

Abstract
The discovery of cell-free DNA fetal (cff DNA) in maternal plasma during pregnancy provides a novel perspective for the development of non-invasive prenatal diagnosis (NIPD). Against the background of maternal DNA, the use of the relatively low concentration of cff DNA is limited in NIPD. Therefore, in order to overcome the complication of the background of maternal DNA and expand the scope of cff DNA application in clinical practice, it is necessary to identify novel universal fetal-specific DNA markers. The GeneChip Human Promoter 1.0R Array set was used in the present study to analyze the methylation status of 12 placental tissue and maternal peripheral blood whole-genome DNA samples. In total, 5 fetus differential hypermethylation regions and 6 fetus differential hypomethylation regions were identified. In order to verify the 11 selected methylation regions and detect the differential CpG sites in these regions, a bisulfate direct sequencing strategy was used. In total, 87 fetal differential methylation CpG sites were identified from 123 CpG sites. The detection of fetal differential methylation DNA regions and CpG sites may be instrumental in the development of efficient NIPD and in the expansion of its application in other disorders.
Keywords: DNA methylation, GeneChip human tiling 2.0R array set, bisulfate direct sequencing
Introduction
Women undergoing high-risk pregnancies typically undergo conventional invasive prenatal diagnosis. The most common invasive prenatal diagnostic techniques are amniocentesis and chorionic villus sampling, which are optimal from the late first trimester to the second trimester. However, conventional invasive prenatal techniques not only raise the risk of fetal loss but also bring pressure to the mothers, and earlier testing results in an increased risk to the fetus and mother (1). Therefore, developing a reliable method for non-invasive prenatal diagnosis (NIPD) which does not confer additional risk to the fetus and mother is of critical importance in prenatal diagnosis.
In 1893, Schmorl et al first described feto-maternal cellular trafficking (2). The presence of fetal cells in the maternal circulation made it possible to promote the development of non-invasive prenatal diagnosis (2). However, there are several problems with using such fetal cells for non-invasive prenatal diagnosis. First of all, the number of fetal cells in the maternal circulation is very small; secondly, it is difficult to isolate these fetal cells from the maternal blood; finally, the enrichment technologies at present are very expensive. Therefore, non-invasive prenatal diagnosis using fetal cells in the maternal circulation is still not widely used in clinical practice. In 1997, Lo et al (3) reported that fetal DNA migrates into maternal peripheral blood during pregnancy and in 1998 also reported that fetal DNA was detectable by polymerase chain reaction (PCR) (4). The discovery of cell-free fetal DNA (cff DNA) in maternal plasma during pregnancy provides a novel perspective for the development of NIPD. The amount of fetal cff DNA is greater than that of the fetal cells in equal amounts of blood (5). At present, NIPD using markers of cff DNA in clinical practice include fetal sex determination for sex-linked disorders, fetal RhD blood group typing, fetal chromosomal aneuploidy detection, and certain fetal single gene genetic diseases (4,6–13). Although the amount of cff DNA increases with the duration of gestation from the seventh gestational week, in the first and second trimester the concentration of cff DNA in maternal plasma only accounts for 3.4 and 6.2% of the total DNA in maternal plasma, respectively (4). Against the background of maternal DNA, the use of the relatively low concentration of cff DNA is limited in NIPD. Therefore, in order to overcome the complication of the background of maternal DNA and expand the scope of application for cff DNA in clinical practice, the identification of novel, universal fetal-specific DNA markers is necessary (14).
The use of epigenetic modifications as fetal-specific markers to detect cff DNA from maternal DNA has been investigated previously (15–18). The cff DNA in maternal plasma originates from the placenta and villus (19,20). Instead of analyzing fetal DNA in the maternal plasma, epigenetic differences between placental and maternal cells have been used in previous studies as a promising strategy for identifying fetal-specific markers for NIPT (19,20). Epigenetic modifications, including DNA methylation, non-coding RNAs and histone modifications, are heritable but are not due to an alteration in genomic sequence (21–23). DNA methylation was the first epigenetic modification to be discovered, and remains the most well studied. In animals, it predominantly involves the addition of a methyl group to the carbon-5 position of cytosine residues of the dinucleotide CpG (24–26). Utilizing fetal disease-specific methylation markers to conduct NIPT to screen for diseases is a promising strategy (15–17).
