

Abstract
Recent theoretical accounts have proposed excitation (E) and inhibition (I) imbalance as a possible mechanistic, network-level hypothesis underlying neural and behavioral dysfunction across neurodevelopmental disorders, particularly autism spectrum disorder (ASD) and schizophrenia (SCZ). These two disorders share some overlap in their clinical presentation as well as convergence in their underlying genes and neurobiology. However, there are also clear points of dissociation in terms of phenotypes and putatively affected neural circuitry. Here we highlight emerging work from the clinical neuroscience literature examining neural correlates of E/I imbalance across children and adults with ASD and adults with both chronic and early-course SCZ. We discuss findings from diverse neuroimaging studies across distinct modalities, conducted with EEG, MEG, 1H-MRS, and fMRI, including effects observed both during task and at rest. Throughout this review we discuss points of convergence and divergence in the ASD and SCZ literature, with a focus on disruptions in neural E/I balance. We also consider these findings in relation to predictions generated by theoretical neuroscience, particularly computational models predicting E/I imbalance across disorders. Finally, we discuss how human non-invasive neuroimaging can benefit from pharmacological challenge studies to reveal mechanisms in ASD and SCZ. Collectively, we attempt to shed light on shared and divergent neuroimaging effects across disorders with the goal of informing future research examining the mechanisms underlying the E/I imbalance hypothesis across neurodevelopmental disorders. We posit that such translational efforts are vital to facilitate development of neurobiologically informed treatment strategies across neuropsychiatric conditions.
Keywords: autism, schizophrenia, E/I balance, review, computational modeling, mechanism
Neural computations rely on balanced excitation and inhibition, predominantly driven by glutamatergic and GABAergic input, respectively. Without excitation, neurons would not fire. Without inhibition, the brain would become epileptogenic. Excitation (E) allows neurons to respond to stimuli, while inhibition (I) tunes their selectivity and enables precise neural representations(1,2). E/I balance is necessary for optimal neural signal formation, synchrony, and transmission, which support information processing driving both simple and complex behaviors. E/I balance breakdowns can have profoundly disabling behavioral effects. Critically, clinical neuroimaging may offer in vivo measurement of E/I balance integrity arising from specific patterns of dysfunction (Table 1).
Table 1.
Consequences of Elevated versus Reduced E/I Ratio.
| Excitation | Inhibition | Predicted Neural Consequences |
|---|---|---|
| Adaptive | Adaptive | Balanced E/I ratio. There is moderate spontaneous baseline activity. Neurons are excitable in response to incoming input, but also well tuned and capable of filtering out irrelevant input. Circuits are well organized and differentiated, capable of synchronization and signal transmission. Circuits are able to support information processing underlying both simple and complex behavior. |
| Increased or Adaptive | Adaptive or Reduced | Elevated E/I ratio. At baseline, circuits may exhibit high levels of random firing and be prone to seizure-like activity. Evoked responses to incoming stimuli can be difficult to obtain. When present, however, evoked responses may be exaggerated. Circuits are poorly ‘tuned’. They may respond to inappropriate stimuli. Functionally-relevant macro-circuitry is hypothesized to be poorly organized, resulting in inefficient and ineffective signal transmission and information processing. Behaviorally, responses to sensory signals may be exaggerated and inappropriate, whereas more complex behavior will be impaired. |
| Adaptive | Increased | Reduced E/I ratio. Spontaneous baseline activity is low and evoked responses to incoming stimuli are limited or blunted. Circuits will be narrowly tuned, to the extent that they are unable to respond to a full range of stimuli. Circuitry will be poorly organized and integrated due to limited opportunities for tuning and synchronization among signals. |
E/I imbalance has been hypothesized as one broad, microcircuit-based alteration underlying brain dysfunction across neurodevelopmental disorders (NDs), including autism spectrum disorder (ASD) and schizophrenia (SCZ)(3–6) (Box S1 for operationalization and commentary regarding this hypothesis). Across disorders, the underlying assumption is that increased E/I ratio (i.e., increased excitation and/or decreased inhibition) drives core symptoms. High epilepsy rates in ASD support this notion(7). ASD and SCZ overlap in their clinical presentation (e.g., social dysfunction, sensory abnormalities)(8), genetics, and neurobiology(9,10) (Fig. 1). However, clear dissociations in clinical phenotype (e.g., hallucinations in SCZ, hand-flapping in ASD), neural alterations, and developmental timing of ASD and SCZ exist. Recently, Gao and Penzes(11) discussed overlapping genetic and molecular evidence implicating E/I imbalance across ASD and SCZ. They highlighted genetic, postmortem, and animal findings suggesting both glutamatergic and GABAergic circuit dysfunction across disorders. These emergent findings emphasize the importance of understanding cross-diagnostic mechanisms affecting E/I balance. A cross-diagnostic approach is consistent with NIMH’s Research Domain Criteria (RDoC) initiative(12), which aims to identify neurobiological processes underlying symptom dimensions spanning psychiatric disorders and testable across analytic levels. Currently, testing within the RDoC framework is limited by few studies incorporating multiple categorical diagnoses for direct comparisons, and absence of transdiagnostic symptom ratings in single disorder studies. Here we take a clinical neuroscience perspective, highlighting emerging evidence from human neuroimaging studies testing markers of E/I imbalance in cortical microcircuits. Though no studies have examined E/I balance cross-diagnostically, we evaluate evidence from parallel ASD and SCZ literatures in considering shared and divergent pathways. We discuss the problem whereby ‘E/I imbalance’ becomes yet another overly-general hypothesis, with minimal mechanistic precision or predictive power, for explaining diverse symptomatology (Box S1). To address this challenge, we discuss where E/I imbalance contributes to specific symptoms that may be constrained developmentally or neuroanatomically. Finally, we argue that refining the E/I imbalance hypothesis should occur cross-diagnostically with an ultimate goal of informing novel treatments targeting related pathways across NDs.
Figure 1. Considering the Complexity of E/I Imbalance Effects on the Cortical Microcircuit Level in the Context of Shared versus Distinct Neurobiology in Schizophrenia and Autism Spectrum Disorder.

1A. Several genes associated with GABAergic and glutamatergic functioning have been implicated across both ASD and SCZ. 1B. The expression of such genes, and the time course at which their expression might go awry, can differ across development. Developmental differences in gene expression affecting E/I balance could contribute to variations in both neural circuitry alterations and ultimate clinical phenotypes. Different colors conceptually highlight distinct time courses and time-dependent patterns of gene expression that may relate to disturbances in each disorder. 1C. Alterations, such as deletions or duplications, of genes altered in SCZ and/or ASD can result in microcircuit dysfunction, characterized by imbalance in E/I neurotransmission, as a result of changes at E->E, E->I, or I->E synapses. 1D. Adapted from(163). The nature of E/I disruption can take any of several different forms (left panel), which in turn would contribute to variable baseline and task-evoked abnormalities in excitatory and inhibitory neural functions. 1E. Based on the complex interactions between the processes depicted in panels 1A–1D, differential neuropathology may emerge from many of the same underlying alterations and may be characterized by regional variability in the degree to which E/I balance is disturbed(101), thereby differentially impacting neural computation at the system level(175). Surface models adapted from Glasser et al.(176).
Magnetic Resonance Spectroscopy
Proton Magnetic Resonance Spectroscopy (1H-MRS) measures total voxel metabolite levels (combined across multiple cellular and extracellular compartments) correlating with neural structure and metabolic alterations(13). Across ASD and SCZ, studies report diagnosis-related alterations in N-acetylaspartate, GABA, glutamate, and glutamine levels (normalized to water or creatine). Particular interest has developed in the glutamine/glutamate combination (Glx), as glutamate released during neurotransmission is taken up by glia and converted to glutamine(14). No uniform increase in glutamate/glutamine or decrease in GABA exists across all ASD or SCZ patients. However, where present, metabolic alterations provide indirect support for cross-diagnostic E/I imbalance. Moreover, 1H-MRS metabolite levels, particularly degree of hyper-glutamatergia, correlate with symptoms, are affected by medication, and, in SCZ, change with illness progression.
In SCZ, increased glutamine in dorsal anterior cingulate cortex (ACC) associates with more psychotic symptoms(15) and worse neuropsychological performance in first-episode (FE-SCZ) patients(16). Higher ACC glutamine/creatine ratio associates with more negative symptoms in early-course (EC-SCZ) patients and correlates with reduced likelihood of remittance(17). Glutamate levels are consistently elevated across striatal(18), frontal, prefrontal, and ACC(19) regions in medication-naïve FE-SCZ patients, and medial prefrontal cortex (mPFC) Glx is elevated in unmedicated patients(20). Increased temporal and frontal Glx relates to particularly severe auditory hallucinations(21), while increased inferior parietal white matter Glx relates to symptom severity and psychotic exacerbations(22). Higher frontal Glx/Cr levels may predict poorer antipsychotic medication response(23). In medicated SCZ patients, glutamate is decreased(24) or unchanged(15) in ACC, decreased in PFC(25), and possibly decreased in hippocampus(24,26). Glx is also reduced in ACC(27) and mPFC of chronic patients, but not EC-SCZ or ultra-high-risk (UHR-SCZ) patients(28). Thus, measured glutamate and glutamine levels, while elevated early in illness, may normalize over illness progression, be sensitive to medication status, and specifically relate to clinical profiles and treatment response. However, possible confounds of long-term polypharmacy remain unresolved(29).
In ASD, less is known about metabolite changes over illness duration, in part because ASD onset occurs before the age when 1H-MRS studies have been implemented. However, ACC Glx is increased in ASD children(30) but reduced and predictive of greater symptom severity in ASD adults(31). These findings point toward a possible progressive shift, as observed in SCZ. Overall, metabolic alterations may correlate with particular symptoms in ASD and be more regionally specific than in SCZ. For example, one study found decreased ACC GABA/creatine ratio corresponding to more impaired social functioning(32), whereas another found no overall changes in ACC GABA, but lower GABA/creatine ratio with more severe symptoms(33). Whereas reduced GABA characterizes auditory and motor regions(34,35), GABA/creatine ratios may be unaffected in visual regions, but aberrantly linked to visual performance(36), Glutamate is increased in ACC(37) and putamen(38) but decreased in medial temporal lobe(37) and unchanged in caudate and thalamus(38). Glx is increased in auditory cortex(39), decreased in basal ganglia, and unchanged in dorsolateral PFC and parietal regions(40). Increased ACC glutamine (32) may relate to symptom severity, specifically emotion recognition deficits. Thus, metabolic evidence for E/I imbalance in ASD points to regional variability. Nonetheless, the direction of disruption in ASD tends toward cortical disinhibition, particularly in childhood. Future cross-diagnostic work should verify the extent and functional consequence of altered GABA and glutamate levels using convergent multi-modal tools. Specifically, studies collecting multi-modal imaging data alongside 1H-MRS will inform mechanistic interpretations of the E/I imbalance hypothesis.
