

Abstract
Nucleosomes impose physical barriers to DNA-templated processes, playing important roles in eukaryotic gene regulation. DNA is packaged into nucleosomes by histone proteins mainly through strong electrostatic interactions that can be modulated by various post-translational histone modifications. Investigating the dynamics of histone dissociation from the nucleosome and how it is altered upon histone modifications is important for understanding eukaryotic gene regulation mechanisms. In particular, histone H2A-H2B dimer displacement in the nucleosome is one of the most important and earliest steps of histone dissociation. Two conflicting hypotheses on the requirement for dimer displacement are that nucleosomal DNA needs to be unwrapped before a dimer can displace and that a dimer can displace without DNA unwrapping. In order to test the hypotheses, we employed three-color single-molecule FRET and monitored in a time-resolved manner the early kinetics of H2AH2B dimer dissociation triggered by high salt concentration and by histone chaperone Nap1. The results reveal that dimer displacement requires DNA unwrapping in the vast majority of the nucleosomes in the salt-induced case, while dimer displacement precedes DNA unwrapping in >60% of the nucleosomes in the Nap1-mediated case. We also found that acetylation at histone H4K16 or H3K56 affects the kinetics of Nap1-mediated dimer dissociation and facilitates the process both kinetically and thermodynamically. On the basis of these results, we suggest a mechanism by which histone chaperone facilitates H2A-H2B dimer displacement from the histone core without requiring another factor to unwrap the nucleosomal DNA.
Graphical abstract
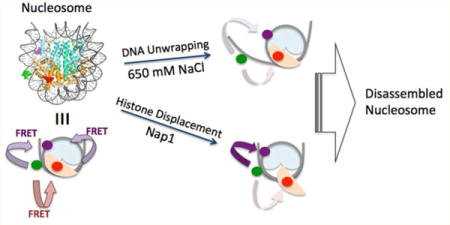
DNA in a eukaryotic cell nucleus is compacted into chromatin by histone proteins. The basic building unit of chromatin is the nucleosome, composed of a histone octamer core wrapped around by 147 bp DNA in 1.67 helical turns.1 A histone octamer core is formed by combining a (H3–H4)2 tetramer and two H2A-H2B dimers.1 These highly positively charged proteins compact negatively charged DNA, providing a platform for versatile changes in gene packaging via post-translational histone modifications. In various nuclear processes involving DNA as a template such as DNA replication, transcription, and DNA repair, the physical barriers imposed by nucleosomes need to be overcome, which must involve nucleosome disassembly. Early steps of nucleosome disassembly include displacement of histones out of their canonical sites, which eventually leads to histone dissociation. In particular, histone H2A-H2B dimer displacement is one of the earliest steps of nucleosome disassembly, the mechanism of which remains largely unclear. Dimer displacement will eventually be followed by dimer dissociation. Dimer dissociation cannot be a simple one-step process because a dimer in the nucleosome contacts DNA, the tetramer, and the other dimer at multiple locations. The dimer-displaced states may be detected by single-molecule assays, whereas they may be too transient to be detected with ensemble-averaging assays. Two conflicting hypotheses postulate that nucleosomal DNA near the DNA–dimer contact region needs to be unwrapped for the dimer to be displaced out of the histone core or that a dimer can spontaneously displace without DNA unwrapping. DNA unwrapping under a physiological condition often involves an enzyme that consumes chemical energy, and therefore, testing these hypotheses will clarify whether or not such an enzyme is required for initiating nucleosome disassembly.
High salt concentrations have often been used to conveniently induce histone displacement and subsequent nucleosome disassembly in vitro.2,3 However, the level of salt used for this purpose is far from being physiological. Moreover, the hydrophobic interactions between histones are also important in stabilizing the nucleosome.1 Therefore, the salt-dependent histone displacement may not properly represent the process under a physiologically relevant condition.
Studies in the past have revealed that histone chaperones participate in nucleosome assembly and disassembly in vivo.4 Nucleosome assembly protein 1 (Nap1) has been utilized and studied in vitro primarily for its nucleosome assembly activity.5,6 Nap1 does not have any function to produce or consume chemical energy such as ATP hydrolysis. Instead, it has a large negatively charged region where histones can bind strongly if they are not a part of a canonical nucleosome, eventually promoting nucleosome assembly.5 Nap1 also has histone H2A-H2B dimer dissociation activity with7 or without FACT (facilitates chromatin transcription),8 possibly facilitating transcription. It has been hypothesized that Nap1 stabilizes spontaneously and transiently displaced histone dimers out of the nucleosome that may eventually trigger dimer dissociation. According to this hypothesis, Nap1 can catalyze dimer displacement without requiring any energy-consuming enzyme to unwrap DNA.
Histone acetylation is a well-conserved chromatin modification that plays crucial roles in gene regulation.9–17 Anomalies in histone acetylation are linked to various types of cancer.18–20 Lysine residues mostly in histone N-terminal tails are targets for acetylation by histone acetyltransferases.13,21,22 Some specific acetylation marks are found critical for transcription elongation.23 Loss of acetylation at H4K16 (H4K16ac) is a distinctive mark of various cancers.24 It has also been reported that H4K16ac inhibits higher order chromatin structure formation in vitro by interfering with the internucleosomal interactions between the N-terminal tail of H4 and the acidic patch of H2AH2B dimer.25,26 Intranucleosomal interactions of DNA with the N-terminal tails of H427 and H3 have been suggested based on cross-linking studies28 which revealed that histone acetylations disrupt the interactions.1,29,30 H3K56 acetylation (H3K56ac) impedes the interaction between histone and DNA near the nucleosome entry/exit sites1,31 and contributes to nucleosome disassembly32 and repositioning33 possibly by facilitating DNA termini unwrapping dynamics.30,34 This function of H3K56ac is well correlated with its roles in transcription regulation.35
Here we investigated salt-induced and Nap1-mediated histone H2A-H2B dimer displacement in the nucleosome in a time-resolved manner based on three-color single-molecule FRET (smFRET). It should be noted that under our conditions histones would unlikely completely dissociate from the nucleosome in a few minutes due to their strong interactions with DNA and other histones. We also investigated the effects of specific histone acetylations at H3K56 and H4K16. We found that dimer displacement requires DNA unwrapping in the salt-induced case, while dimer displacement precedes DNA unwrapping in >60% of the nucleosomes in the Nap1-mediated case. We also found that histone acetylation at H3K56 or H4K16 affects the kinetics and thermodynamics of the process in the Nap1-mediated case. Our results support an early intermediate of dimer dissociation where histone chaperone forms a ternary complex with DNA and a dimer without requiring another factor to unwrap DNA.