Methylated microarray analysis is an efficient technology platform for screening for epigenetic methylation markers. Tiling arrays offer a physical readout of a genome and is used as a discovery tool for mapping sites of DNA interaction in chromatin immunoprecipitation (ChIP) experiments, and for understanding global epigenomic changes including methylation. In the present study, a GeneChip Human Tiling 2.0R Array set designed for ChIP experiments was used to screen for differentially methylated regions in placental and maternal blood cells. This set of 7 arrays contain ~45 million oligonucleotide probes covering all methylation regions in the whole genome. Furthermore, bisulfate direct sequencing was used to verify the results of methylated microarray analysis and to identify the methylation sites in these methylation regions.
Materials and methods
Study samples
Blood samples were collected from 6 healthy Han Chinese pregnant women between 18–24 gestational weeks and placental tissue samples were collected from 6 women with full-term pregnancies in the Prenatal Diagnosis Center of Henan Provincial People's Hospital (Zhengzhou, China) between March 2013 and May 2013. All samples were frozen at −80°C prior to use. Information regarding gestational age, maternal age, numbers of previous pregnancies and abortions and fetal sex was collected (Table I). Written informed consent was obtained from each woman. The present study was performed in-line with the Ethics Committee of Henan Provincial People's Hospital (Zhengzhou, China).
Table I.
General information about the participants.
| Sample ID | Maternal age (years) | Reproductive history | Gestational age | Fetal sex |
|---|---|---|---|---|
| 1 | 28 | G1P0 | 18 week | 46, XX |
| 2 | 37 | G2P0 | 19 week+ 3 day | 46, XX |
| 3 | 33 | G1P0 | 20 week | 46, XX |
| 4 | 24 | G3P0 | 19 week+ 6 day | 46, XY |
| 5 | 32 | G4P1 | 22 week | 46, XY |
| 6 | 27 | G1P0 | 21 week+ 5 day | 46, XX |
| 7 | 26 | G2P0 | 39 week+ 6 day | 46, XY |
| 8 | 27 | G3P1 | 41 week+ 1 day | 46, XX |
| 9 | 29 | G3P1 | 40 week+ 5 day | 46, XY |
| 10 | 31 | G2P0 | 39 week+ 4day | 46, XX |
| 11 | 34 | G2P1 | 40 week+ 2 day | 46, XY |
| 12 | 25 | G2P1 | 38 week+ 6 day | 46, XX |
Exclusion of maternal DNA
The 15 autosomal STR loci and an amelogenin locus were amplified to determine whether there was maternal DNA in the fetal placental tissue by using the PowerPlex 16 system kit (PowerPlex 16; Promega Corporation, Madison, WI, USA) in a multiplex amplification reaction system, according to the manufacturer's protocol, using a 25 µl reaction mixture containing 1.0 µl (0.5–2 ng) genomic DNA, 5.0 µl PowerPlex 16 5xMaster Mix, 5.0 µl PowerPlex 16 5xPrimer Pair Mix and 14.0 µl ddH2O. Thermal cycling was performed using the GeneAmp PCR System 9700 (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The thermocycler conditions for the polymerase chain reaction (PCR) were as follows: Denaturation for 11 min at 95°C, and then 1 min at 96°C, followed by 10 cycles for 30 sec at 94°C, 30 sec at 60°C, 45 sec at 70°C, then 20 cycles for 30 sec at 90°C, 30 sec at 60°C, 45 sec at 70°C and a final elongation step at 60°C for 30 min.
The PCR products were separated and detected by capillary electrophoresis on an ABI 3130 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.). PCR products (2 µl) or PowerPlex 16 allelic ladder (2 µl) was mixed with 15 µl Hi-Di formamide and 0.5 µl internal lane standard 600 (all from Promega Corporation). The loading mixture was denatured at 95°C for 3 min, and then followed by chilling on ice immediately for 3 min. STR alleles were analyzed by comparison with allelic ladders of the kit using the GeneMapper ID 3.2 software (Applied Biosystems; Thermo Fisher Scientific, Inc.). Control DNA from the 9947A female cell line (Promega Corporation) was genotyped as a standard reference in all experiments.
Methylated DNA (mDNA) capture
The Methylamp Methylated DNA Capture kit (P-1015; EpiGentek Group, Inc., Farmingdale, NY, USA) was used to enrich and capture methylated DNA fragments from the blood and placental tissue samples. According to the manufacturer's protocol, methylated DNA enrichment and capture included the following steps: i) DNA isolation; ii) breaking DNA into fragments using a sonicator as follows: Sonication for 10 sec, pause for 30 sec (repeated 10 times) at 100W by an automatically adjusted frequency from 20–25 kHz, with the DNA size subsequently checked by running on 1.2% agarose gel; iii) incubating sonicated DNA at 98°C for 3 min and immediately placing on ice; iv) methylated DNA immunoprecipitation by incubating denatured DNA with an anti-5-methylcytosine antibody (1:100; P-1015; EpiGentek Group, Inc.) at room temperature for 2 h shaking on an orbital shaker at 100 rpm; v) release of mDNA from antibody complex; vi) capturing the mDNA; vii) elution of the mDNA. Following methylated DNA immunoprecipitation (MeDIP), it is possible to analyze the methylation of the DNA using MeDIP-ChIP.
