-Cy5 spots. To quantify the change, we computed the percentage of DNA molecules bound with H1-Cy5 in the presence and absence of Nap1 as shown in Figure 4A. For both 46 and 147 bp DNA, adding Nap1 caused the percentage of DNA bound with H1 to decrease to 5 ± 1% (5.7 ± 1.2 and 4.5 ± 0.2% of the original levels, respectively, for 46 and 147 bp DNA), indicating that Nap1 efficiently removes H1 from DNA. Preincubation of Nap1 with H1 prior to DNA binding also showed a dramatically decreased number of Cy5 spots (<5% of the DNA bound with H1), further supporting the possibility that Nap1 effectively removes H1 from DNA. The small proportion of DNA bound with H1 shows large heterogeneity in the binding stoichiometry (Figure 4B,C). These distributions suggest that Nap1 works more efficiently to dissociate H1 from DNA than from another H1, supporting the role of Nap1 as a competitor for H1 against DNA.28–30 Otherwise, we would have observed a much narrower heterogeneity distribution at this low level of binding. The average H1 stoichiometry calculated from the distribution is 1.9 ± 0.1 H1 molecules per 46 bp DNA and 1.8 ± 0.2 H1 molecules per 147 bp DNA. Counting that this is 5% of the total population on average, we find the overall H1 stoichiometry is <0.1 H1 molecule per 46 or 147 bp DNA, indicating nearly complete removal of H1 from DNA.
Figure 4.
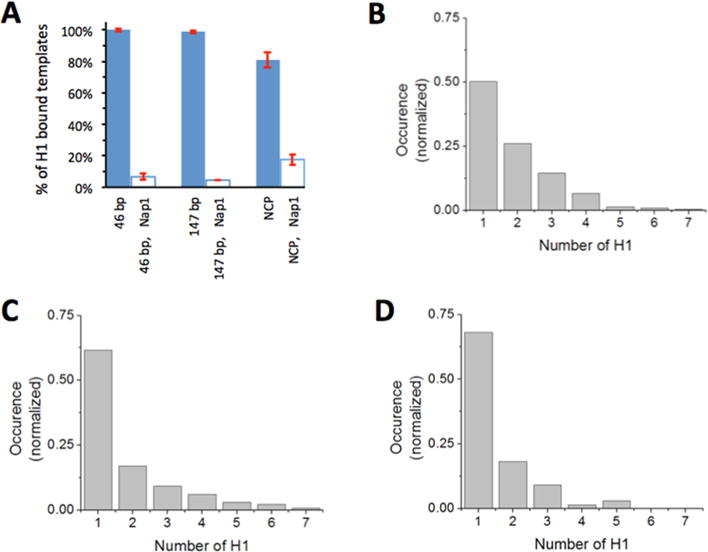
Histone chaperone Nap1 removes H1 from DNA and the nucleosome. (A) Percentages of DNA and the nucleosome with H1 bound in the absence (blue filled bars) and presence (empty bars) of 60 nM Nap1 in imaging buffer. Error bars are the standard deviations from at least three independent measurements. The sets of the total numbers of Cy3 spots analyzed were {830, 451, 684}, {623, 695, 679}, and {170, 297, 357} for 46 bp DNA, 147 bp DNA, and the nucleosome in the absence of Nap1, respectively. The sets of the total numbers of Cy3 spots analyzed were {72, 104, 62}, {39, 46, 44}, and {268, 284, 314, 324, 311} for 46 bp DNA, 147 bp DNA, and the nucleosome in the presence of Nap1, respectively. (B) Histogram of the stoichiometry of binding of H1 to 46 bp ds-DNA in the presence of 60 nM Nap1. The total number of complexes analyzed is 227. (C) Histogram of the stoichiometry of binding of H1 to the 147 bp ds-DNA in the presence of 60 nM Nap1. The total number of complexes analyzed is 130. (D) Histogram of the stoichiometry of binding of H1 to the nucleosome in the presence of 60 nM Nap1. The total number of complexes analyzed is 66.