Electrophysiology and Magnetoencephalography
Modulation of local neuronal activity affects broad electrical neural activity profiles, during rest and task states, as measured with electroencephalography (EEG) and magnetoencephalography (MEG). Greater excitation (and higher glutamate levels(41)) associates with larger event-related potentials (ERPs; i.e., peaks in brain activity time-locked to stimuli). Increased E/I ratio is also reflected in the power spectra of spontaneous, oscillatory brain activity, particularly in high-frequency (gamma; 30–90Hz) bands((42), but see(43)). Neural disinhibition yields higher baseline activity across frequency bands and failure of event-related activity to modulate, adapt, or be suppressed over repeated events(44). Pharmacological models altering GABAergic and glutamatergic neurotransmission confirm excitatory and inhibitory signaling contribute significantly to high-frequency oscillations(45,46). Thus, higher baseline excitation and altered stimulus-related evoked response can result from increased excitation, decreased inhibition, or a combination. Though oscillatory findings below largely describe high-frequency abnormalities, phase-amplitude coupling must be considered(47). Whether gamma abnormalities are downstream consequences of primary low-frequency (e.g., delta, theta) deficits should be explored empirically. Additionally, when comparing gamma-related ASD and SCZ findings, the influence of developmental changes in gamma rhythms on observed differences between child and adult samples must be considered(48).
In SCZ, both resting and stimulus-evoked gamma-band alterations are well documented and indicative of cortical disinhibition(49,50). The prevailing view indicates SCZ is characterized by elevated baseline gamma alongside reduced stimulus-evoked gamma response. The latter set of findings could either indicate reduced task-related excitatory signal(51) or result from an interaction between baseline alterations and task responses, resulting in difficulty detecting task-related signal given high baseline power(52). Heightened resting gamma power(53,54) supports a hyper-excited baseline state. Additionally, resting oscillatory connectivity (increased delta, theta, low beta, and gamma) is altered in SCZ. Delta and gamma hyper-connectivity is most pronounced in EC-SCZ(55), whereas alpha connectivity is decreased regardless of illness duration(55,56).
Task-based EEG has provided further support for elevated E/I ratio in SCZ. Amplitude of visual evoked potentials (VEPs) characterizes visual stream integrity (Fig. 2A), where specific early peaks reflect glutamatergic and GABAergic functioning. VEP alterations in SCZ are well established(57). For instance, a recent study comparing binocular VEPs to summed VEPs under monocular deprivation found reduced VEP plasticity, indexed by this binocular effect(58). Other EEG and MEG evidence in the visual modality offers convergent support for increased E/I ratio. Deviance detection during low-level visual feature (orientation) perception is reduced in SCZ(59), suggesting inhibitory failure over repeated stimuli. Furthermore, increased sustained gamma power to visual gratings characterizes schizoaffective disorder(60), wherein mood disturbance coincides with psychosis. Even medication-naïve EC-SCZ patients show atypical EEG patterns during tests of perceptual closure. These findings include widespread elevations in MEG responses, poor response modulation with stimulus repetitions, decreased high-gamma (60–120Hz) power, and reduced gamma-beta coupling(61,62). Collectively, EEG findings in SCZ suggest abnormal excitatory activity spreading, failure to gate (inhibit) responses, impaired high-frequency oscillation generation, and failure to down-regulate task-irrelevant activity during visual perception. These deficits reflect neural disinhibition.
Figure 2. Neuroimaging Paradigms Tapping into E/I Balance in Sensory and Associative Circuits.
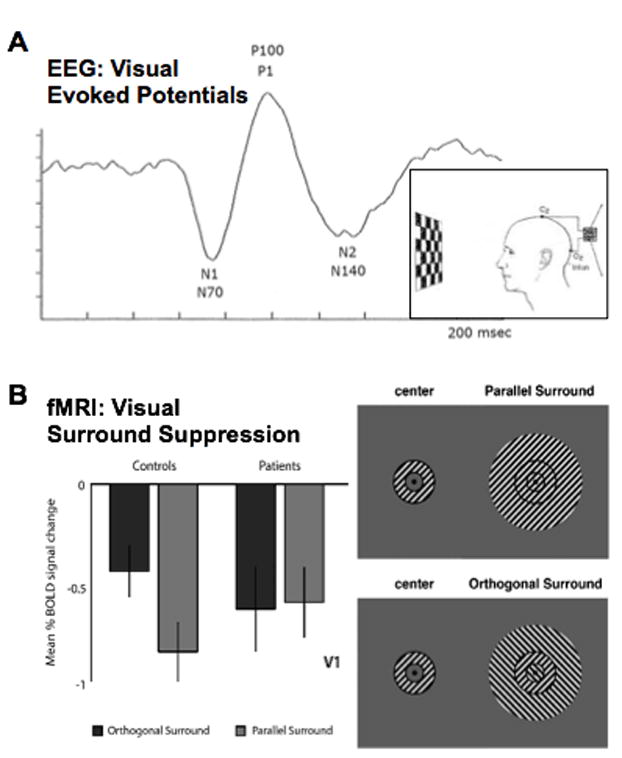
2A. Visual Evoked Potential (VEP) paradigm, in which EEG is recorded over occipital cortex to contrast-reversing checkerboard stimuli. This paradigm results in a canonical waveform, wherein successive peaks reflect glutamatergic and GABAergic activity. It has been used in both SCZ and ASD. 2B. Adapted from Seymour et al.(123). Surround suppression stimuli, wherein activation elicited by a grating-filled annulus is suppressed due to lateral inhibition in the context of parallel (top right panel) but not perpendicular (bottom right panel) surround. Seymour and colleagues(123) showed that patients with SCZ exhibit reduced surround suppression effects (left panel). This type of paradigm is also being used in studies of individuals with ASD.
Strong evidence for atypical auditory processing and higher cognitive functioning in SCZ(63,64) also implicate E/I imbalance(65–67). Reduced auditory steady-state responses (ASSR) entrained to periodic stimuli are a hallmark SCZ feature(68,69). This alteration corresponds to reduced gamma phase-locking, suggesting GABAergic dysregulation(68). Indeed, GABA levels are associated with theta, alpha, and beta activity gating during auditory tasks(70). However, in a separate study, higher induced gamma power during 40Hz ASSR was found in SCZ and associated with more auditory hallucinations(71). The direction of alterations may be a function of NMDA transmission(72). Reduced fronto-central alpha activity, alongside increased parietal-occipital alpha, suggests deficient long-range inhibition of task-irrelevant auditory information, perhaps driving SCZ-related perceptual alterations(73). De-synchronization of event-related alpha during auditory perception is also reported in UHR-SCZ(74). Reduced gamma response and phase-locking in similar tasks characterize EC-SCZ and UHR-SCZ patients who had not converted(75). These results suggest electrophysiological markers of E/I imbalance may precede full-blown illness, indicating neural markers could be effective for tracking risk. This idea is further supported by findings of altered ASSR in unaffected relatives(76,77). Mechanistically, premorbid damage to superior temporal gyrus cortical thickness may underlie downstream alterations in gamma oscillations during auditory entrainment(78).
Finally, during higher-order cognition, SCZ patients show reduced gamma during working memory, which requires active maintenance of information over time. Reduced task-evoked gamma over DLPFC correlates with reduced GABA, measured with 1H-MRS(79), suggesting a shared mechanism. Interestingly, repeated transcranial magnetic stimulation (TMS) over DLPFC can normalize excessive gamma oscillations and improve cognitive performance(80). Combined, these findings imply a specific link between disinhibition and impaired cognition and a possible therapeutic approach for targeting E/I imbalance-associated alterations.
In ASD, resting-state EEG and MEG findings paint a somewhat different picture. As in SCZ, resting delta, theta, and gamma oscillatory power are elevated(81). However, in contrast to decreased resting alpha connectivity in SCZ, in ASD, resting-state alpha activity is increased(82). Moreover, though long-range alpha connectivity is increased in ASD, signal complexity in this range is decreased, which could reflect decreased predictability, regularity, and repeatability of neuronal signals(83). Thus, resting-state EEG signatures of E/I imbalance across ASD and SCZ may be, at least in part, dissociable, particularly for alpha.
Task-based ERP signatures of E/I imbalance have been characterized in genetic syndromes causing ASD and impacting excitatory or inhibitory synaptic functioning. Individuals with mutations of NLGN4X-coding genes, expressed in inhibitory and excitatory synapses(84), were more likely to exhibit ASD and showed abnormal ERP to auditory deviance detection. In Fragile X syndrome, both auditory and visual ERP amplitudes were increased(85). This finding suggests hyper-excitable neural responses, most prominent in audition, in a genetic syndrome with known glutamate disruption and causing ASD. In idiopathic ASD, task-based EEG correlates of E/I imbalance have been tested less extensively. Largely using low-level sensory tasks, these studies yield mixed results. During tactile perception, target finger stimulation activates both its representation in somatosensory cortex and representations of adjacent fingers due to local intra-cortical connections. Cortical inhibition controls neural response amplitude to adjacent finger stimulation. In ASD, MEG revealed local hypo-connectivity and enhanced inhibition comparing individual versus concurrent tactile stimulation of adjacent fingers(86,87), suggesting reduced E/I ratio. In audition, reduced gamma phase locking during ASSR parallels SCZ findings(88). Though this potential E/I imbalance biomarker has been studied less extensively in ASD, unaffected relatives show similar phase locking decreases(89). Thus, as in SCZ, ASSR alterations may be useful for tracking risk. Mechanistically, there are cortical thickness deficits in superior temporal regions in ASD, but these tend to be greater in adults than children(90). Therefore, whereas cortical thickness reductions may precede illness onset and E/I imbalance in SCZ, ASSR gamma abnormalities in ASD may be driven by a different mechanism, such as auditory cortex GABA reductions (described above).