DNA methylation analysis
The GeneChip Human Promoter 1.0R Array set (Affymetrix; Thermo Fisher Scientific, Inc.) was used to detect the promoter regions across the whole genome. GeneChip Fluidics Station 400 and GeneChip Scanner 3,000 7 G (Affymetrix; Thermo Fisher Scientific, Inc.) were used for analysis. According to the Affymetrix chip-on-chip procedures, immunoprecipitated DNA targets were tagged with primer A (5′-GTTTCCCAGTCACGGTCNNNNNNNNN-3′) using DNA Sequenase (Affymetrix; Thermo Fisher Scientific, Inc). Tagged DNA was amplified with primer B (5′-GTTTCCCAGTCACGGTC-3′) using LA Taq polymerase (Takara Bio, Inc., Otsu, Japan) with the following cycling conditions: 15 cycles of 30 sec at 95°C, 30 sec at 45°C, 30 sec at 55°C, 1 min at 72°C; then 15 cycles of 30 sec at 95°C, 30 sec at 45°C, 30 sec at 55°C (for every subsequent cycle, add 5 sec), 1 min at 72°C, and 4°C hold. Next, the PCR products were purified using MiniElute PCR purification kit (Qiagen, Inc., Valencia, CA, USA) and a fragmentation mix was obtained. Then, the GeneChip WT Double-Stranded DNA Terminal Labeling kit was used to fragment and label the double-stranded DNA, and the GeneChip Hybridization, Wash, and Stain kit (both from Affymetrix; Thermo Fisher Scientific, Inc.) was used to hybridize, wash and stain the labeled target DNA according to the manufacturer's protocol. Finally, the chip was scanned using the GeneChip Scanner 3,000 7G controlled by GeneChip® Command Console® Software (AGCC) (both from Affymetrix; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol.
Bisulfate direct sequencing
Genomic DNA was isolated using a DNA isolation kit (Invitrogen; Thermo Fisher Scientific, Inc.) from blood and placental tissue samples according to the manufacturer's protocol. Bisulfate direct sequencing was conducted to verify the selected methylation regions by microarray analysis and to detect the methylation sites in these selected methylation regions. Prior to conduction of bisulfate direct sequencing, the following steps were performed: i) Bisulfate modification of the sample genomic DNA, performed using the EpiTect Bisulfate kit (Qiagen, Inc.), according to the manufacturer's protocol; ii) PCR amplification, using the primer sequences in Table II and the reaction conditions in Table III; iii) electrophoresis and recycling of the PCR products; iv) ligating purified PCR products into the vector pUC18-T (Sangon Biotech Co., Ltd., Shanghai, China); v) the preparation of competent DH5α E. coli cells (Takara Biotechnology Co., Ltd., Dalian, China) using a Rapid Competent Cell Preps kit (B529307) and a One Step Cell Preparation kit (SK9307) (both from Sangon Biotech Co., Ltd.); vi) colony screening (inoculation and selection of monoclonal colonies) and plasmid extraction using a Miniprep Plasmid Purification kit (SK8161; Sangon Biotech Co., Ltd.); vii) sequencing, using a BigDye® Terminator v3.1 Cycle Sequencing kit (Applied Biosystems; Thermo Fisher Scientific, Inc.), the primers listed in Table II and the reaction conditions listed in Table III, and performed with an ABI 3130 Genetic Analyzer. In total, 5 fetal hypermethylation regions and 6 fetal hypomethylation regions were tested.
Table II.