Effect of Nap1 on Binding of H1 to the Nucleosome
To study the effect of the chaperone on binding of H1 to a nucleosome, we first incubated H1 with the nucleosome for 15 min in TE buffer with 50 mM NaCl and then imaged the nucleosome in 150 mM NaCl TE buffer with 60 nM Nap1. Adding Nap1 dramatically decreased the population density of the nucleosome bound with H1 from 80.7 ± 4.8 to 17.5 ± 1.7% (Figure 4A). On the basis of these results, we conclude that Nap1 at this concentration level leaves 22 ± 2% of the nucleosome population still bound with H1, or a population approximately 4-fold larger than the naked DNA case (5 ± 1%). Moreover, the fraction of nucleosomes bound by a single H1 is increased from 33 to 62% due to the presence of Nap1. These results suggest that histone chaperone removes H1 from DNA preferentially over the nucleosome and that the histone chaperone removes H1 from the nucleosome more efficiently when it is bound in a noncanonical manner.
DISCUSSION
To investigate binding of H1 to DNA and the nucleosome, we developed a new hybrid surface combining a supported lipid bilayer and a biotin-PEG submonolayer. The supported lipid bilayer prevents H1 from nonspecifically binding to the sample chamber, and the biotin-PEG submonolayer provides anchor points for conjugating biotin-tagged DNA or nucleosomes. The lipid bilayer was necessary to block the strong electrostatic interaction between the highly positively charged H1 and the negatively charged microscope slide surface. Many DNA- and RNA-interacting proteins are highly positively charged, and therefore, this hybrid surface will open up a wide window of opportunity to investigate such proteins at the single-molecule level.
The new surface allowed us to investigate binding of H1 to DNA and the nucleosome at the single-molecule level. The measurements resulted in an apparent average H1 footprint on DNA of 27–44 bp. These values are consistent with previously reported values.13,14 Surprisingly, however, the stoichiometry of binding of H1 to DNA and the nucleosome is very much heterogeneous and widely distributed, which may provide an explanation for the discrepancies among the previously reported values ranging from 10 to 40 bp per H1 footprint on DNA.13,14 The wide heterogeneity is due to self-association among H1 molecules that might be related to polydisperse nucleosome repeat lengths (NRLs) in cells. Researchers have suggested that the electrostatic interactions between H1 and DNA/nucleosome are a major determinant of the stoichiometry of direct binding of H1 to the nucleosome based on a theoretical study.49 They estimated the parameters of their electrostatic model for direct binding of H1 to DNA with experimental data showing a linear dependence of NRL on H1 stoichiometry.50 It is widely accepted that NRL is very much heterogeneous and polydisperse. Therefore, our results suggest that the heterogeneous H1 DNA and nucleosome binding stoichiometries due to H1 self-association may be correlated with the heterogeneous polydisperse NRLs in cells.
We also found that Nap1 can act as an effective H1 chaperone by removing H1 preferentially from DNA over the nucleosome and by dissociating H1 from the nucleosome more efficiently when it is bound noncanonically. Nap1 is a chaperone for core histones, forming a stable complex with H2A–H2B dimers or (H3–H4)2 tetramers.51,52 Shintomi et al. found that Nap1 forms a stable complex with linker histone B4 (H1X or H1M) in Xenopus eggs.29 Kepert et al. found that Nap1 removes H1 bound on chromatin fiber prepared from HeLa cells.28 We found that Nap1 reduces the percentage of DNA bound with H1 by 20-fold from 100 to 5% and nucleosome bound with H1 by ~4-fold from 81 to 18%. Although Nap1 removes H1 from both DNA and the nucleosome, the function is 5 times more effective with naked DNA. This result suggests Nap1 functions to selectively remove H1 from nucleosome free regions (NFRs) and longer linkers over the nucleosome. Moreover, Nap1 preferentially removes H1 from nucleosomes bound in superstoichiometric ratios. Interaction of H1 with chromatin in vivo is dynamic on a time scale of a few to tens of seconds.46 Given the high efficiency of Nap1 in removing H1 from DNA, one expects H1 to be dominantly associated with nucleosomes in the presence of this histone chaperone, as was indeed previously reported.53 Therefore, we conclude that Nap1 might be a very effective linker histone chaperone in vivo, removing H1 from longer linkers and NFRs preferentially over the nucleosome and ensuring a stoichiometry of nucleosome binding close to 1:1.
A potential mechanism for Nap1 to remove H1 from DNA and the nucleosome should involve the formation of at least a transient Nap1–H1–DNA (or the nucleosome) ternary complex because we did not observe any spontaneous dissociation of H1 from DNA or the nucleosome within our observation time (25 min). Consequently, Nap1 must interact with H1 while H1 is still bound to the DNA or the nucleosome. To this end, one may hypothesize that transient and repetitive exposure of a region bound with H1, which can strongly interact with Nap1, is required for ternary complex formation. Investigating fast and spontaneous H1 structural dynamics with a single-molecule method at a higher temporal resolution54 would be a straightforward way to test this hypothesis.