In the visual domain, ASD findings are mixed. Two studies suggest elevated E/I ratio, showing reduced steady-state gamma and orientation-specific contextual modulation and increased neural noise to VEP stimuli(91,92). Conversely, in parallel studies of ASD and controls, higher peak gamma frequency and lower orientation discrimination thresholds (both associated with more precise circuit tuning and greater inhibition) were found in both ASD and neurotypical individuals with fewer autistic traits(93,94). While these findings may seem contradictory, they could suggest the direction of E/I imbalance plays a role in determining clinical phenotype and functional impairment. Findings in neurotypical individuals with more autistic traits may relate to ‘state’ versus ‘trait’ effects or to severity level needed to ‘unmask’ task-evoked deficits. Findings showing modulation of MEG response to repeated sounds was only reduced in ASD individuals with clinical auditory hypersensitivity(95) support this hypothesis. Overall, patterns of E/I imbalance may manifest more heterogeneously in ASD than SCZ. Specifically, disinhibition may be constrained to distinct processes and circuitry, perhaps underlying discrete phenotypes in ASD subsets.
Functional Neuroimaging
Functional MRI offers an additional tool for mapping neural correlates of E/I disruption, exploring their regional constraints, and testing functional correlates of computationally-based circuit predictions. The following section highlights a selection of recently published fMRI papers. It is not meant to be exhaustive or exclusionary. Rather, it highlights illustrative studies reporting findings that can be computationally modeled, translated, or integrated in multimodal neuroimaging approaches.
Resting-State fMRI
Resting-state functional neuroimaging (rs-fMRI) enables mapping the macro-organization of large-scale functional brain networks(96–99). Network-level disruptions should be prominent when local microcircuit function is disrupted, since precise E/I balance is critical for formation and maintenance of organized local and large-scale circuits. In that sense, rs-fMRI reflects large-scale network consequences of local circuit disruptions(100,101). In SCZ, studies have mapped resting network dysconnectivity, which may relate to altered E/I balance. Recent work identified altered resting thalamo-cortical connectivity, including reduced thalamic-prefrontal-cerebellar connectivity and elevated thalamic-sensory-motor connectivity(102–105). Similarly, resting hyper-connectivity characterizes association cortices, including frontal-parietal control network, but not sensory networks(101). Bi-directional findings suggest cortical disinhibition specifically altering top-down control, thereafter differentially affecting communication between thalamus and frontal/sensory regions. Cortico-striatal functional connectivity alterations also support neural disinhibition in SCZ(106). There are known inhibitory glutamatergic projections onto dorsal striatum, which exhibits altered functional connectivity in EC-SCZ(97,107). In healthy adults, increased mPFC glutamate associates with greater resting functional connectivity between mPFC and both thalamic and striatal regions(108). This finding converges with increased glutamate (and elevated E/I balance) being functionally linked with striatal and thalamic rs-fMRI connectivity differences in SCZ.
Echoing the 1H-MRS literature, illness progression represents an important emerging variable in SCZ rs-fMRI studies. This picture is complicated by treatment confounds: studies in EC-SCZ report rs-fMRI alterations, but the direction of reported effects varies depending on treatment. For example, in unmedicated SCZ, hippocampus and precuneus connectivity is decreased(109). In EC-SCZ, altered effective connectivity across ‘default-mode’ network nodes are reported(110), consistent with data-driven studies(107). Others report PFC hyper-connectivity is specific to EC-SCZ, whereas PFC connectivity reductions characterize chronic SCZ(111). Hyper-connectivity in medication-naïve EC-SCZ was most prominent in PFC circuits and default-mode regions and predicted positive symptom severity(107). Interestingly, in EC-SCZ patients followed longitudinally for 12 months, this functionally ‘hyper-connected’ state attenuated and correlated with symptom improvements after treatment initiation(107). This effect underscores the need to consider illness duration, medication status, and symptom severity during study participation when assessing E/I imbalance markers. Symptom improvement with PFC connectivity normalization suggests potential therapeutic value of normalizing E/I imbalance. However, precise upstream mechanisms driving this neuroimaging observation remain unknown. Combining animal, pharmacological and computational studies will be critical to mechanistically characterize these findings.
Fewer rs-fMRI studies have examined functional connectivity in ASD as a possible neural correlate of E/I imbalance. Adults with ASD, most closely age-matched to SCZ samples, show local hyper-connectivity in superior and middle frontal gyrus, local hypo-connectivity in fusiform and middle temporal gyri, and no alterations in long-range connections(112). A second study in ASD adults also found no evidence for whole-brain alterations in connectivity. Conversely, however, evidence here supported decreased functional connectivity in specific frontal and temporal brain regions(113), with no evidence for hyper-connectivity. Another study reported frontal-striatal connectivity reductions, alongside alterations in the developmental trajectory of striatum-putamen connections, where these connections increased with age in ASD(114) but declined with age in controls.
In ASD children, a mixed pattern of long-range hyper- and hypo-connectivity has been identified, with directionally disparate alterations across regions and networks(115–118). Importantly, all studies suggested relationships between connectivity alterations and social symptom severity. Alterations in resting-state connectivity also associated with change in symptoms and adaptive impairments over time(98). This work collectively emphasizes the importance of considering age and developmental stage, both when designing studies and when comparing findings across studies and across disorders where onset ages differ. Moreover, it highlights the need to examine correlations between rs-fMRI connectivity and continuous measures of symptomatology ttranscending diagnostic category. In general, functional connectivity measured via rs-fMRI may most specifically differentiate SCZ from controls, whereas it may be less reliably altered in ASD, particularly by adulthood. If this hypothesis holds, it suggests dissociation in the expression of E/I imbalance-related neural alterations for large-scale networks in clinical populations with notable differences in core phenotypes.
Task-Based fMRI
Few task-based fMRI assays of E/I imbalance have been conducted in ASD or SCZ, despite behavioral studies suggesting alterations in processes modulated by E/I balance(119–122). In SCZ, orientation-specific context modulation during visual perception was reduced, suggesting inhibitory mechanism failure((123) (Fig. 2B). Hyper-activation, consistent with disinhibition, was observed in auditory cortex following presentation of single-tone stimuli(124). Higher glutamate levels (measured at rest by 1H-MRS) associated with greater inferior parietal BOLD signal during auditory cognitive control in SCZ, whereas this correlation was negative in controls(125). These task-based findings converge with the E/I imbalance hypothesis in SCZ, with the third study suggesting a functional link between task-related hyper-excitability and altered glutamate neurotransmission.
In ASD, task-based neuroimaging studies testing for E/I imbalance support disinhibition in local cortical circuits. During visual motion perception, greater activation and faster hemodynamic decay was seen in V5/MT, suggesting reduced inhibitory modulation(126). During passive language processing, negative BOLD responses were reduced in ASD, suggesting failure of inhibitory processes to induce regional deactivation(127). In controls, reduced orientation-specific surround suppression, reliant on lateral inhibition in visual receptive fields, associated with more autistic traits(128). Thus, task-based fMRI studies suggest neural disinhibition may associate with autism symptomatology in a continuous fashion, spanning into non-clinical populations.
In both ASD and SCZ literatures, there is significant need to expand approaches using task-based fMRI to test for E/I imbalance. In particular, fMRI may be used best in conjunction with EEG. Given EEG’s poor spatial resolution alongside clues from EEG studies that disinhibition may be regionally specific in ASD, multimodal studies including fMRI may be particularly important for testing E/I imbalance in ASD. Our ability to draw conclusions from fMRI studies will be maximized by (and may be of only incremental return without) simultaneous or parallel EEG and/or MEG studies to explain how oscillatory activity and neurotransmitter alterations contribute to BOLD signal abnormalities(129). This process will be key to understanding how task-based neuroimaging findings reflect specific alterations in excitatory and inhibitory neurotransmission and ratio.
Looking Forward: Pharmacological, TMS, Genetic Disorders, and Computational Studies Informing Probes of the Cross-diagnostic E/I Imbalance Hypothesis
All of the reviewed neuroimaging work, while critical to our understanding of E/I imbalance across ASD and SCZ, cannot ultimately reveal mechanisms by which E/I balance becomes disrupted (Box S1). Inconsistencies across the SCZ and ASD literatures likely reflect remaining uncertainty regarding underlying disease mechanisms. However, particularly for SCZ, defined models of synaptic and microcircuit dysfunction have been helpful in articulating predictions of both functional impairment and potential treatment targets. Viewing SCZ as resulting from disrupted glutamatergic activity affecting only specific forms of inhibition yields testable hypotheses regarding which neural functions ought to reflect E/I imbalance and which not. For example, SCZ genes tend to be expressed throughout cortex and affect both somatostatin and parvalbumin inhibitory cells(130). Somatostatin cells are more densely expressed in superficial cortical levels and synapse on dendritic shafts and spines of pyramidal neurons, whereas fast-spiking parvalbumin interneurons are expressed in deeper cortical layers and target pyramidal neuron cell bodies(131,132). Therefore, whereas reduced parvalbumin interneuron cell function results in increased resting and reduced evoked gamma oscillations(133), altered somatostatin cell function may contribute to functional hyper-connectivity due to these cells spreading horizontally across neighboring cortical columns.
With this backdrop, pharmacological neuroimaging in SCZ has identified potential mechanisms driving E/I imbalance that affect described neuroimaging assays. In particular, transient administration of the N-methyl-D-aspartate-receptor (NMDAR) antagonist ketamine to healthy controls increases global functional connectivity(134), similar to observations in EC-SCZ(107). Moreover, acute ketamine induces aspects of the SCZ clinical phenotype and results in reduced task-dependent activation and connectivity during spatial working memory(134,135). Interestingly, a study combining ketamine administration with other potential glutamate release modulators suggests that, at least with ketamine, connectivity changes occur due to NMDAR blockade rather than downstream glutamatergic effects(136). This finding is consistent with arterial spin labeling studies on this topic(137). It may have important implications for identifying drug targets related to altered NMDAR signaling in SCZ, which likely contribute to E/I imbalance(138). While pharmacological models of SCZ in healthy controls have provided important tests of mechanistic predictions for explaining the SCZ disease state, as yet, no parallel model exists for ASD. In ASD, gaps in our knowledge regarding the underlying pathology make articulating clear directional hypotheses more difficult, such that research has pursued testing E/I imbalance in a more haphazard way. At this time, no pharmacological agent has been identified that replicates key features of the ASD phenotype or hallmark experimental findings. Thus, ASD research lags behind SCZ in its utilization of pharmacological models to test precise mechanistic hypotheses.
Across disorders, pharmacological trials in patient populations have been helpful in informing whether manipulating E/I balance directly can alter neural signal and clinical presentation. To this end, in ASD, long-term bumetanide administration, which decreases intracellular chloride and reinforces GABA’s inhibitory effect, enhanced BOLD activation during emotional face perception(139). Acute administration of benzodiazepines, which are GABA agonists, increased occipital and prefrontal BOLD signal while viewing emotional images in SCZ, though it decreased signal in the same regions in controls(140,141). These pharmacological studies are consistent with alterations in GABAergic and glutamatergic functioning across ASD and SCZ. They further implicate disinhibited cortical circuits that can be normalized using specific pharmacological agents. By pairing drug trials with bench work to understand specific targets and action mechanisms for successful pharmacological agents, we stand to learn more about both treatment and underlying dysfunction. Interestingly, recent work suggests that cognitive training can normalize neural markers of altered auditory gating in SCZ, suggesting non-pharmacological interventions may also be successful in modulating and normalizing neural responses associated with cortical disinhibition(142).