Primer sequences used for bisulfate direct sequencing.
| Primer name | Forward primers (5′-3′) | Reverse primers (5′-3′) | Amplicon size (bp) |
|---|---|---|---|
| FHypeMR 1 | GGGTGTTAGGGTGGTGTAGAG | TACAAAACAAACCCAAAATTCTAC | 355 |
| FHypeMR 2 | TTTATTTGTTTAGTTTATTGTTTGAG | TAAAATCCCTTTTCAAAACAAC | 416 |
| FHypeMR 3 | AAAATTTAGGTAAGGAAATGTATTAG | TTTTCCTACTCCAACCATCC | 578 |
| FHypeMR 4 | TTTTGAAATAAATTTGTGAGGG | CTAACAACCCAACCACAACTAC | 437 |
| FHypeMR 5 | GAAAGTTTTTAGGTGGTTTTAGAG | CAAAACAAAAAAATATCTCTAAACC | 581 |
| FHypoMR 1 | GTTTATTGTTGTTTTTATTTGGG | CTAATAATCTTACTTCCCATTTCAC | 602 |
| FHypoMR 2 | GGGATAAAGAAATTAATATTGATG | CTCAATAAAACACTTCTAAAACATC | 509 |
| FHypoMR 3 | AAATAGATTTAAATAATTTGTGTTATTG | AAAAACCACTCCTCTTATAAAATC | 472 |
| FHypoMR 4 | GATTTTATTTTTGTTGGGGTG | AACAAACACATCATAACACAAAAC | 398 |
| FHypoMR 5 | TTAGTTAAGATTTTTTTTAGAATTGTAG | TAAAAACTTTATACAAATAATTTAATTTC | 405 |
| FHypoMR 6 | TGAATATAGTTTTGTTGGTTGTG | TAAATCCTTAATCACTAAACAACAC | 343 |
FHypeMR, fetus hypermethylation regions; FHypoMR, fetus hypomethylation regions.
Table III.
Amplification reaction conditions for bisulfate direct sequencing.
| Name | Amplification reaction conditions |
|---|---|
| FHypeMR 1 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 56°C, 1 min at 72°C; 10 min at 72°C |
| FHypeMR 2 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54°C, 1 min at 72°C; 10 min at 72°C |
| FHypeMR 3 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54°C, 1 min at 72°C; 10 min at 72°C |
| FHypeMR 4 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54.5°C, 1 min at 72°C; 10 min at 72°C |
| FHypeMR 5 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 55°C, 1 min at 72°C; 10 min at 72°C |
| FHypoMR 1 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54.5°C, 1 min at 72°C; 10 min at 72°C |
| FHypoMR 2 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 53°C, 1 min at 72°C; 10 min at 72°C |
| FHypoMR 3 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54°C, 1 min at 72°C; 10 min at 72°C |
| FHypoMR 4 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54.5°C, 1 min at 72°C; 10 min at 72°C |
| FHypoMR 5 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 54°C, 1 min at 72°C; 10 min at 72°C |
| FHypoMR 6 | 4 min at 98°C; 30 cycles for 45 sec at 94°C, 45 sec at 53°C, 1 min at 72°C; 10 min at 72°C |
FHypeMR, fetus hypermethylation regions; FHypoMR, fetus hypomethylation regions.
Statistical analysis
Affymetrix Tiling Analysis Software Version 1.1 (TAS; Affymetrix; Thermo Fisher Scientific, Inc.) was used for analysis and quality control of the GeneChip Tiling Arrays. TAS analyzed feature intensity data stored in the GeneChip Operating Software (GCOS), output CEL files and produced signals and P-values for each genomic position examined. Affymetrix Power Tools Software 1.15.1 (Affymetrix; Thermo Fisher Scientific, Inc.) was used to convert CEL files to text files. The methylation regions of the fetus group and the mother control group were compared using the Model-based Analysis of Tiling-array (MAT) calculation (P<0.0001). MAT (build: 3.07312009) was available from http://liulab.dfci.harvard.edu/MAT/. CisGenome 2.0 software (http://www.biostat.jhsph.edu/~hji/cisgenome/) (27) was used for annotating the gene names which were close to the methylation regions. Scatter plots were constructed utilizing plot function of R Software Package 3.0.1 (www.r-project.org). Two sets of results (from the fetus and the mother) of bisulfate direct sequencing were analyzed using the Quantification Tool for Methylation Analysis (QUMA) (web server version, http://quma.cdb.riken.jp/) (28) and compared by statistical software SPSS 13.0 (SPSS, Inc., Chicago, IL, USA) using two-way analysis of variance method followed by the boferroni post hoc test. P<0.01 was considered to indicate a statistically significant difference.