MATERIALS AND METHODS
Nucleosomal DNA Preparation
Nucleosomal DNA labeled with a FRET pair (Cy3 and Cy5.5) was constructed by ligating oligonucleotides as described previously.6 Briefly, five oligonucleotides were annealed by linearly decreasing the temperature from 95 to 5 °C for 45 min to construct 147 bp Widom 601 nucleosomal DNA36 with a 20 nucleotides linker that contains biotin at one end (see Figure S1 for the sequence of the DNA). The oligonucleotides including the one labeled with Cy3 were purchased from Integrated DNA Technologies (Coralville, IA). A Cy5.5 fluorophore functionalized with NHS ester (GE Healthcare Life Sciences, Pittsburgh, PA) was used to label an oligonucleotide via a Unilink amino modifier (Integrated DNA Technologies) with a six-carbon spacer (Integrated DNA Technologies). The annealed DNA was cleaned up with a PCR purification kit (Qiagen, Valencia, CA), and ligated with T4 ligase (New England BioLabs, Ipswich, MA) at 16 °C for 16 h. The ligated DNA construct was purified on a 2% agarose gel.
Protein Preparation
6XHis-Yeast Nap1 histone chaperone (yNap1, or Nap1 in this report) was expressed and purified with Ni-NTA resin (Thermo Fisher Scientific, Waltham, MA) as reported elsewhere.37 Xenopus laevis core histones were separately prepared as detailed elsewhere,38 and H2B T112C39 was labeled with an Atto647N fluorophore functionalized with maleimide (Sigma-Aldrich, St. Louis, MO). As for H3K56ac and H4K16ac preparation, the thiol–ene coupling reaction was performed on H3K56C/C110A and H4K16C, respectively, as reported earlier,30,40 which was modified from earlier publications.41,42 This modification strategy has been utilized to reproduce results obtained with specific acetylations via native chemical ligation.40,42 H3K56ac and H4K16ac in our nucleosomes reported here refer to H3Ks56ac and H4Ks16ac. Briefly, H3K56ac and H4K16ac were prepared by reacting the cysteine thiols with N-vinyl-acetamide (NVA). Mass-spectrometric analyses of the acetylated histones confirm near complete acetylation (Figure S2). H2A-H2B dimer and (H3–H4)2 tetramer were refolded and purified as described in ref 38.
Nucleosome Reconstitution
Nucleosomes were assembled by combining DNA, 2× H2A-H2B dimer, and (H3–H4)2 tetramer at a high salt concentration (2.0 M NaCl) and then dialyzing against sequentially decreasing salt concentrations, from 850 to 2.5 mM NaCl in five steps in a TE buffer for 1 h in each step. Nucleosome assembly was confirmed on a 5% native PAGE (Figure S3). The labeling efficiency of H2A-H2B dimer was controlled by mixing labeled and unlabeled dimers. The labeling efficiency was adjusted to ~50%, which was confirmed by UV absorption values at 260 and 647 nm. Residual absorbance of Atto647N at 260 nm has been taken into account for the calculation.
Three-Color smFRET Experiment
Quartz microscope slides were passivated as follows. First, slides were functionalized with 5k Mw biotin-PEG-silane (Laysan Bio, Inc., Arab, AL) as previously published,43 and then coated with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, Avanti Polar Lipids, Alabaster, AL) lipid bilayer following the manufacturer’s protocol prior to immobilizing nucleosomes via biotin–streptavidin conjugation. Nap1 was introduced onto the immobilized nucleosomes in 10 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 3% glycerol in the presence of protocatechuate dioxygenase (0.02 unit/μL, Sigma-Aldrich), protocatechuic acid (1 mM, Sigma-Aldrich), and Trolox (1 mM, Sigma-Aldrich) for elevated photostability and prolonged lifetime of fluorophores.
We employed a one-donor and two-acceptor FRET scheme with Cy3 (donor), Atto647N (acceptor 1), and Cy5.5 (acceptor 2) on a prism coupled total internal reflection fluorescence microscope (home-built based on TE2000 from Nikon, Japan), where the FRET donor was excited with a 532 nm laser (GCL-150-L, CrystaLaser, Reno NV). Fluorescence signals from the three fluorophores were collected through a water-immersion objective lens (CFI Plan Apochromat, 60x/1.20, Nikon, Japan), and spectrally split into three regions by dichroic mirrors (Chroma Technology, Bellows Falls, VT) with a cutoff wavelength of 620 nm to separate Cy3 emission from the other two, and of 690 nm to separate Atto647N emission from Cy5.5 emission. Additional emission filters, HQ590/70m (Chroma Technology) and HQ655LP (Chroma Technology) for Cy3 and Atto647N/Cy5.5 channels respectively, were also employed to remove the laser beam and to reduce leaks between channels. Spatially resolved fluorescence signals from multiple nucleosome particles on a surface area of 35 × 100 μm2 in the three spectral regions were integrated for 250 ms and recorded continuously as a time series of fluorescence images with an electron multiplying CCD camera (iXon+897, Andor Technology, Belfast, Ireland) at a 4 Hz recording rate.
Single-Molecule FRET Data Analysis
Time traces of intensities of Cy3, Atto647N, and Cy5.5 fluorescence were obtained from multiple series of fluorescence images taken as described in the previous section. Fluorescence intensities were corrected for interchannel leaks that were determined by measuring intensities in the three channels when only a single fluorophore was present. With the predetermined leak parameters, α, β, γ, and δ, the leak-corrected intensities, I3, I647N, and I5.5 of Cy3, Atto647N, and Cy5.5, respectively, were obtained by solving the following set of three linear equations.
where α, β, γ, and δ are leak ratios for I3 to Atto647N channel, I647N to Cy5.5 channel, I5.5 to Atto647N channel, and I3 to Cy5.5 channel, respectively. I3′, I647N′, and I5.5′ are the fluorescence intensities obtained at a time point from the brightness of the corresponding pixels on the movie frame imaged on an electron multiplying CCD camera (iXon+897, Andor Technology, Belfast, Ireland). The FRET efficiencies were calculated with the formula FRET647N = I647N/(I3 + I647N + I5.5) and FRET5.5 = I5.5/(I3 + I647N + I5.5). For the following hidden Markov model (HMM) kinetics analysis on DNA dynamics, FRET5.5 = I5.5/(I3 + I5.5) was used instead.