Another complementary experimental method to unpack the described neuroimaging effects involves TMS. TMS is a noninvasive technique that induces electrical current in the brain. This current causes depolarization of neurons and generation of action potentials(143), which can produce a hyper-excitable state. Indeed, repeated TMS over right DLPFC in healthy controls increased network connectivity among areas supporting working memory function(144). This finding suggests induced cortical disinhibition can result in alterations in both cognition reminiscent of SCZ and in its underlying neural circuitry. Modeling SCZ and ASD disease states in healthy controls using TMS will inform our understanding of underlying mechanisms, particularly as related to clarifying affected brain regions.
Yet another important approach involves studies in NDs caused by known genetic alterations in which a substantial proportion of affected individuals have a clinical phenotype closely resembling idiopathic ASD or SCZ. As compared to pharmacological modeling, which has been more illuminating for SCZ, work in rare genetic disorders thus far has offered more insight into the neurobiology of ASD. Phelan-McDermid Syndrome is a genetic disorder caused by terminal deletions of the chromosomal region 22q13.3, which includes the SHANK3 gene that encodes structural components of excitatory synapses(145). Affected patients have many features of ASD. Using clinical neuroscience techniques to test for specific markers of E/I imbalance in this and other genetic populations (e.g., Fragile X, Rett syndromes) may increase understanding of the pathway from genetic abnormality, to neural microcircuit alteration, to differences in neural signaling detectable via neuroimaging, to specific clinical phenotypes. 22q11.2 syndrome presents an interesting population for probing experimental correlates of E/I imbalance, as ASD and SCZ are both common in 22q11.2 individuals(146,147). Thus, studies in 22q11.2 could be useful in teasing apart which aspects of E/I imbalance correspond to which aspects of shared or divergent clinical phenotypes among individuals sharing a known genetic alterations.
Finally, emerging ‘computational psychiatry’ focuses on biophysically-grounded modeling of neural networks. This field has begun to generate mechanistic behavioral and neuroimaging predictions relevant for SCZ and ASD(148–153). This approach incorporates the relevant synaptic detail necessary to generate downstream neural predictions after upstream perturbations to parameters regulating E/I balance. For instance, a computational model altering inhibitory signals in auditory cortex replicates auditory entrainment deficits observed in SCZ patients(154). Computational modeling studies have also demonstrated that elevating E/I balance in a microcircuit capable of working memory computations results in a specific pattern of behavioral errors(2). This hypothesized ‘disinhibited’ behavioral and neural regime was confirmed experimentally following NMDAR antagonism(138,153). Finally, a computational perturbation simulating disinhibition in a large-scale functional connectivity model predicted neural activity patterns confirmed in resting state scans from SCZ patients. Critically, observed alterations in patients were diagnosis-specific, not seen in either controls or bipolar disorder patients(100,101). In ASD, computational models reducing circuit inhibition(155) have been useful in modeling predictions for perceptual alterations identified in behavioral studies(156), but have not yet modeled neuroimaging predictions. Importantly, however, biophysically-grounded models are capable of modeling synaptic compensations that can generate cross-diagnostic predictions.
Collectively, these cross-disciplinary methods highlight how pairing pharmacological manipulations in healthy and affected individuals with TMS, studies in rare genetic disorders, and computational modeling, can offer promising approaches to test specific mechanistic hypotheses of E/I balance disruptions across disorders (Fig. 3). Such studies will help inform how E/I imbalance may be pervasive or localized to specific brain regions, relate to specific aspects of perceptual, social, and cognitive task performance altered in SCZ and/or ASD, and disrupt particular features of underlying brain activation and connectivity.
Figure 3. Computational and Pharmacological Studies Informing E/I Imbalance Cross-diagnostically.

Figure adapted from(177). 3A. Computational modeling of microcircuit E/I balance predicts neural activity that is disrupted when E/I ratio is elevated (i.e. disinhibition induced via reduction of feedback inhibition shown with red arrow). This computational manipulation generates predictions relevant for E/I balance both at rest and during task states(100, 135, 178). 3B. Pharmacological models, such as NMDAR antagonism via ketamine, known to disrupt E/I balance can be used to test computational models to determine whether well understood in vivo disruptions result in predicted alterations in neural activity. 3C. Findings from patients with SCZ or ASD can then be compared to results generated by computational and pharmacological models to gain a better understanding of the underlying mechanisms driving the disease state. This iterative ‘computational psychiatric’ framework can help deepen insight into the links between circuit mechanism, neural system deficits, and symptoms across diagnoses(179).
Summary and Future Directions
This review highlights recent multi-modal neuroimaging work across ASD and SCZ implicating alterations in E/I balance (Box S2) - a complex property of microcircuits that can take many forms and result from many underlying alterations. However, as knowledge increases regarding the basic cellular, molecular, and circuit-level alterations in SCZ and ASD, very specific predictions associated with particular patterns of E/I imbalance may be generated and tested using multi-modal neuroimaging techniques (Fig. 1). More importantly, as evidence emerges regarding patterns of E/I imbalance in vivo, results can be translated back to animal models, where E/I imbalance can be replicated and pharmacological agents affecting GABAergic and/or glutamatergic signaling can be brought into preclinical trials. Already, this work has begun in ASD using mouse models of monogenic causes of ASD. Both insulin-like growth factor(157) and GABA-receptor agonist Arbaclofen(158) administration rescue the clinical phenotype in ASD mouse models. These initial findings are already being translated to targeted clinical trials of new treatment compounds(159).
Despite a clear need for translational research, some consistent themes emerged across this complex neuroimaging literature (e.g., progressive effects in SCZ apparent in both 1H-MRS and rs-fMRI studies). Additionally, correlations between neural metrics of E/I imbalance and symptom levels were often reported (albeit studies are often underpowered). This pattern suggests the magnitude of E/I imbalance may relate to clinical severity in a graded manner. That said, fMRI, MRS, and EEG/MEG literatures remain equivocal regarding the precise spatial pattern and direction of E/I alterations, particularly in ASD. Cross-diagnostic comparisons must be interpreted cautiously because they emerge from independent literatures. However, it is likely that, within and across ASD and SCZ, regional specificity of affected circuits, cell and receptor types impacted, and developmental timeframe for altered E/I imbalance contribute to heterogeneity in the clinical phenotype.
Several specific, important areas for future study emerged. New cross-diagnostic research, taking an RDoC approach in well clinically characterized groups, will be impactful. The aim here is to determine whether we garner added precision about the presence and consequence of E/I imbalance when we consider its relation to specific cognitive, behavioral, and psychiatric variables across diagnoses. Differences observed over the course of illness in SCZ and between children and adults with ASD point to the importance of understanding whether E/I alterations are temporally stable or fluctuate over development. Some proactive strategies may help address this important challenge for cross-diagnostic studies: i) including careful characterization of typically developing E/I balance patterns; ii) capitalization on high-risk samples and unaffected relatives with shared biomarkers; iii) studies that map ‘state’ versus progressive ‘trait’ markers that change over time (e.g., with development, treatment, disease progression, or symptom exacerbation or remittance); iv) systematic matching of age and medication history in adult ASD and SCZ samples. Next, given variable patterns of E/I disturbance (or lack thereof) across cortical regions and neuroimaging platforms, forthcoming multi-modal studies need to clarify the precise spatial patterns of E/I balance disruptions across brain areas in SCZ and ASD (e.g. combined EEG/fMRI applied cross-diagnostically). Also, focus on rare genetic variants associated with both disorders (e.g., 22q11.2, CNTAP2) and conferring known impact on synaptic function may constrain genetic, biological, and phenotypic heterogeneity within study samples. This approach will enable more power to reveal causal mechanisms. Finally, given the general lack of specificity and robustness of most findings to date, computational modeling and pharmacological challenges can help to test specific experimental hypotheses and precise circuit mechanisms related to putative E/I imbalance in SCZ and ASD. Collectively embracing these cross-diagnostic challenges is critical to guiding targeted treatment development for disorders of neurodevelopment.
Supplementary Material
Acknowledgments
The authors wish to acknowledge the following sources of funding support: NARSAD Young Investigator Award (PI: Foss-Feig), Autism Science Foundation Accelerator Award (PI: Foss-Feig), Seaver Foundation funding (Foss-Feig) NIMH R01 MH107426 (PI: McPartland, Srihari), NIH R01MH103831 (PI: Srihari), NIH RC1 MH088971 (PI: Srihari), NIH TL1 TR000141 (PI: Murray); NIH 1DP5-OD012109 (PI: Anticevic); NIH R01 MH108590 (PI: Anticevic); NARSAD Independent Investigator Award (PI: Anticevic); NIH 1R03MH105765-02 (PI: Anticevic); NIH 1R01MH107426 (Co-I: Anticevic).