Given that Nap1 induces dissociation of H1 from DNA and competes weakly against DNA for core histones, DNA may also be able to dissociate H1 from its substrate, which suggests that the H1 profile may be equilibrated automatically within a dense chromatin region where H1 can interact simultaneously with two or more DNA regions. Under this hypothesis, histone chaperone function in regulating the H1 profile would be effective only in a loose chromatin region, where H1 might be used only to maintain the structural integrity of nucleosomes, but not to fold the array. A high-resolution microscopic study on large-scale phase changes in heterogeneous chromatin with fluorescently labeled H1 would be a straightforward way to test this hypothesis.
Our single-molecule investigations of binding of H1 to DNA and the nucleosome revealed that the binding is static and has an apparently fixed ensemble-averaged stoichiometry with a wide distribution at the single-molecule level due to self-association. We also report a potential function of the histone chaperone Nap1 in controlling the higher-order chromatin structure by selectively removing linker histones from NFRs and longer linker regions.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants GM097286 to T.-H.L. and GM052426 to J.J.H.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b01247.
Figure S1 (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Hewish DR, Burgoyne LA. Chromatin substructure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem Biophys Res Commun. 1973;52:504–510. doi: 10.1016/0006-291x(73)90740-7. [DOI] [PubMed] [Google Scholar]
- 2.Sahasrabuddhe CG, Van Holde KE. The Effect of trypsin on nuclease-resistant chromatin fragments. J Biol Chem. 1974;249:152–156. [PubMed] [Google Scholar]
- 3.Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. J Cell Biol. 2006;175:869–880. doi: 10.1083/jcb.200604005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thoma F, Koller T. Influence of histone H1 on chromatin structure. Cell. 1977;12:101–107. doi: 10.1016/0092-8674(77)90188-x. [DOI] [PubMed] [Google Scholar]
- 5.Robinson PJJ, Rhodes D. Structure of the 30 nm chromatin fibre: A key role for the linker histone. Curr Opin Struct Biol. 2006;16:336–343. doi: 10.1016/j.sbi.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Thomas JO. Histone H1: location and role. Curr Opin Cell Biol. 1999;11:312–317. doi: 10.1016/S0955-0674(99)80042-8. [DOI] [PubMed] [Google Scholar]
- 7.Bondarenko VA, Steele LM, Ujvari A, Gaykalova DA, Kulaeva OI, Polikanov YS, Luse DS, Studitsky VM. Nucleosomes can form a polar barrier to transcription elongation by RNA polymerase II. Mol Cell. 2006;24:469–479. doi: 10.1016/j.molcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Lu MJ, Mpoke SS, Dadd CA, Allis CD. Phosphorylated and dephosphorylated linker histone H1 reside in distinct chromatin domains in Tetrahymena macronuclei. Mol Biol Cell. 1995;6:1077–1087. doi: 10.1091/mbc.6.8.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 10.Robinson PJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol. 2008;381:816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zlatanova J, van Holde K. Linker histones versus HMG1/2: a struggle for dominance? BioEssays. 1998;20:584–588. doi: 10.1002/(SICI)1521-1878(199807)20:7<584::AID-BIES10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 12.Alekseev OM, Bencic DC, Richardson RT, Widgren EE, O’Rand MG. Overexpression of the linker histone-binding protein tNASP affects progression through the cell cycle. J Biol Chem. 2003;278:8846–8852. doi: 10.1074/jbc.M210352200. [DOI] [PubMed] [Google Scholar]
- 13.Mamoon NM, Song Y, Wellman SE. Histone H10 and its carboxyl-terminal domain bind in the major groove of DNA. Biochemistry. 2002;41:9222–9228. doi: 10.1021/bi020199n. [DOI] [PubMed] [Google Scholar]
- 14.Machha VR, Waddle JR, Turner AL, Wellman S, Le VH, Lewis EA. Calorimetric studies of the interactions of linker histone H1° and its carboxyl (H1°-C) and globular (H1°-G) domains with calf-thymus DNA. Biophys Chem. 2013;184:22–28. doi: 10.1016/j.bpc.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Ramakrishnan V. Histone structure and the organization of the nucleosome. Annu Rev Biophys Biomol Struct. 1997;26:83–112. doi: 10.1146/annurev.biophys.26.1.83. [DOI] [PubMed] [Google Scholar]