HMM Analysis
The FRET5.5 traces were subjected to hidden Markov model (HMM) analysis to extract the kinetics information as described previously.44 We used four-state models (HF, MF, LF1, and LF0 for high-, mid-, low1-, and low0-FRET) because it is the minimum number of states that properly model the data based on visual inspection. FRET5.5 is calculated with a formula I5.5/(I3 + I5.5). The FRET5.5 time trajectories report mainly DNA motion. The starting point is the fully wrapped DNA, and the ending point is nearly completely unwrapped DNA in the dimer contact region. Dimer partial displacement from the intact nucleosomal state may increase FRET5.5 slightly. According to the analysis results, however, this increased FRET5.5 does not form a separate state likely because the difference is too small. Therefore, we assigned the HF state to the intact nucleosome where DNA is fully wrapped regardless of whether the dimer is fully intact or partially displaced (Figure S5). On the basis of the fact that dimer partial displacement without DNA change does not result in a noticeable FRET5.5 change, we assigned the midrange FRET states (MF and LF1) to the intermediates with partially unwrapped DNA with various extents of dimer displacement (Figure S5). Finally, the lowest FRET state, or LF0, should be assigned to a state further unwrapped at the dimer contact region (Figure S5). See Figure S5 for a depiction of these kinetic states. The errors of the results were estimated by repeating the analysis five times, where each analysis was performed on a randomly chosen 80% subset of the entire data. The standard deviation of the results was taken as the errors of the HMM analysis. The errors were mostly within 20% of the average values, which agrees well with previously published results.44
Data Filtering Criteria
Typically 40–60% of the samples with an apparent sign of single Cy3 (i.e., single step photobleaching in the Cy3 spectral region) show both FRET5.5 and FRET647N signals indicating nearly or fully assembled nucleosomes. The rest is aggregated and nearly or fully disassembled nucleosomes that were excluded from further analysis. We also excluded nucleosomes with premature fluorophore photobleaching. Among the assembled nucleosomes, there were three different groups of populations showing three different levels of Atto647N fluorescence intensity. In one group, the fluorescence level of Cy5.5 is higher than Atto647N, which is a sign that the distal H2A-H2B was labeled with an Atto647N. In another group, FRET647N is higher than 0.6, which is a sign that both dimers were labeled with the dye or that histones were aggregated on DNA. We excluded these nucleosomes and aggregates from further analysis. In summary, we included in the analysis single nucleosomes only with initial FRET647N < 0.6 and Atto647N intensity higher than Cy5.5 intensity. These criteria are independent from nucleosome dynamics, and therefore, do not bias the results in any systematic way.
RESULTS
Three-Color smFRET Measurements Enabled Detection of DNA Unwrapping and H2A-H2B Dimer Displacement in a Time-Resolved Manner during the Early Steps of Dimer Dissociation from the Nucleosome
To investigate the early steps of dimer dissociation from the nucleosome in the context of DNA unwrapping, we employed a three-color smFRET system. The FRET donor Cy3 was labeled at the +54th nucleotide from the dyad of the Widom 601 sequence,36 a FRET acceptor Cy5.5 was labeled at the −15th nucleotide from the dyad, and the other acceptor Atto647N was labeled at H2B T112C. The labeling positions were chosen so that FRET5.5 occurs moderately efficiently (0.4–0.6 FRET efficiency) in a nucleosome with the DNA fully wrapped, and FRET647N occurs moderately efficiently in a nucleosome with the labeled H2A-H2B dimer intact (Figure 1A). The donor position on the DNA is where mainly H2A-H2B interacts with DNA. This position was selected to avoid the DNA region showing fast dynamics on a millisecond time scale (~15 bp from a terminus).30,45 When the DNA unwraps near this dimer contact region while the dimer is still intact, the fluorescence intensities from both Cy5.5 (I5.5) and Atto647N (I647N) would decrease, while the Cy3 (I3) intensity would increase (Figure 1B, Figure 2A). When both the dimer and DNA dissociate simultaneously from the nucleosome, I647N would increase, I5.5 would decrease, and I3 would remain nearly unchanged (Figure 1C). In the case of dimer displacement with the DNA wrapped in the dimer contact region, I647N would decrease, and I5.5 and I3 would increase (Figure 1D, Figure 2B). In the first case (Figure 1B), I3 could slightly increase or decrease depending on the distance between Cy3 and Atto647N. Either way, the I3 change does not matter to the analysis because the three cases (Figure 1B–D) can be distinguished from one another solely based on the changes in I647N and I5.5. These predictions are based on an assumption that Atto647N is labeled on the same side as Cy3 is (Figure 1A). In order to avoid nucleosomes where this assumption is invalid, nucleosomes showing higher I5.5 than I647N were excluded from further analysis because this intensity pattern indicates that the Atto647N labeled dimer is on the other side of the Cy3 labeled DNA region. By analyzing the sequence of FRET changes in the nucleosomes according to these guidelines, we were able to investigate the kinetics and thermodynamics of DNA unwrapping and H2A-H2B dimer displacement during the early steps of dimer dissociation.
Figure 1.
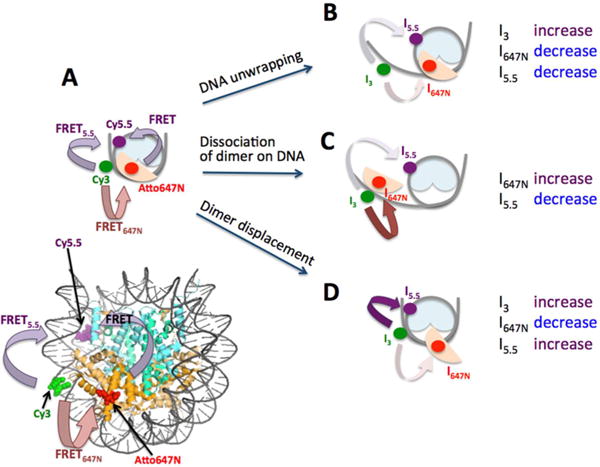
Three-color FRET experimental setup to monitor the early steps of histone H2A-H2B dimer dissociation. (A) A schematic representation of a complete nucleosome core particle with intact DNA and H2A-H2B dimer reporting moderately high FRET5.5 and FRET647N. Fluorescence intensities from Cy3, Atto647N, and Cy5.5 are denoted by I3, I647N, and I5.5, respectively. (B) A nucleosome with unwrapped DNA and intact dimer would report decreasing I5.5 and I647N and increasing I3. (C) A nucleosome with DNA and dimer dissociating simultaneously would show decreasing I5.5 and increasing I647N. This mode of dissociation can be via gyre opening or DNA unwrapping, which cannot be distinguished in our setup. (D) A nucleosome with displaced dimer with intact DNA would report increasing I5.5 and I3, and decreasing I647N.
Figure 2.