Footnotes
Financial Disclosures
Dr. McPartland reported he is an investigator on a biomarker development grant funded by Janssen Research & Development. Dr. Anticevic disclosed that he consults and is a member of the Scientific Advisory Board for and a consultant for BlackThorn Therapeutics. Dr. Krystal disclosed having received consultant fees from AMGEN, AstraZeneca Pharmaceuticals, Biogen, Idec, MA, Biomedisyn Corporation, Forum Pharmaceuticals, Janssen Research & Development, Otsuka America Pharmaceutical, Inc., Sunovion Pharmaceuticals, Inc., Takeda Industries, Taisho Pharmaceutical Co., Ltd; Dr. Krystal also serves on the Scientific Advisory Board at Biohaven Pharmaceuticals, Blackthorn Therapeutics, Inc., Lohocla Research Corporation, Luc Therapeutics, Inc., Pfizer Pharmaceuticals, and TRImaran Pharma, has stock in ArRETT Neuroscience, Inc. and Biohaven Pharmaceuticals Medical Sciences, and has stock options in Blackthorn Therapeutics, Inc. and Luc Therapeutics, Inc. Dr. Krystal reported his patents on “Dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia” (US Patent #:5,447,948), “Glutamate Modulating Agents in the Treatment of Mental Disorders” (US Patent No. 8,778,979 B2), “Intranasal Administration of Ketamine to Treat Depression” (US Application No. 14/197,767; US application or PCT International application No. 14/306,382), “Composition and methods to treat addiction” (Provisional Use Patent Application no.61/973/961), and “Treatment Selection for Major Depressive Disorder” (USPTO docket number Y0087.70116US00), and reported that he received research funding from AstraZeneca Pharmaceuticals, who provides the drug, Saracatinib, for research related to NIAAA grant “Center for Translational Neuroscience of Alcoholism [CTNA-4], and from Pfizer Pharmaceuticals, who provides an investigational drug, PF-03463275, for research related to NIH grant “Translational Neuroscience Optimization of GlyT1 Inhibitor.” Dr. Krystal is also the editor of Biological Psychiatry. All other authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–494. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray JD, Anticevic A, Corlett PR, Gancsos M, Krystal JH, Wang X-J. Linking Microcircuit Dysfunction to Cognitive Impairment: Effects of Disinhibition Associated with Schizophrenia in a Cortical Working Memory Model. Cerebral Cortex. 2014;24:859–872. doi: 10.1093/cercor/bhs370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Global Burden of Disease Study 2010. Lancet 2012 [Google Scholar]
- 4.Lisman J. Excitation, inhibition, local oscillations, or large-scale loops: what causes the symptoms of schizophrenia? Current Opinion in Neurobiology. 2012;22:537–544. doi: 10.1016/j.conb.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes, Brain, & Behavior. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spence SJ, Schneider MT. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatric Research. 2009;65:599–606. doi: 10.1203/01.pdr.0000352115.41382.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstantareas MM, Hewitt T. Autistic disorder and schizophrenia: diagnostic overlaps. Journal of autism and developmental disorders. 2001;31:19– 28. doi: 10.1023/a:1005605528309. [DOI] [PubMed] [Google Scholar]
- 9.State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.State MW, Sestan N. Neuroscience. The emerging biology of autism spectrum disorders. Science. 2012;337:1301–1303. doi: 10.1126/science.1224989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao R, Penzes P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Current molecular medicine. 2015;15:146–167. doi: 10.2174/1566524015666150303003028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC) toward a new classification framework for research on mental disorders. American Journal of Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 13.Mason GF, Krystal JH. MR spectroscopy: its potential role for drug development for the treatment of psychiatric diseases. NMR Biomedicine. 2006;19:690–701. doi: 10.1002/nbm.1080. [DOI] [PubMed] [Google Scholar]
- 14.Yüksel C, Öngür D. Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biological Psychiatry. 2010;68:785–794. doi: 10.1016/j.biopsych.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bustillo JR, Rediske N, Jones T, Rowland LM, Abbott C, Wijtenburg SA. Reproducibility of phase rotation stimulated echo acquisition mode at 3T in schizophrenia: Emphasis on glutamine. Magnetic Resonance in Medicine. 2015 doi: 10.1002/mrm.25638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dempster K, Norman R, Théberge J, Densmore M, Schaefer B, Williamson P. Glutamatergic metabolite correlations with neuropsychological tests in first episode schizophrenia. Psychiatry Research: Neuroimaging. 2015;233:180–185. doi: 10.1016/j.pscychresns.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2012;37:2515–2521. doi: 10.1038/npp.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plitman E, de la Fuente-Sandoval C, Reyes-Madrigal F, Chavez S, Gómez-Cruz G, León-Ortiz P, et al. Elevated Myo-Inositol, Choline, and Glutamate Levels in the Associative Striatum of Antipsychotic-Naive Patients With First-Episode Psychosis: A Proton Magnetic Resonance Spectroscopy Study With Implications for Glial Dysfunction. Schizophrenia bulletin. 2015:sbv118. doi: 10.1093/schbul/sbv118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smesny S, Gussew A, Biesel NJ, Schack S, Walther M, Rzanny R, et al. Glutamatergic dysfunction linked to energy and membrane lipid metabolism in frontal and anterior cingulate cortices of never treated first-episode schizophrenia patients. Schizophrenia research. 2015;168:322–329. doi: 10.1016/j.schres.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, et al. Elevated prefrontal cortex γ-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Archives of general psychiatry. 2012;69:449–459. doi: 10.1001/archgenpsychiatry.2011.1519. [DOI] [PubMed] [Google Scholar]
- 21.Hugdahl K, Craven AR, Nygård M, Løberg E-M, Berle JØ, Johnsen E, et al. Glutamate as a mediating transmitter for auditory hallucinations in schizophrenia: A 1 H MRS study. Schizophrenia research. 2015;161:252–260. doi: 10.1016/j.schres.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 22.Ota M, Ishikawa M, Sato N, Hori H, Sasayama D, Hattori K, et al. Glutamatergic changes in the cerebral white matter associated with schizophrenic exacerbation. Acta Psychiatrica Scandinavica. 2012;126:72–78. doi: 10.1111/j.1600-0447.2012.01853.x. [DOI] [PubMed] [Google Scholar]
- 23.Szulc A, Konarzewska B, Galinska-Skok B, Lazarczyk J, Waszkiewicz N, Tarasow E, et al. Proton magnetic resonance spectroscopy measures related to short-term symptomatic outcome in chronic schizophrenia. Neuroscience letters. 2013;547:37–41. doi: 10.1016/j.neulet.2013.04.051. [DOI] [PubMed] [Google Scholar]
- 24.Gallinat J, McMahon K, Kühn S, Schubert F, Schaefer M. Cross-sectional Study of Glutamate in the Anterior Cingulate and Hippocampus in Schizophrenia. Schizophrenia bulletin. 2015:sbv124. doi: 10.1093/schbul/sbv124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsman A, Mandl RC, Klomp DW, Bohlken MM, Boer VO, Andreychenko A, et al. GABA and glutamate in schizophrenia: A 7 T 1 H-MRS study. NeuroImage: Clinical. 2014;6:398–407. doi: 10.1016/j.nicl.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stan A, Ghose S, Zhao C, Hulsey K, Mihalakos P, Yanagi M, et al. Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Molecular psychiatry. 2014 doi: 10.1038/mp.2014.54. [DOI] [PubMed] [Google Scholar]
- 27.Rowland LM, Kontson K, West J, Edden RA, Zhu H, Wijtenburg SA, et al. In vivo measurements of glutamate, GABA, and NAAG in schizophrenia. Schizophrenia bulletin. 2012:sbs092. doi: 10.1093/schbul/sbs092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Natsubori T, Inoue H, Abe O, Takano Y, Iwashiro N, Aoki Y, et al. Reduced frontal glutamate+ glutamine and N-acetylaspartate levels in patients with chronic schizophrenia but not in those at clinical high risk for psychosis or with first-episode schizophrenia. Schizophrenia bulletin. 2014;40:1128–1139. doi: 10.1093/schbul/sbt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsman A, van den Heuvel MP, Klomp DWJ, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in Schizophrenia: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophrenia Bulletin. 2013;39:120–129. doi: 10.1093/schbul/sbr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bejjani A, O’Neill J, Kim JA, Frew AJ, Yee VW, Ly R, et al. Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PloS one. 2012;7:e38786. doi: 10.1371/journal.pone.0038786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Elst LT, Maier S, Fangmeier T, Endres D, Mueller G, Nickel K, et al. Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: evidence in support of the excitatory/inhibitory imbalance hypothesis. Molecular psychiatry. 2014;19:1314–1325. doi: 10.1038/mp.2014.62. [DOI] [PubMed] [Google Scholar]
- 32.Cochran DM, Sikoglu EM, Hodge SM, Edden RA, Foley A, Kennedy DN, et al. Relationship among Glutamine, γ-Aminobutyric Acid, and Social Cognition in Autism Spectrum Disorders. Journal of child and adolescent psychopharmacology. 2015;25:314–322. doi: 10.1089/cap.2014.0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brix MK, Ersland L, Hugdahl K, Grüner R, Posserud M-B, Hammar Å, et al. Brain MR spectroscopy in autism spectrum disorder—the GABA excitatory/inhibitory imbalance theory revisited. Frontiers in human neuroscience. 2015:9. doi: 10.3389/fnhum.2015.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaetz W, Bloy L, Wang D, Port R, Blaskey L, Levy S, et al. GABA estimation in the brains of children on the autism spectrum: measurement precision and regional cortical variation. NeuroImage. 2014;86:1–9. doi: 10.1016/j.neuroimage.2013.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rojas DC, Singel D, Steinmetz S, Hepburn S, Brown MS. Decreased left perisylvian GABA concentration in children with autism and unaffected siblings. NeuroImage. 2014;86:28–34. doi: 10.1016/j.neuroimage.2013.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robertson CE, Ratai EM, Kanwisher N. Reduced GABAergic Action in the Autistic Brain. Current Biology. 2016;26:80–85. doi: 10.1016/j.cub.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 37.Joshi G, Biederman J, Wozniak J, Goldin RL, Crowley D, Furtak S, et al. Magnetic resonance spectroscopy study of the glutamatergic system in adolescent males with high-functioning autistic disorder: a pilot study at 4T. European archives of psychiatry and clinical neuroscience. 2013;263:379–384. doi: 10.1007/s00406-012-0369-9. [DOI] [PubMed] [Google Scholar]