- 16.Brown DT, Izard T, Misteli T. Mapping the interaction surface of linker histone H10 with the nucleosome of native chromatin in vivo. Nat Struct Mol Biol. 2006;13:250–255. doi: 10.1038/nsmb1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Widom J. Chromatin structure: Linking structure to function with histone H1. Curr Biol. 1998;8:R788–R791. doi: 10.1016/s0960-9822(07)00500-3. [DOI] [PubMed] [Google Scholar]
- 18.Fang H, Clark DJ, Hayes JJ. DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res. 2012;40:1475–1484. doi: 10.1093/nar/gkr866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cattini PA, Harborne N, Allan J. The linker histone stoichiometry of chicken erythrocyte chromatin in situ. J Electron Microsc. 1988;37:31–37. [PubMed] [Google Scholar]
- 20.Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014;344:376–380. doi: 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- 21.Carter GJ, van Holde K. Self-association of linker histone H5 and of its globular domain: evidence for specific self-contacts. Biochemistry. 1998;37:12477–12488. doi: 10.1021/bi980716v. [DOI] [PubMed] [Google Scholar]
- 22.Maman JD, Yager TD, Allan J. Self-association of the globular domain of histone H5. Biochemistry. 1994;33:1300–1310. doi: 10.1021/bi00172a003. [DOI] [PubMed] [Google Scholar]
- 23.Bernas T, Brutkowski W, Zarebski M, Dobrucki J. Spatial heterogeneity of dynamics of H1 linker histone. Eur Biophys J. 2014;43:287–300. doi: 10.1007/s00249-014-0962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richardson RT, Batova IN, Widgren EE, Zheng LX, Whitfield M, Marzluff WF, O’Rand MG. Characterization of the histone H1-binding Protein, NASP, as a cell cycle-regulated somatic protein. J Biol Chem. 2000;275:30378–30386. doi: 10.1074/jbc.M003781200. [DOI] [PubMed] [Google Scholar]
- 25.Finn RM, Browne K, Hodgson KC, Ausio J. sNASP, a histone H1-specific eukaryotic chaperone dimer that facilitates chromatin assembly. Biophys J. 2008;95:1314–1325. doi: 10.1529/biophysj.108.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Ge Z, Walsh ST, Parthun MR. The human histone chaperone sNASP interacts with linker and core histones through distinct mechanisms. Nucleic Acids Res. 2012;40:660–669. doi: 10.1093/nar/gkr781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richardson RT, Alekseev OM, Grossman G, Widgren EE, Thresher R, Wagner EJ, Sullivan KD, Marzluff WF, O’Rand MG. Nuclear autoantigenic sperm protein (NASP), a linker histone chaperone that is required for cell proliferation. J Biol Chem. 2006;281:21526–21534. doi: 10.1074/jbc.M603816200. [DOI] [PubMed] [Google Scholar]
- 28.Kepert JF, Mazurkiewicz J, Heuvelman GL, Toth KF, Rippe K. NAP1 modulates binding of linker histone H1 to chromatin and induces an extended chromatin fiber conformation. J Biol Chem. 2005;280:34063–34072. doi: 10.1074/jbc.M507322200. [DOI] [PubMed] [Google Scholar]
- 29.Shintomi K, Iwabuchi M, Saeki H, Ura K, Kishimoto T, Ohsumi K. Nucleosome assembly protein-1 is a linker histone chaperone in Xenopus eggs. Proc Natl Acad Sci U S A. 2005;102:8210–8215. doi: 10.1073/pnas.0500822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saeki H, Ohsumi K, Aihara H, Ito T, Hirose S, Ura K, Kaneda Y. Linker histone variants control chromatin dynamics during early embryogenesis. Proc Natl Acad Sci U S A. 2005;102:5697–5702. doi: 10.1073/pnas.0409824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Giebler HA, Isaacson MK, Nyborg JK. Eviction of linker histone H1 by NAP-family histone chaperones enhances activated transcription. Epigenet Chromatin. 2015;8:30. doi: 10.1186/s13072-015-0022-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George EM, Brown DT. Prothymosin α is a component of a linker histone chaperone. FEBS Lett. 2010;584:2833–2836. doi: 10.1016/j.febslet.2010.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gadad SS, Senapati P, Syed SH, Rajan RE, Shandilya J, Swaminathan V, Chatterjee S, Colombo E, Dimitrov S, Pelicci PG, Ranga U, Kundu TK. The multifunctional protein nucleophosmin (NPM1) is a human linker histone H1 chaperone. Biochemistry. 2011;50:2780–2789. doi: 10.1021/bi101835j. [DOI] [PubMed] [Google Scholar]