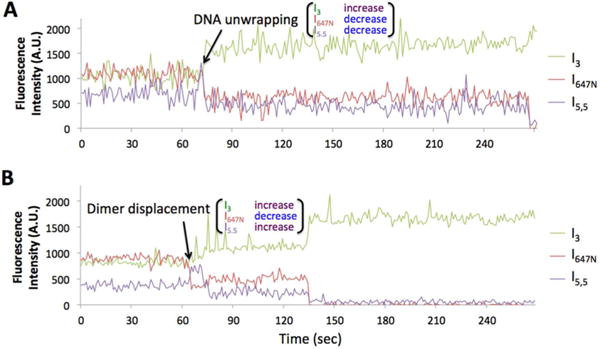
Representative time traces of fluorescence intensities during the early steps of dimer dissociation (A.U. = arbitrary unit). More traces are shown in Figure S4. (A) DNA unwrapping initiates dimer dissociation as characterized by simultaneously decreasing I5.5 and I647N with increasing I3. This trace is from the wild type nucleosome under the high salt condition. The acceptor fluocesnce signals are eventually extingiuished at the end of the time trace by either dimer dissociation or photobleaching within 1−2 frames at ~270 s. (B) Dimer displacement initiates dissociation as characterized by increasing I5.5 and simultaneously decreasing I647N and increasing I3. This trace is from the wild type nucleosome under the Nap1 condition. The increased donor intensity and the decreased acceptor intensities at 135 s are due to either Atto647N photobleaching or dimer dissociation.
In order to verify that the nucleosomes immobilized on the surface are specifically bound to biotin and functioning properly, we counted the number of surface immobilized nucleosomes with a fluorescently labeled H2A-H2B dimer considerably displaced in the absence and presence of Nap1. We counted a nucleosome to have a dimer considerably displaced if it shows no or negligible FRET647N signal (<0.1 FRET efficiency). In order to simplify the analysis, we excluded the nucleosomes containing two fluorescently labeled dimers. The numbers are plotted against time in Figure S6. In the absence of Nap1 at 50 mM NaCl for 30 min (Figure S6A), we did not observe any noticeable dimer displacement. At 650 mM NaCl, however, we observed displacement within 15 min as described in the following sections. In the presence of Nap1 at 50 mM NaCl, we observed that a considerable number of nucleosomes have the dimers displaced during the first 35 min and eventually only 13% of the nucleosomes retained the fluorescently labeled dimer fully intact after 65 min of incubation. Since we monitored only one of the two dimers, we concluded that 2% (= 13% × 13%) of the nucleosomes retained both dimers fully intact after 65 min incubation in the presence of Nap1. These results support that the experimental system functions properly for our measurements.
DNA Unwrapping Is Required for Dimer Displacement in the Salt-Induced Case, but Not in the Nap1-Mediated Case
We initiated dimer displacement by injecting a buffer containing a high level of salt (650 mM NaCl) or Nap1 (600 nM + 50 mM NaCl) and recorded fluorescence intensities from nucleosomes for 15 min. We analyzed the sequence of FRET changes from the individual nucleosomes undergoing dimer displacement and DNA unwrapping as outlined in the previous section. According to our measurements, we did not observe any intact nucleosomes showing FRET changes indicating simultaneous dissociation of the dimer and DNA (Figure 1C), and therefore, excluded this path from further analysis. A small number of nucleosomes showing this signature of FRET always start with the fluorescence levels indicating partially disassembled nucleosomes that were already excluded from the analysis. We counted the numbers of nucleosomes showing (1) decreasing I647N and I5.5 with increasing I3 and (2) decreasing I647N with increasing I5.5 and I3 (Table 1). These intensity changes indicate that, respectively, (1) DNA unwraps first (Figure 1B, Figure 2A) and (2) dimer displaces first (Figure 1D, Figure 2B). The results clearly show that the first step of dimer dissociation depends on the experimental condition for inducing nucleosome disassembly (Table 1 and Figure 3). In salt-induced dissociation, 95% of nucleosomes started the process by DNA unwrapping prior to dimer displacement (Table 1A, Figure 3A), whereas only 38% of nucleosomes took the same path in Nap1-mediated dissociation (Table 1B, Figure 3B). The percentage of nucleosomes that show dimer displacement prior to DNA unwrapping was 5% in salt-induced dissociation (Table 1A, Figure 3A), which increased to 63%, or by >12-fold in Nap1-mediated dissociation (Table 1B, Figure 3B). These results suggest that Nap1 leads dimer dissociation to a path where a dimer displaces in the nucleosome before DNA unwraps. A high salt condition, on the other hand, leads the dissociation to the other path where DNA unwraps before dimer displaces. This difference is likely because a high salt level at 650 mM NaCl destabilizes the interaction between DNA and histones considerably, but not to the extent where dimers substantially displace from the nucleosome before DNA unwraps.
Table 1.
(A) The Number of Nucleosomes Grouped by the Initial Step of Salt-Induced Dimer Dissociation, (B) The Number of Nucleosomes Categorized by the Initial Step of Nap1-Mediated Dimer Dissociation, (C) Statistical Analysis on the Numbers of Nucleosomes Showing Dimer Displacement First in the Three Cases of Nap1-Mediated Dimer Dissociation
| (A) | ||||||
|---|---|---|---|---|---|---|
| Salt-induced dimer dissociation | ||||||
| DNA unwrapping first | Dimer displacement first | |||||
| WT | H4K16ac | H3K56ac | WT | H4K16ac | H3K56ac | |
| # of events | 160 | 117 | 69 | 9 | 8 | 4 |
| in % | 95 ± 2 | 94 ± 2 | 95 ± 3 | 5 ± 2 | 6 ± 2 | 5 ± 3 |
| (B) | ||||||
|---|---|---|---|---|---|---|
| NAP1-mediated dimer dissociation | ||||||
| DNA unwrapping first | Dimer displacement first | |||||
| WT | H4K16ac | H3K56ac | WT | H4K16ac | H3K56ac | |
| #of events | 78 | 38 | 71 | 130 | 39 | 59 |
| in % | 37 ± 3 | 49 ± 6 | 55 ± 4 | 62.5 ± 3.4 | 51 ± 6 | 45 ± 4 |
| (C) | |||
|---|---|---|---|
| NAP1-mediated dimer dissociation | |||
| Dimer displacement first | |||
| WT | H4K16ac | H3K56ac | |
| in % | 62.5 ± 3.4 | 51 ± 6 | 45 + 4 |
| Δ % from WT | – | −12 ± 7 | −18 ± 5 |
| p-value (one-tailed) | – | <0.00001 | <0.00001 |
Figure 3.