- 38.Doyle-Thomas KA, Card D, Soorya LV, Wang AT, Fan J, Anagnostou E. Metabolic mapping of deep brain structures and associations with symptomatology in autism spectrum disorders. Research in autism spectrum disorders. 2014;8:44–51. doi: 10.1016/j.rasd.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown MS, Singel D, Hepburn S, Rojas DC. Increased Glutamate Concentration in the Auditory Cortex of Persons With Autism and First - Degree Relatives: A 1H - MRS Study. Autism Research. 2013;6:1–10. doi: 10.1002/aur.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horder J, Lavender T, Mendez M, O’Gorman R, Daly E, Craig M, et al. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [1H] MRS study. Translational psychiatry. 2014;4:e364. doi: 10.1038/tp.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall M-H, Jensen JE, Du F, Smoller JW, O’Connor L, Spencer KM, et al. Frontal P3 event-related potential is related to brain glutamine/glutamate ratio measured in vivo. NeuroImage. 2015;111:186–191. doi: 10.1016/j.neuroimage.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annual Review of Neuroscience. 2012;35:203–225. doi: 10.1146/annurev-neuro-062111-150444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cousijn H, Haegens S, Wallis G, Near J, Stokes MG, Harrison PJ, et al. Resting GABA and glutamate concentrations do not predict visual gamma frequency or amplitude. Proceedings of the National Academy of Sciences. 2014;111:9301–9306. doi: 10.1073/pnas.1321072111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ayala YA, Pérez-González D, Malmierca MS. Stimulus-specific adaptation in the inferior colliculus: The role of excitatory, inhibitory and modulatory inputs. Biological psychology. 2015 doi: 10.1016/j.biopsycho.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 45.Muthukumaraswamy SD, Routley B, Droog W, Singh KD, Hamandi K. The effects of AMPA blockade on the spectral profile of human early visual cortex recordings studied with non-invasive MEG. Cortex; a journal devoted to the study of the nervous system and behavior. 2016 doi: 10.1016/j.cortex.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Magazzini L, Muthukumaraswamy SD, Campbell AE, Hamandi K, Lingford-Hughes A, Myers JF, et al. Significant reductions in human visual gamma frequency by the gaba reuptake inhibitor tiagabine revealed by robust peak frequency estimation. Human brain mapping. 2016 doi: 10.1002/hbm.23283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annual review of neuroscience. 2012;35:203. doi: 10.1146/annurev-neuro-062111-150444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rojas DC, Maharajh K, Teale PD, Kleman MR, Benkers TL, Carlson JP, et al. Development of the 40Hz steady state auditory evoked magnetic field from ages 5 to 52. Clinical neurophysiology. 2006;117:110–117. doi: 10.1016/j.clinph.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Current psychiatry reports. 2010;12:335–344. doi: 10.1007/s11920-010-0124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez-Burgos G, Lewis DA. NMDA Receptor Hypofunction, Parvalbumin-Positive Neurons and Cortical Gamma Oscillations in Schizophrenia. Schizophrenia Bulletin. 2012;38:950–957. doi: 10.1093/schbul/sbs010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gandal MJ, Edgar JC, Klook K, Siegel SJ. Gamma synchrony: towards a translational biomarker for the treatment-resistant symptoms of schizophrenia. Neuropharmacology. 2012;62:1504–1518. doi: 10.1016/j.neuropharm.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirano Y, Oribe N, Kanba S, Onitsuka T, Nestor PG, Spencer KM. Spontaneous gamma activity in schizophrenia. JAMA psychiatry. 2015 doi: 10.1001/jamapsychiatry.2014.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pittman-Polletta BR, Kocsis B, Vijayan S, Whittington MA, Kopell NJ. Brain Rhythms Connect Impaired Inhibition to Altered Cognition in Schizophrenia. Biological psychiatry. 2015 doi: 10.1016/j.biopsych.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uhlhaas PJ, Singer W. Oscillations and neuronal dynamics in schizophrenia: the search for basic symptoms and translational opportunities. Biological psychiatry. 2014 doi: 10.1016/j.biopsych.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 55.Di Lorenzo G, Daverio A, Ferrentino F, Santarnecchi E, Ciabattini F, Monaco L, et al. Altered resting-state EEG source functional connectivity in schizophrenia: the effect of illness duration. Frontiers in human neuroscience. 2015:9. doi: 10.3389/fnhum.2015.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lehmann D, Faber PL, Pascual-Marqui RD, Milz P, Herrmann WM, Koukkou M, et al. Functionally aberrant electrophysiological cortical connectivities in first episode medication-naive schizophrenics from three psychiatry centers. Frontiers in human neuroscience. 2014:8. doi: 10.3389/fnhum.2014.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Javitt DC. Meeting overview: Sensory perception and schizophrenia, Lausanne, Switzerland June 31–July 1, 2014. Schizophrenia Research: Cognition. 2015 doi: 10.1016/j.scog.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foxe JJ, Yeap S, Leavitt VM. Brief monocular deprivation as an assay of short-term visual sensory plasticity in schizophrenia–“the binocular effect”. Frontiers in psychiatry. 2013:4. doi: 10.3389/fpsyt.2013.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farkas K, Stefanics G, Marosi C, Csukly G. Elementary sensory deficits in schizophrenia indexed by impaired visual mismatch negativity. Schizophrenia research. 2015;166:164–170. doi: 10.1016/j.schres.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 60.Brealy J, Shaw A, Richardson H, Singh KD, Muthukumaraswamy S, Keedwell PA. Increased visual gamma power in schizoaffective bipolar disorder. Psychological medicine. 2015;45:783–794. doi: 10.1017/S0033291714001846. [DOI] [PubMed] [Google Scholar]
- 61.Rivolta D, Castellanos NP, Stawowsky C, Helbling S, Wibral M, Grützner C, et al. Source-reconstruction of event-related fields reveals hyperfunction and hypofunction of cortical circuits in antipsychotic-naive, first-episode schizophrenia patients during mooney face processing. The Journal of Neuroscience. 2014;34:5909–5917. doi: 10.1523/JNEUROSCI.3752-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun L, Castellanos N, Grützner C, Koethe D, Rivolta D, Wibral M, et al. Evidence for dysregulated high-frequency oscillations during sensory processing in medication-naïve, first episode schizophrenia. Schizophrenia research. 2013;150:519–525. doi: 10.1016/j.schres.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 63.Barch DM. The cognitive neuroscience of schizophrenia. Annual review of clinical psychology. 2005;1:321–353. doi: 10.1146/annurev.clinpsy.1.102803.143959. [DOI] [PubMed] [Google Scholar]
- 64.Javitt DC, Sweet RA. Auditory dysfunction in schizophrenia: integrating clinical and basic features. Nature Reviews Neuroscience. 2015;16:535–550. doi: 10.1038/nrn4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen I-W, Helmchen F, Lütcke H. Specific Early and Late Oddball-Evoked Responses in Excitatory and Inhibitory Neurons of Mouse Auditory Cortex. The Journal of Neuroscience. 2015;35:12560–12573. doi: 10.1523/JNEUROSCI.2240-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kirkwood A. Balancing Excitation and Inhibition. Neuron. 2015;86:348–350. doi: 10.1016/j.neuron.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 67.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Donnell BF, Vohs JL, Krishnan GP, Rass O, Hetrick WP, Morzorati SL. The auditory steady-state response (ASSR): a translational biomarker for schizophrenia. Supplements to Clinical neurophysiology. 2013:101–112. doi: 10.1016/b978-0-7020-5307-8.00006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thuné H, Recasens M, Uhlhaas PJ. The 40-Hz Auditory Steady-State Response in Patients With Schizophrenia: A Meta-analysis. JAMA psychiatry. 2016;73:1145–1153. doi: 10.1001/jamapsychiatry.2016.2619. [DOI] [PubMed] [Google Scholar]
- 70.Rowland LM, Edden RA, Kontson K, Zhu H, Barker PB, Hong LE. GABA predicts inhibition of frequency-specific oscillations in schizophrenia. The Journal of neuropsychiatry and clinical neurosciences. 2013;25:83–87. doi: 10.1176/appi.neuropsych.11120368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hirano Y, Oribe N, Kanba S, Onitsuka T, Nestor PG, Spencer KM. Spontaneous Gamma Activity in Schizophrenia. JAMA Psychiatry. 2015;72:813–821. doi: 10.1001/jamapsychiatry.2014.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sivarao DV, Chen P, Senapati A, Yang Y, Fernandes A, Benitex Y, et al. 40 Hz Auditory Steady-State Response Is a Pharmacodynamic Biomarker for Cortical NMDA Receptors. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Başar-Eroğlu C, Schmiedt-Fehr C, Mathes B. Auditory-evoked alpha oscillations imply reduced anterior and increased posterior amplitudes in schizophrenia. Supplements to Clinical neurophysiology. 2012;62:121–129. doi: 10.1016/b978-0-7020-5307-8.00008-9. [DOI] [PubMed] [Google Scholar]
- 74.Kayser J, Tenke CE, Kroppmann CJ, Alschuler DM, Fekri S, Ben-David S, et al. Auditory event-related potentials and alpha oscillations in the psychosis prodrome: neuronal generator patterns during a novelty oddball task. International Journal of Psychophysiology. 2014;91:104–120. doi: 10.1016/j.ijpsycho.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez VB, Roach BJ, Woods SW, Srihari VH, McGlashan TH, Ford JM, et al. Early auditory gamma-band responses in patients at clinical high risk for schizophrenia. Supplements to Clinical neurophysiology. 2013;62:147. doi: 10.1016/b978-0-7020-5307-8.00010-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rass O, Forsyth JK, Krishnan GP, Hetrick WP, Klaunig MJ, Breier A, et al. Auditory steady state response in the schizophrenia, first-degree relatives, and schizotypal personality disorder. Schizophrenia research. 2012;136:143–149. doi: 10.1016/j.schres.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hong LE, Summerfelt A, McMahon R, Adami H, Francis G, Elliott A, et al. Evoked gamma band synchronization and the liability for schizophrenia. Schizophrenia research. 2004;70:293–302. doi: 10.1016/j.schres.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 78.Edgar JC, Chen Y-H, Lanza M, Howell B, Chow VY, Heiken K, et al. Cortical thickness as a contributor to abnormal oscillations in schizophrenia? NeuroImage: Clinical. 2014;4:122–129. doi: 10.1016/j.nicl.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen C-MA, Stanford AD, Mao X, Abi-Dargham A, Shungu DC, Lisanby SH, et al. GABA level, gamma oscillation, and working memory performance in schizophrenia. NeuroImage: Clinical. 2014;4:531–539. doi: 10.1016/j.nicl.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Farzan F, Barr MS, Sun Y, Fitzgerald PB, Daskalakis ZJ. Transcranial magnetic stimulation on the modulation of gamma oscillations in schizophrenia. Annals of the New York Academy of Sciences. 2012;1265:25–35. doi: 10.1111/j.1749-6632.2012.06543.x. [DOI] [PubMed] [Google Scholar]
- 81.Cornew L, Roberts TP, Blaskey L, Edgar JC. Resting-state oscillatory activity in autism spectrum disorders. Journal of autism and developmental disorders. 2012;42:1884–1894. doi: 10.1007/s10803-011-1431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edgar JC, Heiken K, Chen Y-H, Herrington JD, Chow V, Liu S, et al. Resting-state alpha in autism spectrum disorder and alpha associations with thalamic volume. Journal of autism and developmental disorders. 2015;45:795–804. doi: 10.1007/s10803-014-2236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ghanbari Y, Bloy L, Edgar JC, Blaskey L, Verma R, Roberts TP. Joint analysis of band-specific functional connectivity and signal complexity in autism. Journal of autism and developmental disorders. 45:444–460. doi: 10.1007/s10803-013-1915-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonnet-Brilhault F, Alirol S, Blanc R, Bazaud S, Marouillat S, Thépault R, et al. GABA/Glutamate synaptic pathways targeted by integrative genomic and electrophysiological explorations distinguish autism from intellectual disability. Molecular psychiatry. 2015 doi: 10.1038/mp.2015.75. [DOI] [PubMed] [Google Scholar]