- 34.Kato K, Okuwaki M, Nagata K. Role of template activating factor-I as a chaperone in linker histone dynamics. J Cell Sci. 2011;124:3254–3265. doi: 10.1242/jcs.083139. [DOI] [PubMed] [Google Scholar]
- 35.Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee TH. DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JY, Wei S, Lee TH. Effects of histone acetylation by Piccolo NuA4 on the structure of a nucleosome and the Interactions between two Nucleosomes. J Biol Chem. 2011;286:11099–11109. doi: 10.1074/jbc.M110.192047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JY, Lee J, Yue H, Lee TH. Dynamics of nucleosome assembly and effects of DNA methylation. J Biol Chem. 2015;290:4291–4303. doi: 10.1074/jbc.M114.619213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graneli A, Yeykal CC, Prasad TK, Greene EC. Organized arrays of individual DNA molecules tethered to supported lipid bilayers. Langmuir. 2006;22:292–299. doi: 10.1021/la051944a. [DOI] [PubMed] [Google Scholar]
- 39.Luger K, Rechsteiner TJ, Richmond TJ. Methods in Enzymology. Vol. 304. Academic Press; San Diego: 1999. Preparation of nucleosome core particle from recombinant histones; pp. 3–19. [DOI] [PubMed] [Google Scholar]
- 40.Chafin DR, Hayes JJ. Site-directed cleavage of DNA by linker histone-Fe(II) EDTA conjugates. In: Moss T, editor. DNA-Protein Interactions. Vol. 148. Humana Press; Totowa, NJ: 2001. pp. 275–290. [DOI] [PubMed] [Google Scholar]
- 41.Toth KF, Mazurkiewicz J, Rippe K. Association states of nucleosome assembly protein 1 and its complexes with histones. J Biol Chem. 2005;280:15690–15699. doi: 10.1074/jbc.M413329200. [DOI] [PubMed] [Google Scholar]
- 42.Thastrom A, Lowary PT, Widom J. Measurement of histone-DNA interaction free energy in nucleosomes. Methods. 2004;33:33–44. doi: 10.1016/j.ymeth.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 43.Castellana ET, Cremer PS. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf Sci Rep. 2006;61:429–444. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brocard MP, Triebe S, Peretti M, Doenecke D, Khochbin S. Characterization of the two H1°-encoding genes from Xenopus laevis. Gene. 1997;189:127–134. doi: 10.1016/s0378-1119(96)00845-1. [DOI] [PubMed] [Google Scholar]
- 45.Fazio T, Visnapuu ML, Wind S, Greene EC. DNA curtains and nanoscale curtain rods: High-throughput tools for single molecule imaging. Langmuir. 2008;24:10524–10531. doi: 10.1021/la801762h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408:877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- 47.Allan J, Staynov DZ, Gould H. Reversible dissociation of linker histone from chromatin with preservation of internucleosomal repeat. Proc Natl Acad Sci U S A. 1980;77:885–889. doi: 10.1073/pnas.77.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark DJ, Thomas JO. Salt-dependent cooperative interaction of histone H1 with linear DNA. J Mol Biol. 1986;187:569–580. doi: 10.1016/0022-2836(86)90335-9. [DOI] [PubMed] [Google Scholar]
- 49.Cherstvy AG, Teif VB. Electrostatic effect of H1-histone protein binding on nucleosome repeat length. Phys Biol. 2014;11:044001. doi: 10.1088/1478-3975/11/4/044001. [DOI] [PubMed] [Google Scholar]
- 50.Woodcock CL, Skoultchi AI, Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006;14:17–25. doi: 10.1007/s10577-005-1024-3. [DOI] [PubMed] [Google Scholar]
- 51.Ishimi Y, Kikuchi A. Identification and molecular cloning of yeast homolog of nucleosome assembly protein I which facilitates nucleosome assembly in vitro. J Biol Chem. 1991;266:7025–9. [PubMed] [Google Scholar]
- 52.Fujii-Nakata T, Ishimi Y, Okuda A, Kikuchi A. Functional analysis of nucleosome assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J Biol Chem. 1992;267:20980–20986. [PubMed] [Google Scholar]
- 53.Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160:1145–1158. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 54.Wei S, Falk SJ, Black BE, Lee TH. A novel hybrid single molecule approach reveals spontaneous DNA motion in the nucleosome. Nucleic Acids Res. 2015;43:e111. doi: 10.1093/nar/gkv549. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.