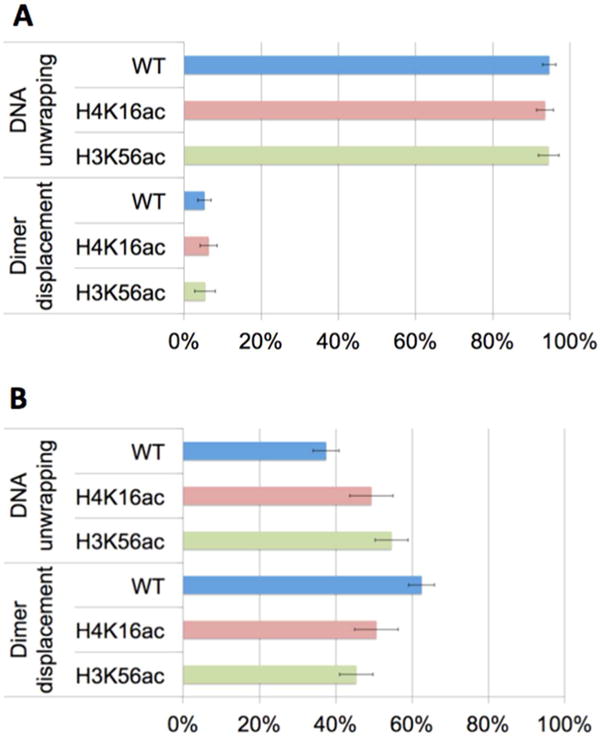
Population densities of nucleosomes grouped by the initial dimer dissociation step. The two populations are denoted by “DNA unwrapping” and “Dimer displacement”. Nucleosomes in the “DNA unwrapping” group start dimer dissociation by DNA unwrapping prior to dimer displacement. Nucleosomes in the “Dimer displacement” group start dimer dissociation by dimer displacement prior to DNA unwrapping. Dimer displacement was induced by (A) a high salt level and (B) Nap1. WT, H4K16ac, and H3K56ac denote wild-type nucleosome (i.e., no acetylation), nucleosome acetylated at H4K16, and nucleosome acetylated at H3K56, respectively. The error bars represent the standard deviations of the counts assuming a binomial distribution whose variance is given by np(1 − p), where n is the sample size and p is the probability. (A) In the 95% of the population, dimer dissociation induced by salt starts with DNA unwrapping and the effect of acetylation on this statistics is negligible. (B) In the 63% of the population, dimer dissociation mediated by Nap1 starts with dimer displacement (wild-type, WT). This population density decreases significantly upon histone acetylation at H4K16 or H3K56 (Table 1C).
It should be noted that no noticeable change in any FRET signal was detected for 30 min at 50 mM NaCl in the absence of Nap1 (Figure S6A, measurements were done for 1 min at every 10 min time point for 30 min). This result indicates that DNA unwrapping we observed in the presence of Nap1 was catalyzed by Nap1. These results also indicate that Nap1 does not require large-scale DNA unwrapping to bind H2A-H2B in the nucleosome. Therefore, the observed dimer displacement with fully wrapped DNA must require transient small-scale displacement during which Nap1 can bind and stabilize the displaced dimer. This conclusion is further supported by our observation that, in the absence of Nap1, this transient small-scale displacement leads rarely to observable displacement even at 650 mM NaCl unless the DNA is unwrapped. Taken altogether, these results support that DNA unwrapping is required for dimer displacement during salt-induced dissociation, but not during Nap1-mediated dissociation.
Histone Acetylation State Affects the Early Dimer Dissociation Paths in the Nap1-Mediated Case
We also investigated the early dimer dissociation steps in histone-acetylated nucleosomes at H4K16 and at H3K56 under a high salt condition (650 mM NaCl) in the absence of Nap1 and under a moderate salt condition (50 mM NaCl) in the presence of Nap1. The results are shown in Table 1 and Figure 3. In the high-salt case, no noticeable change is induced by H4K16ac or H3K56ac. In the presence of Nap1, however, the percentage of nucleosomes where the dissociation is initiated by dimer displacement decreased considerably upon these acetylations (Table 1B, Figure 3B). Statistical analysis revealed that the difference is significant (Table 1C). This result indicates that upon these acetylations a dimer dissociates more via the other route where DNA unwraps before dimer displaces. Dissociation via this route would be accelerated when DNA unwrapping is facilitated. Therefore, we suggest that this effect of acetylation is likely due to facilitated DNA unwrapping. Assuming that Nap1-mediated dissociation is physiologically more relevant than salt-induced dissociation is, our results suggest that these histone acetylations may impact on the kinetic paths of dimer dissociation in vivo.
Kinetics Analysis Reveals That Histone Acetylation Kinetically Facilitates the Early Steps of Dimer Dissociation Mediated by Nap1
We performed kinetics analysis on the FRET5.5 signal to investigate the DNA dynamics during Nap1-mediated dimer dissociation. This DNA dynamics represents the unwrapping and rewrapping motions of the DNA in the dimer contact region (Figure S5). The FRET5.5 traces were analyzed with hidden Markov models to extract the kinetics information.44 We found that at least four FRET5.5 states were necessary to optimize the data. More states optimize the data by increasing the number of states in the Mid-FRET range (FRET5.5 near 0.25–0.45) to varying extents in the three cases (wild-type and the two acetylated nucleosomes). Here, we use the 4-state modeling results to extract the rates to and from the Low0-, Low1-, Mid-, and High-FRET states, assuming that the Mid-FRET state may be a convolution of two or more states among which the transitions cannot be analyzed clearly. The results from four-state modeling (Low0-, Low1-, Mid-, and High-FRET; LF0, LF1, MF, and HF in the order of increasing FRET5.5) are shown in Tables 2 and 3. The analysis allowed transitions only between two adjacent states, and did not include photobleached regions. The FRET efficiencies of the wild-type nucleosome in the 4 states are 0.090, 0.17, 0.29, and 0.56, those of the H4K16ac nucleosome are 0.11, 0.21, 0.33, and 0.54, and those of the H3K56ac nucleosome are 0.070, 0.17, 0.31, and 0.53, respectively for LF0, LF1, MF, and HF (Table 2). These FRET values are very close to each other and within the error of the analysis (Table 2), suggesting that the intermediate states of DNA unwrapping are not significantly altered by the acetylations. The kinetic rates, however, depend on the acetylations. In particular, the initial unwrapping rate (kHF→MF) was increased with the acetylations by ~4-fold (Table 3, Figure 4), suggesting that the DNA region where it contacts a dimer becomes more dynamic upon acetylation. Once unwrapped, the rewrapping rate (MF→HF) did not depend on the acetylation status. Further DNA fluctuations (kMF→LF1 and kLF1→MF) did not display much difference with the acetylations. However, H3K56 acetylation accelerated the last step flapping motion as shown in higher kLF1→LF0 and kLF0→LF1 (Table 3, Figure 4). The last unwrapping rate was increased by more than 3-fold, and the last rewrapping rate was increased by about 2-fold. It should be noted that the fluorescence signal change from the dimer motion (I647N) could be imbedded in the Cy5.5 intensity, which is a main reason for the convolved MF state. Regardless, the first (kHF→MF) and the last (kLF1→LF0) steps of FRET5.5 change should report DNA unwrapping motion because when we consider all possible intermediates (see Figure S5 for details) it is evident that these FRET changes cannot occur without DNA unwrapping in the dimer contact region.