- 85.Knoth IS, Vannasing P, Major P, Michaud JL, Lippé S. Alterations of visual and auditory evoked potentials in fragile X syndrome. International Journal of Developmental Neuroscience. 2014;36:90–97. doi: 10.1016/j.ijdevneu.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Coskun MA, Loveland KA, Pearson DA, Papanicolaou AC, Sheth BR. Functional assays of local connectivity in the somatosensory cortex of individuals with autism. Autism Research. 2013;6:190–200. doi: 10.1002/aur.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Coskun MA, Loveland KA, Pearson DA, Papanicolaou AC, Sheth BR. Interaction of finger representations in the cortex of individuals with autism: a functional window into cortical inhibition. Autism Research. 2013;6:542–549. doi: 10.1002/aur.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gandal MJ, Edgar JC, Ehrlichman RS, Mehta M, Roberts TP, Siegel SJ. Validating γ oscillations and delayed auditory responses as translational biomarkers of autism. Biological psychiatry. 2010;68:1100–1106. doi: 10.1016/j.biopsych.2010.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rojas DC, Teale PD, Maharajh K, Kronberg E, Youngpeter K, Wilson LB, et al. Transient and steady-state auditory gamma-band responses in first-degree relatives of people with autism spectrum disorder. Molecular autism. 2011;2:1. doi: 10.1186/2040-2392-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wallace GL, Dankner N, Kenworthy L, Giedd JN, Martin A. Age-related temporal and parietal cortical thinning in autism spectrum disorders. Brain: a journal of neurology. 2010:awq279. doi: 10.1093/brain/awq279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Snijders TM, Milivojevic B, Kemner C. Atypical excitation–inhibition balance in autism captured by the gamma response to contextual modulation. Neuroimage: Clinical. 2013;3:65–72. doi: 10.1016/j.nicl.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weinger PM, Zemon V, Soorya L, Gordon J. Low-contrast response deficits and increased neural noise in children with autism spectrum disorder. Neuropsychologia. 2014;63:10–18. doi: 10.1016/j.neuropsychologia.2014.07.031. [DOI] [PubMed] [Google Scholar]
- 93.Dickinson A, Bruyns-Haylett M, Jones M, Milne E. Increased peak gamma frequency in individuals with higher levels of autistic traits. European Journal of Neuroscience. 2015;41:1095–1101. doi: 10.1111/ejn.12881. [DOI] [PubMed] [Google Scholar]
- 94.Dickinson A, Bruyns-Haylett M, Smith R, Jones M, Milne E. Superior orientation discrimination and increased peak gamma frequency in autism spectrum conditions. Journal of Abnormal Psychology. 2016;125:412–422. doi: 10.1037/abn0000148. [DOI] [PubMed] [Google Scholar]
- 95.Matsuzaki J, Kagitani-Shimono K, Sugata H, Hirata M, Hanaie R, Nagatani F, et al. Progressively increased M50 responses to repeated sounds in autism spectrum disorder with auditory hypersensitivity: a magnetoencephalographic study. 2014 doi: 10.1371/journal.pone.0102599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anticevic A, Cole MW, Repovs G, Savic A, Driesen NR, Yang G, et al. Connectivity, pharmacology, and computation: toward a mechanistic understanding of neural system dysfunction in schizophrenia. Frontiers in Psychiatry. 2013:4. doi: 10.3389/fpsyt.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fornito A, Harrison BJ, Goodby E, Dean A, Ooi C, Nathan PJ, et al. Functional Dysconnectivity of Corticostriatal Circuitry as a Risk Phenotype for Psychosis. JAMA psychiatry (Chicago, Ill) 2013 doi: 10.1001/jamapsychiatry.2013.1976. [DOI] [PubMed] [Google Scholar]
- 98.Plitt M, Barnes KA, Wallace GL, Kenworthy L, Martin A. Resting-state functional connectivity predicts longitudinal change in autistic traits and adaptive functioning in autism. Proceedings of the National Academy of Sciences. 2015 doi: 10.1073/pnas.1510098112. 201510098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uhlhaas PJ. Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Current Opinion in Neurobiology. 2013;23:283–290. doi: 10.1016/j.conb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 100.Yang GJ, Murray JD, Repovs G, Cole MW, Savic A, MFG, et al. Altered Global Brain Signal in Schizophrenia. Proceedings of the National Academy of Science USA. 2014;111:7438–7443. doi: 10.1073/pnas.1405289111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang GJ, Murray JD, Wang X-J, Glahn DC, Pearlson GD, Repovs G, et al. Functional hierarchy underlies preferential connectivity disturbances in schizophrenia. Proceedings of the National Academy of Science USA. 2016;113:E219–228. doi: 10.1073/pnas.1508436113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klingner CM, Langbein K, Dietzek M, Smesny S, Witte OW, Sauer H, et al. Thalamocortical connectivity during resting state in schizophrenia. European archives of psychiatry and clinical neuroscience. 2014;264:111–119. doi: 10.1007/s00406-013-0417-0. [DOI] [PubMed] [Google Scholar]
- 103.Woodward ND, Karbasforoushan H, Heckers S. Thalamocortical dysconnectivity in schizophrenia. American Journal of Psychiatry. 2012;169:1092–1099. doi: 10.1176/appi.ajp.2012.12010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Woodward ND, Heckers S. Mapping thalamocortical functional connectivity in chronic and early stages of psychotic disorders. Biological psychiatry. 2015 doi: 10.1016/j.biopsych.2015.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Anticevic A, Cole MW, Repovs G, Murray JD, Brumbaugh MS, Winkler AM, et al. Characterizing Thalamo-Cortical Disturbances in Schizophrenia and Bipolar Illness. Cerebral cortex. 2013 doi: 10.1093/cercor/bht165. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anticevic A, Hu X, Xiao Y, Hu J, Li F, Bi F, et al. Early-Course Unmedicated Schizophrenia Patients Exhibit Elevated Prefrontal Connectivity Associated with Longitudinal Change. The Journal of Neuroscience. 2015;35:267–286. doi: 10.1523/JNEUROSCI.2310-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duncan NW, Wiebking C, Tiret B, Marjanska M, Hayes DJ, Lyttleton O, et al. Glutamate concentration in the medial prefrontal cortex predicts resting-state cortical-subcortical functional connectivity in humans. PloS one. 2013:8. doi: 10.1371/journal.pone.0060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kraguljac NV, White DM, Hadley J, Reid MA, Lahti AC. Hippocampal - parietal dysconnectivity and glutamate abnormalities in unmedicated patients with schizophrenia. Hippocampus. 2014;24:1524–1532. doi: 10.1002/hipo.22332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bastos-Leite AJ, Ridgway GR, Silveira C, Norton A, Reis S, Friston KJ. Dysconnectivity within the default mode in first-episode schizophrenia: a stochastic dynamic causal modeling study with functional magnetic resonance imaging. Schizophrenia bulletin. 2014:sbu080. doi: 10.1093/schbul/sbu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Anticevic A, Corlett PR, Cole MW, Savic A, Gancsos M, Tang Y, et al. N-Methyl-D-Aspartate receptor antagonist effects on prefrontal cortical connectivity better model early than chronic schizophrenia. Biological psychiatry. 2015;77:569–580. doi: 10.1016/j.biopsych.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 112.Itahashi T, Yamada T, Watanabe H, Nakamura M, Ohta H, Kanai C, et al. Alterations of local spontaneous brain activity and connectivity in adults with high-functioning autism spectrum disorder. Molecular Autism. 2015;6:30. doi: 10.1186/s13229-015-0026-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tyszka JM, Kennedy DP, Paul LK, Adolphs R. Largely typical patterns of resting-state functional connectivity in high-functioning adults with autism. Cerebral cortex. 2014;24:1894–1905. doi: 10.1093/cercor/bht040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Padmanabhan A, Lynn A, Foran W, Luna B, O’Hearn K. Age related changes in striatal resting state functional connectivity in autism. Frontiers in human neuroscience. 2013;7 doi: 10.3389/fnhum.2013.00814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Abbott AE, Nair A, Keown CL, Datko M, Jahedi A, Fishman I, et al. Patterns of Atypical Functional Connectivity and Behavioral Links in Autism Differ Between Default, Salience, and Executive Networks. Cerebral cortex. 2015:bhv191. doi: 10.1093/cercor/bhv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yerys BE, Gordon EM, Abrams DN, Satterthwaite TD, Weinblatt R, Jankowski KF, et al. Default mode network segregation and social deficits in autism spectrum disorder: Evidence from non-medicated children. Neuroimage Clinical. 2015;18:223–232. doi: 10.1016/j.nicl.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jann K, Hernandez LM, Beck-Pancer D, McCarron R, Smith RX, Dapretto M, et al. Altered resting perfusion and functional connectivity of default mode network in youth with autism spectrum disorder. Brain and behavior. 2015;5 doi: 10.1002/brb3.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lynch CJ, Uddin LQ, Supekar K, Khouzam A, Phillips J, Menon V. Default mode network in childhood autism: posteromedial cortex heterogeneity and relationship with social deficits. Biological psychiatry. 2013;74:212–219. doi: 10.1016/j.biopsych.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Javitt DC. Sensory processing in schizophrenia: neither simple nor intact. Schizophrenia Bulletin. 2009;35:1059–1064. doi: 10.1093/schbul/sbp110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Serrano-Pedraza I, Romero-Ferreiro V, Read JC, Diéguez-Risco T, Bagney-Lifante A, Caballero-González M, et al. Reduced visual orientation-surround suppression in schizophrenia shown by measuring contrast detection thresholds. Journal of Vision. 2014;14:1406–1406. doi: 10.3389/fpsyg.2014.01431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tadin D, Kim J, Doop ML, Gibson C, Lappin JS, Blake R, et al. Weakened center-surround interactions in visual motion processing in schizophrenia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:11403–11412. doi: 10.1523/JNEUROSCI.2592-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yang E, Tadin D, Glasser DM, Hong SW, Blake R, Park S. Visual context processing in schizophrenia. Clinical psychological science: a journal of the Association for Psychological Science. 2013;1:5–15. doi: 10.1177/2167702612464618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seymour K, Stein T, Sanders LL, Guggenmos M, Theophil I, Sterzer P. Altered contextual modulation of primary visual cortex responses in schizophrenia. Neuropsychopharmacology. 2013;38:2607–2612. doi: 10.1038/npp.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mayer AR, Ruhl D, Merideth F, Ling J, Hanlon FM, Bustillo J, et al. Functional imaging of the hemodynamic sensory gating response in schizophrenia. Human brain mapping. 2013;34:2302–2312. doi: 10.1002/hbm.22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Falkenberg LE, Westerhausen R, Craven AR, Johnsen E, Kroken RA, Specht K, et al. Impact of glutamate levels on neuronal response and cognitive abilities in schizophrenia. NeuroImage: Clinical. 2014;4:576–584. doi: 10.1016/j.nicl.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Takarae Y, Luna B, Minshew NJ, Sweeney JA. Visual motion processing and visual sensorimotor control in autism. Journal of the International Neuropsychological Society. 