Table 2.
FRET5.5 Values from HMM Analysis (Four States)
| FRET5.5 | WT | H4K16ac | H3K56ac |
|---|---|---|---|
| Low0 | 0.090 ± 0.010 | 0.11 ± 0.01 | 0.070 ± 0.004 |
| Low1 | 0.17 ± 0.02 | 0.21 ± 0.01 | 0.17 ± 0.01 |
| Mid | 0.29 ± 0.03 | 0.33 ± 0.01 | 0.31 ± 0.01 |
| High | 0.56 ± 0.04 | 0.54 ± 0.01 | 0.53 ± 0.02 |
Table 3.
Kinetic Rates (/s) of DNA Motions Observed with FRET5.5
| FRET5.5 Transition | WT | H4K16ac | H3K56ac |
|---|---|---|---|
| 1. Low0 → Low1 | 0.10 ± 0.03 | 0.099 ± 0.024 | 0.20 ± 0.01 |
| 2. Low1 → Low0 | 0.082 ± 0.02 | 0.080 ± 0.010 | 0.25 ± 0.05 |
| 3. Low1 → Mid | 0.25 ± 0.07 | 0.19 ± 0.03 | 0.26 ± 0.05 |
| 4. Mid → Low1 | 0.078 ± 0.045 | 0.13 ± 0.02 | 0.14 ± 0.03 |
| 5. Mid → High | 0.090 ± 0.137 | 0.14 ± 0.02 | 0.17 ± 0.01 |
| 6. High → Mid | 0.028 ± 0.031 | 0.11 ± 0.02 | 0.11 ± 0.03 |
Figure 4.
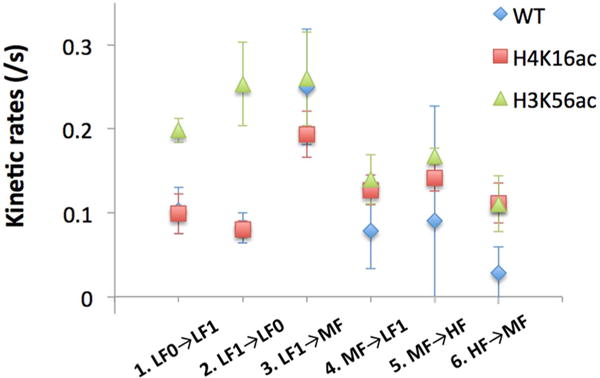
Kinetic rates between two adjacent states in the four FRET states analyzed by hidden Markov models. WT denotes wild-type, or no acetylation. Acetylations at H4K16 (H4K16ac) and H3K56 (H3K56ac) increase the kinetic rate from High to Mid FRET. H3K56 acetylation facilitates the DNA fluctuations between the Low1 and Low0 FRET states. The errors are the standard deviations obtained from five HMM analyses per case (see Materials and Methods). The rates and errors are listed in Table 3.
H4K16ac and H3K56ac Lower the Energy Barriers for the Early Dimer Dissociation Steps by Destabilizing Histone–DNA Contacts
On the basis of the kinetic rates of the early steps of Nap1-mediated dimer dissociation (Table 3), we estimated the thermodynamic stabilities of the intermediates by numerical simulations. We used a freely accessible software package for this simulation (Tenua, a kinetics simulator for Java, http://bililite.com/tenua). From our smFRET experiments, we already know that the vast majority of the nucleosomes do not complete dimer dissociation within 10 min of Nap1 incubation (Figure S6B). According to the simulations, the population densities of the intermediates reach a steady state within 10 min. Therefore, it should be reasonable to assume that these steady-state population densities represent their relative thermodynamic stabilities. The relative free energy values of the four FRET states as obtained from the state population densities are shown in Table 4. The ΔG value for state i was obtained from Ni/N0 = e−ΔG/RT, where R is the gas constant, T is the temperature of the experiment (= 298 K), Ni and N0 are the population densities of states i and LF0 respectively (Figure 5). We set the LF0 state as the reference state because the most DNA-unwrapped state has a thermodynamic stability least likely affected by the acetylation states. The thermodynamics of the initial nucleosomal DNA unwrapping shows overall an uphill process indicating that nucleosomes become unstable during the early steps of dimer dissociation. The results indicate that histone acetylation facilitates histone dissociation thermodynamically by decreasing the slope of the ΔG uphill in the H4K16ac and H3K56ac cases (Figure 5). This effect arises mainly from destabilization of the intact nucleosome (i.e., elevated free energy of the HF state upon acetylation) likely due to weakened histone-DNA contacts.
Table 4.
Relative Free Energies (ΔG) of the Early Dimer Dissociation Intermediates
| (kJ/mol) | WT | H4K16ac | H3K56ac |
|---|---|---|---|
| High FRET5.5 | −4.8 ± 0.8 | −3.0 ± 0.5 | −2.6 ± 0.8 |
| Mid FRET5.5 | −1.5 ± 0.9 | −2.1 ± 0.6 | −1.2 ± 0.9 |
| Low1 FRET5.5 | 0.28 ± 0.87 | −1.1 ± 0.5 | 0.24 ± 0.86 |
| Low0 FRET5.5 | 0 | 0 | 0 |
Figure 5.
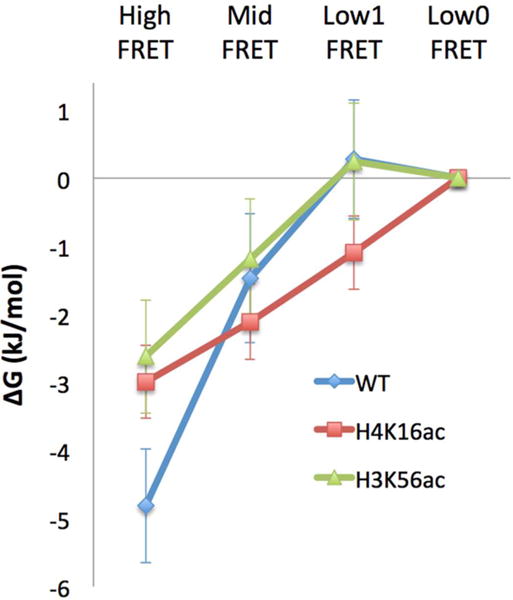
Relative thermodynamic stabilities of the early intermediates of dimer dissociation. WT, H4K16ac, and H3K56ac denote wild-type nucleosome (i.e., no acetylation), nucleosome acetylated at H4K16, and nucleosome acetylated at H3K56, respectively. The relative free energies (ΔG) were derived from the equilibrium population densities obtained by numerical simulations based on the kinetic rates listed in Table 3. The free energy values were aligned to the Low0 FRET state assuming that this state has a thermodynamic stability unaffected by the acetylation state. The error bars represent the standard deviations of five simulations based on five different sets of kinetic rates estimated from five different sets of HMM analysis results (see Materials and Methods). The ΔG values are listed in Table 4. RT at 298 K is 2.48 kJ/mol.
DISCUSSION
Histone dissociation from the nucleosome is an essential process for nucleosome disassembly that should precede or coincide with many eukaryotic genome transactions involving DNA as a substrate. Histone chaperones interfere with histone-DNA interactions and thus have been hypothesized to mediate histone dissociation during nucleosome disassembly. In particular, the chronology of DNA unwrapping and histone H2A-H2B dimer displacement has significant implications in the roles of histone chaperone in nucleosome disassembly and gene regulation.7,46 If DNA unwrapping is required for dimer displacement by a chaperone, nucleosome disassembly can be initiated only by an enzyme that can unwrap DNA, which limits the function of the chaperone. In order to monitor the chronology of DNA unwrapping and dimer displacement, we employed a three-color smFRET system. Our results support that a high salt condition (650 mM NaCl) perturbs mostly the electrostatic interactions between DNA and histones, and thereby helping initiate dimer dissociation by DNA unwrapping and subsequent dimer displacement. In the absence of a chaperone, DNA unwrapping is required for dimer displacement even at this extremely high salt level. In the presence of Nap1, however, dimer displaces before DNA unwraps in 63% of the nucleosomes, indicating that Nap1 creates a new path for dimer displacement that does not require prior DNA unwrapping. This result suggests that physiological nucleosome disassembly may be initiated either by histone dimer displacement first or by DNA unwrapping first, both of which can be catalyzed by histone chaperone.
On the basis of our results, we suggest a mechanism by which histone chaperone mediates histone dissociation from an intact nucleosome. When we attempted to observe spontaneous DNA unwrapping and dimer displacement in the absence of Nap1 at a moderate salt concentration (50 mM NaCl), no visible change in the FRET signals was observed for 30 min. This result indicates that DNA or dimer must stay fully intact on the time scale of 250 ms or longer for at least 30 min. Taken together, our results support a hypothesis where Nap1 must interfere with DNA–histone contacts that may be transiently displaced due to high frequency thermal fluctuations,45 eventually resulting in a ternary complex comprising DNA–Nap1–histone. This mechanism would not require any additional energy-consuming factor such as a chromatin remodeler to unwrap DNA prior to histone dissociation. For efficient ternary complex formation, there must be a threshold lifetime of the histone displaced out of the nucleosome. If the high frequency structural dynamics is on the time scale similar to this threshold lifetime, Nap1 is essentially to amplify a small difference in the structural dynamics to a large change in the histone dissociation efficiency.
According to the above hypothesis, the recently reported high-frequency DNA motion in the nucleosome must have a critical implication in Nap1-histone binding.30,47 This high-frequency motion should depend on changes modulating DNA–histone interactions such as histone variation and modification.30,47 Histone acetylation at H4K16 or H3K56 drives dimer dissociation more toward a path where DNA unwraps before a dimer is displaced. The results from HMM analysis on FRET5.5 dynamics (i.e., DNA dynamics) indicate that H4K16ac and H3K56ac facilitate nucleosome disassembly mainly by destabilizing the fully intact nucleosomal state. These effects are likely due to facilitated DNA dynamics upon these acetylations, which would increase both Nap1 binding and DNA unwrapping rates. As for H3K56ac, several reports support a hypothesis where removal of the positive charge at H3K56 upon acetylation facilitates nucleosomal DNA unwrapping near the dimer contact region.33–35 This hypothesis is also supported by our recent report on facilitated dynamics of nucleosome termini upon H3K56ac.30 As for H4K16ac, it has not been shown to affect DNA unwrapping dynamics near the dimer contact region. Recent crystal structure of acetylated nucleosomes suggested that acetylations on H4 tail destabilize intranucleosomal interactions at the ±1.5 superhelical location (SHL) where (H3–H4)2 tetramer interacts with DNA.48 At this point, the only reasonable hypothesis for the H4K16ac effect on DNA unwrapping near the dimer contact region is that the DNA dynamics we report here may depend on the DNA flexibility at the ±1.5 SHL. This suggestion is based on the fact that the short span of DNA from ±1.5 SHL to the dimer contact region (40–50 bp) is far shorter than the persistent length of ds-DNA (~150 bp). Within this short span of DNA, a change in the flexibility or structure at one region may have a strong impact on the structural dynamics at another region. In summary, we suggest that these acetylations facilitate the high-frequency DNA motion near the dimer contact region, which eventually accelerate dimer displacement by increasing the rates of DNA unwrapping and Nap1 binding.
By employing a three-color smFRET system, we monitored the early steps of histone H2A-H2B dimer dissociation in the context of DNA unwrapping in a time-resolved manner. This study revealed the heterogeneous paths of dimer dissociation mediated by histone chaperone that are distinct from the salt-induced one. On the basis of the results, we suggest a mechanism by which histone chaperone mediates histone dissociation from an intact nucleosome without requiring another factor to unwrap DNA. Our system can be further utilized to investigate many other kinetic and thermodynamic aspects of nucleosome assembly and disassembly intermediates. For example, with both dimers labeled in one nucleosome under alternating multicolor excitation, one can measure the kinetic and thermodynamic stabilities of the nucleosome lacking one dimer, the hexasome,49 induced by various conditions.
Supplementary Material
Acknowledgments
Funding
This research was supported by NIH [GM097286].
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b01252.