2014;20:113–122. doi: 10.1017/S1355617713001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Karten A, Hirsch J. Brief Report: Anomalous Neural Deactivations and Functional Connectivity During Receptive Language in Autism Spectrum Disorder: A Functional MRI Study. Journal of autism and developmental disorders. 2015;45:1905–1914. doi: 10.1007/s10803-014-2344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Flevaris AV, Murray SO. Orientation-specific surround suppression in the primary visual cortex varies as a function of autistic tendency. Frontiers in human neuroscience. 2014;8 doi: 10.3389/fnhum.2014.01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Singh KD. Which “neural activity” do you mean? fMRI, MEG, oscillations and neurotransmitters. NeuroImage. 2012;62:1121–1130. doi: 10.1016/j.neuroimage.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 130.Hoftman GD, Volk DW, Bazmi HH, Li S, Sampson AR, Lewis DA. Altered cortical expression of GABA-related genes in schizophrenia: illness progression vs developmental disturbance. Schizophrenia Bulletin. 2015;41:180–191. doi: 10.1093/schbul/sbt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tuncdemir SN, Wamsley B, Stam FJ, Osakada F, Goulding M, Callaway EM, et al. Early Somatostatin Interneuron Connectivity Mediates the Maturation of Deep Layer Cortical Circuits. Neuron. 2016;89:521–535. doi: 10.1016/j.neuron.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Developmental Neurobiology. 2011;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cardin JA, Carlen M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Driesen NR, McCarthy G, Bhagwagar Z, Bloch MH, Calhoun VD, D'Souza DC, et al. The Impact of NMDA Receptor Blockade on Human Working Memory-Related Prefrontal Function and Connectivity. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38:2613–2622. doi: 10.1038/npp.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Anticevic A, Gancsos M, Murray JD, Repovs G, Driesen NR, Ennis DJ, et al. NMDA Receptor Function in Large-Scale Anti-Correlated Neural Systems with Implications for Cognition and Schizophrenia. Proceedings of the National Academy of Science USA. 2012;109:16720–16725. doi: 10.1073/pnas.1208494109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Joules R, Doyle O, Schwarz A, O’Daly O, Brammer M, Williams S, et al. Ketamine induces a robust whole-brain connectivity pattern that can be differentially modulated by drugs of different mechanism and clinical profile. Psychopharmacology. 2015:1–14. doi: 10.1007/s00213-015-3951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shcherbinin S, Doyle O, Zelaya FO, de Simoni S, Mehta MA, Schwarz AJ. Modulatory effects of ketamine, risperidone and lamotrigine on resting brain perfusion in healthy human subjects. Psychopharmacology. 2015;232:4191–4204. doi: 10.1007/s00213-015-4021-z. [DOI] [PubMed] [Google Scholar]
- 138.Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biological psychology. 2015 doi: 10.1016/j.biopsycho.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 139.Hadjikhani N, Zürcher NR, Rogier O, Ruest T, Hippolyte L, Ben-Ari Y, et al. Improving emotional face perception in autism with diuretic bumetanide: a proof-of-concept behavioral and functional brain imaging pilot study. Autism. 2015;19:149–157. doi: 10.1177/1362361313514141. [DOI] [PubMed] [Google Scholar]
- 140.Taylor SF, Demeter E, Phan KL, Tso IF, Welsh RC. Abnormal GABAergic function and negative affect in schizophrenia. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2014;39:1000–1008. doi: 10.1038/npp.2013.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tso IF, Fang Y, Phan KL, Welsh RC, Taylor SF. Abnormal GABAergic function and face processing in schizophrenia: A pharmacologic-fMRI study. Schizophrenia research. 2015;168:338–344. doi: 10.1016/j.schres.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Popov T, Rockstroh B, Weisz N, Elbert T, Miller GA. Adjusting brain dynamics in schizophrenia by means of perceptual and cognitive training. PloS one. 2012;7:e39051. doi: 10.1371/journal.pone.0039051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cole JC, Bernacki CG, Helmer A, Pinninti N, O’reardon JP. Efficacy of Transcranial Magnetic Stimulation (TMS) in the Treatment of Schizophrenia: A Review of the Literature to Date. Innovations in clinical neuroscience. 2014;12:12. [PMC free article] [PubMed] [Google Scholar]
- 144.Esslinger C, Schüler N, Sauer C, Gass D, Mier D, Braun U, et al. Induction and quantification of prefrontal cortical network plasticity using 5 Hz rTMS and fMRI. Human brain mapping. 2014;35:140–151. doi: 10.1002/hbm.22165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Uppal N, Puri R, Yuk F, Janssen WG, Bozdagi-Gunal O, Harony-Nicolas H, et al. Ultrastructural analyses in the hippocampus CA1 field in Shank3-deficient mice. Molecular Autism. 2015;6 doi: 10.1186/s13229-015-0036-x. eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Archives of General Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- 147.Vorstman JA, Morcus ME, Duijff SN, Klaassen PW, Heineman-de Boer JA, Beemer FA, et al. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. Journal of the American Academy of Child and Adolescent Psychiatry. 2006;45:1104–1113. doi: 10.1097/01.chi.0000228131.56956.c1. [DOI] [PubMed] [Google Scholar]
- 148.Stephan KE, Iglesias S, Heinzle J, Diaconescu AO. Translational perspectives for computational neuroimaging. Neuron. 2015;87:716–732. doi: 10.1016/j.neuron.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 149.Wang X-J, Krystal JH. Computational Psychiatry. Neuron. 2014;84:638–654. doi: 10.1016/j.neuron.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stephan KE, Mathys C. Computational approaches to psychiatry. Current opinion in neurobiology. 2014 doi: 10.1016/j.conb.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 151.Rolls ET, Deco G. A computational neuroscience approach to schizophrenia and its onset. Neurosci Biobehav Rev. 2010 doi: 10.1016/j.neubiorev.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 152.Montague PR, Dolan RJ, Friston KJ, Dayan P. Computational psychiatry. Trends In Cognitive Sciences. 2012;16:72–80. doi: 10.1016/j.tics.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Anticevic A, Murray JD, Barch DM. Bridging Levels of Understanding in Schizophrenia Through Computational Modeling. Clinical psychological science. 2015;3:433–459. doi: 10.1177/2167702614562041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vierling-Claassen D, Siekmeier P, Stufflebeam S, Kopell N. Modeling GABA alterations in schizophrenia: a link between impaired inhibition and altered gamma and beta range auditory entrainment. Journal of neurophysiology. 2008;99:2656–2671. doi: 10.1152/jn.00870.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rosenberg A, Patterson JS, Angelaki DE. A computational perspective on autism. Proceedings of the National Academy of Science USA. 2015;112:9185–9165. doi: 10.1073/pnas.1510583112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Foss-Feig JH, Tadin D, Schauder KB, Cascio CJ. A substantial and unexpected enhancement of motion perception in autism. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2013;33:8243–8249. doi: 10.1523/JNEUROSCI.1608-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bozdagi O, Tavassoli T, Buxbaum JD. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Molecular autism. 2013;4:1. doi: 10.1186/2040-2392-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Silverman JL, Pride M, Hayes J, Puhger K, Butler-Struben H, Baker S, et al. GABAB receptor agonist R-baclofen reverses social deficits and reduces repetitive behavior in two mouse models of autism. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2015;40:2228–2239. doi: 10.1038/npp.2015.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kolevzon A, Bush L, Wang AT, Halpern D, Frank Y, Grodberg D, et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Molecular autism. 2014;5:1. doi: 10.1186/2040-2392-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. Journal of Neuroscience. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 162.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. Journal of Neuroscience. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Logothetis NK. What we can do and what we cannot do with fMRI. Nature. 2008;453:869–878. doi: 10.1038/nature06976. [DOI] [PubMed] [Google Scholar]
- 164.Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–679. doi: 10.1126/science.1247190. [DOI] [PubMed] [Google Scholar]
- 165.Do KQ, Cuenod M, Hensch TK. Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophrenia bulletin. 2015:sbv065. doi: 10.1093/schbul/sbv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Meredith R. Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neurodevelopmental disorders. Neuroscience & Biobehavioral Reviews. 2015;50:180–188. doi: 10.1016/j.neubiorev.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 167.Krystal JH, Anticevic A. Toward illness phase-specific pharmacotherapy for schizophrenia. Biological Psychiatry. 2015;78:738–740. doi: 10.1016/j.biopsych.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 168.Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- 169.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends in Neurosciences. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26:365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Moghaddam B, Javitt D. From Revolution to Evolution: The Glutamate Hypothesis of Schizophrenia and its Implication for Treatment. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2011 doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain research. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 173.Dickinson A, Jones M, Milne E. Measuring neural excitation and inhibition in autism: Different approaches, different findings and different interpretations. Brain Research. 2016;1648:277–289. doi: 10.1016/j.brainres.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 174.Cole MW, Yang GJ, Murray JD, Repovš G, Anticevic A. Functional connectivity change as shared signal dynamics. Journal of Neuroscience Methods. 2016;259:22–39. doi: 10.1016/j.jneumeth.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cole MW, Repovš G, Anticevic A. The frontoparietal control system: a central role in mental health. Neuroscientist. 2014;20:652–664. doi: 10.1177/1073858414525995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Glasser MF, Coalson TS, Robinson EC, Hacker CD, Harwell J, Yacoub E, et al. A multi-modal parcellation of human cerebral cortex. Nature. 2016;536:171–178. doi: 10.1038/nature18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Anticevic A, Cole MW, Murray JD, Corlett PR, Wang XJ, Krystal JH. The role of default network deactivation in cognition and disease. Trends in Cognitive Science. 2012;16:584–592. doi: 10.1016/j.tics.2012.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Murray JD, Anticevic A, Ichinose M, Gancsos M, Corlett PR, Krystal JH, et al. Effects of disinhibition in a cortical working memory circuit model with relevance to schizophrenia. Society for Neuroscience Abstract Viewer and Itinerary Planner. 2012:42. [Google Scholar]
- 179.Anticevic A, Murray JD, Barch DM. Bridging Levels of Understanding in Schizophrenia Through Computational Modeling. Clinical Psychological Science. 2015;3:433459. doi: 10.1177/2167702614562041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.