Nucleosomal DNA sequences (Figure S1), mass-spectra of acetylated histones (Figure S2), a gel image verifying nucleosome reconstitution reactions (Figure S3), additional sample FRET time traces (Figure S4), FRET changes linked to schematized nucleosome structural changes (Figure S5), and histone dimer retention histograms (Figure S6) (PDF)
ORCID
Tae-Hee Lee: 0000-0003-2034-6394
Author Contributions
J.L. and T.L. designed and performed the measurements and wrote the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Hoch DA, Stratton JJ, Gloss LM. Protein-protein Forster resonance energy transfer analysis of nucleosome core particles containing H2A and H2A.Z. J Mol Biol. 2007;371:971–988. doi: 10.1016/j.jmb.2007.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yager TD, McMurray CT, van Holde KE. Salt-induced release of DNA from nucleosome core particles. Biochemistry. 1989;28:2271–2281. doi: 10.1021/bi00431a045. [DOI] [PubMed] [Google Scholar]
- 4.Okuwaki M, Kato K, Shimahara H, Tate S, Nagata K. Assembly and disassembly of nucleosome core particles containing histone variants by human nucleosome assembly protein I. Mol Cell Biol. 2005;25:10639–10651. doi: 10.1128/MCB.25.23.10639-10651.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell. 2010;37:834–842. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JY, Lee J, Yue H, Lee TH. Dynamics of nucleosome assembly and effects of DNA methylation. J Biol Chem. 2015;290:4291–4303. doi: 10.1074/jbc.M114.619213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh FK, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, Studitsky VM. Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci U S A. 2013;110:7654–7659. doi: 10.1073/pnas.1222198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park YJ, Chodaparambil JV, Bao Y, McBryant SJ, Luger K. Nucleosome assembly protein 1 exchanges histone H2A-H2B dimers and assists nucleosome sliding. J Biol Chem. 2005;280:1817–1825. doi: 10.1074/jbc.M411347200. [DOI] [PubMed] [Google Scholar]
- 9.Allfrey VG, Faulkner R, Mirsky AE. Acetylation Methylation of Histones Their Possible Role in Regulation of RNA Synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vidali G, Boffa LC, Bradbury EM, Allfrey VG. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc Natl Acad Sci U S A. 1978;75:2239–2243. doi: 10.1073/pnas.75.5.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chahal SS, Matthews HR, Bradbury EM. Nature. 1980;287:76–79. doi: 10.1038/287076a0. [DOI] [PubMed] [Google Scholar]
- 12.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 13.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Utley RT, Lacoste N, Jobin-Robitaille O, Allard S, Cote J. Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Mol Cell Biol. 2005;25:8179–8190. doi: 10.1128/MCB.25.18.8179-8190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, Christman MF. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature. 2002;419:411–415. doi: 10.1038/nature01035. [DOI] [PubMed] [Google Scholar]
- 16.Tse C, Sera T, Wolffe AP, Hansen JC. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol. 1998;18:4629–4638. doi: 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hebbes TR, Clayton AL, Thorne AW, Crane-Robinson C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 1994;13:1823–1830. doi: 10.1002/j.1460-2075.1994.tb06451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 19.Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 20.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 21.Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Cote J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999;18:5108–5119. doi: 10.1093/emboj/18.18.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choy JS, Kron SJ. NuA4 subunit Yng2 function in intra-S-phase DNA damage response. 2002;22:8215–8225. doi: 10.1128/MCB.22.23.8215-8225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al. Combinatorial patterns of histone acetylations and methyl-ations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 25.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Lu C, Yang Y, Fan Y, Yang R, Liu CF, Korolev N, Nordenskiold L. Influence of histone tails and H4 tail acetylations on nucleosome-nucleosome interactions. J Mol Biol. 2011;414:749–764. doi: 10.1016/j.jmb.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 27.Ebralidse KK, Grachev SA, Mirzabekov AD. Nature. 1988;331:365–367. doi: 10.1038/331365a0. [DOI] [PubMed] [Google Scholar]
- 28.Zheng C, Lu X, Hansen JC, Hayes JJ. Salt-dependent intra- and internucleosomal interactions of the H3 tail domain in a model oligonucleosomal array. J Biol Chem. 2005;280:33552–33557. doi: 10.1074/jbc.M507241200. [DOI] [PubMed] [Google Scholar]
- 29.Morales V, Richard-Foy H. Role of histone N-terminal tails and their acetylation in nucleosome dynamics. Mol Cell Biol. 2000;20:7230–7237. doi: 10.1128/mcb.20.19.7230-7237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Lee J, Lee TH. Lysine Acetylation Facilitates Spontaneous DNA Dynamics in the Nucleosome. J Phys Chem B. 2015;119:15001–15005. doi: 10.1021/acs.jpcb.5b09734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. J Mol Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 32.Xu F, Zhang Q, Zhang K, Xie W, Grunstein M. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc Natl Acad Sci U S A. 2008;105:9000–9005. doi: 10.1073/pnas.0800057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J Mol Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 36.Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 37.Lee JY, Lee TH. Effects of DNA methylation on the structure of nucleosomes. J Am Chem Soc. 2012;134:173–175. doi: 10.1021/ja210273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 39.D’Arcy S, Martin KW, Panchenko T, Chen X, Bergeron S, Stargell LA, Black BE, Luger K. Chaperone Nap1 shields histone surfaces used in a nucleosome and can put H2A-H2B in an unconventional tetrameric form. Mol Cell. 2013;51:662–677. doi: 10.1016/j.molcel.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhall A, Wei S, Fierz B, Woodcock CL, Lee TH, Chatterjee C. Sumoylated human histone H4 prevents chromatin compaction by inhibiting long-range internucleosomal interactions. J Biol Chem. 2014;289:33827–33837. doi: 10.1074/jbc.M114.591644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoyle CE, Bowman CN. Thiol-ene click chemistry. Angew Chem, Int Ed. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 42.Li F, Allahverdi A, Yang R, Lua GB, Zhang X, Cao Y, Korolev N, Nordenskiold L, Liu CF. A direct method for site-specific protein acetylation. Angew Chem, Int Ed. 2011;50:9611–9614. doi: 10.1002/anie.201103754. [DOI] [PubMed] [Google Scholar]
- 43.Yue H, Fang H, Wei S, Hayes JJ, Lee TH. Single-Molecule Studies of the Linker Histone H1 Binding to DNA and the Nucleosome. Biochemistry. 2016;55:2069–2077. doi: 10.1021/acs.biochem.5b01247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee TH. Extracting kinetics information from single-molecule fluorescence resonance energy transfer data using hidden markov models. J Phys Chem B. 2009;113:11535–11542. doi: 10.1021/jp903831z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Wei S, Lee J, Yue H, Lee TH. Single-Molecule Observation Reveals Spontaneous Protein Dynamics in the Nucleosome. J Phys Chem B. 2016;120:8925. doi: 10.1021/acs.jpcb.6b06235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuryan BG, Kim J, Tran NN, Lombardo SR, Venkatesh S, Workman JL, Carey M. Histone density is maintained during transcription mediated by the chromatin remodeler RSC and histone chaperone NAP1 in vitro. Proc Natl Acad Sci U S A. 2012;109:1931–1936. doi: 10.1073/pnas.1109994109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei S, Falk SJ, Black BE, Lee TH. A novel hybrid single molecule approach reveals spontaneous DNA motion in the nucleosome. Nucleic Acids Res. 2015;43:e111. doi: 10.1093/nar/gkv549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakamori M, Fujii Y, Suka N, Shirouzu M, Sakamoto K, Umehara T, Yokoyama S. Intra- and inter-nucleosomal interactions of the histone H4 tail revealed with a human nucleosome core particle with genetically-incorporated H4 tetra-acetylation. Sci Rep. 2015;5:17204. doi: 10.1038/srep17204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arimura Y, Tachiwana H, Oda T, Sato M, Kurumizaka H. Structural Analysis of the Hexasome, Lacking One Histone H2A/H2B Dimer from the Conventional Nucleosome. Biochemistry. 2012;51:3302–3309. doi: 10.1021/bi300129b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.