Results
The fetal and maternal DNA samples were all amplified using the PowerPlex 16 system kit in a multiplex amplification reaction system, and there was no maternal DNA in the placental tissue. DNA methylation detection of the 12 placental tissues and maternal peripheral blood DNA samples were conducted using the GeneChip Human Tiling 2.0R Array set. Across the whole genome, probes were tiled at ~35-base pair intervals as measured from the central position of adjacent oligonucleotides, leaving ~10-base pair gaps between oligonucleotides. In total, the values of the DNA methylation levels of all the DNA methylation regions in the whole genome were obtained for each sample. In order to obtain the fetus differential methylation regions, the DNA methylation levels in the methylation regions of fetuses were compared with those in mothers by using MAT calculation (P<0.0001). From the results, 383 fetus hypermethylation DNA regions and 458 fetus hypomethylation DNA regions were identified. The number of fetus hypermethylation DNA regions and fetus hypomethylation DNA regions in each chromosome are presented in Table IV. In order to identify the methylation differences between samples, scatter plots were constructed using R Software 3.0.1 (29), including the fetus hypomethylation region on chromosome X (Fig. 1A) and the fetus hypermethylation region on chromosome 7 (Fig. 1B). Red solid circles, which referred to the DNA methylation level of the fetus in the specified chromosomal location were gathered together, blue solid circles which referred to the DNA methylation level of the mother in this chromosomal location gathered together, and the red solid circle groups were visibly separated from the blue solid circle groups. If this was the case for any generated scatter plot, there were deemed to be differences in DNA methylation levels between fetuses and mothers. According to the above principle, 22 fetus hypermethylation regions and 26 fetus hypomethylation regions were selected. To further understand the sequence structure and function of these 48 methylation regions, the genes which these regions were associated with were identified using the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). A total of 5 fetus hypermethylation regions and 6 fetus hypomethylation regions were identified according to their functions. These were FHypeMR 1 (Chr4:81,219,256-81,219, 713; Table V), FHypeMR 2 (Chr6:50,894,411-50,894,837; Table V), FHypeMR 3 (Chr21:38,791,187-38,792, 106; Table V), FHypeMR 4 (Chr7:35,261,345-35,261,771; Table V), FHypeMR 5 (Chr22:25,205,384-25,205, 989; Table V), FHypoMR 1 (Chr1:41,215,030-41,215,734; Table V), FHypoMR 2 (ChrX:28,517,115-28,517, 628; Table V), FHypoMR 3 (Chr6:132,312,025-132,312,573; Table V), FHypoMR 4 (Chr1:10,194,437-10,194,830; Table V), FHypoMR 5 (Chr6:6267573-6268106; Table V) and FHypoMR 6 (Chr13:72,527,950-72,528,544; Table V).
Table IV.
Number of FHypeMR and FHypoMR in each chromosome.
| Chr No. | FHypeMR | FHypoMR | Total FDMR | % FHypeMR | % FHypoMR |
|---|---|---|---|---|---|
| Chr 1 | 48 | 47 | 95 | 50.53 | 49.47 |
| Chr 2 | 29 | 21 | 50 | 58.00 | 42.00 |
| Chr 3 | 23 | 25 | 48 | 47.92 | 52.08 |
| Chr 4 | 26 | 19 | 45 | 57.78 | 42.22 |
| Chr 5 | 30 | 18 | 48 | 62.50 | 37.50 |
| Chr 6 | 27 | 17 | 44 | 61.36 | 38.64 |
| Chr 7 | 15 | 18 | 33 | 45.45 | 54.55 |
| Chr 8 | 19 | 30 | 49 | 38.78 | 61.22 |
| Chr 9 | 21 | 11 | 32 | 65.63 | 34.37 |
| Chr 10 | 11 | 13 | 24 | 45.83 | 54.17 |
| Chr 11 | 18 | 28 | 46 | 39.13 | 60.87 |
| Chr 12 | 23 | 15 | 38 | 60.53 | 39.47 |
| Chr 13 | 10 | 14 | 24 | 41.67 | 58.33 |
| Chr 14 | 12 | 19 | 31 | 38.71 | 61.29 |
| Chr 15 | 13 | 22 | 35 | 37.14 | 62.86 |
| Chr 16 | 7 | 9 | 16 | 43.75 | 56.25 |
| Chr 17 | 17 | 26 | 43 | 39.53 | 60.47 |
| Chr 18 | 6 | 9 | 15 | 40.00 | 60.00 |
| Chr 19 | 9 | 25 | 34 | 26.47 | 73.53 |
| Chr 20 | 6 | 14 | 20 | 30.00 | 70.00 |
| Chr 21 | 7 | 2 | 9 | 77.78 | 22.22 |
| Chr 22 | 6 | 7 | 13 | 46.15 | 53.85 |
| Chr X | – | 49 | 49 | – | 100.00 |
FHypeMR, fetus hypermethylation regions; FHypoMR, fetus hypomethylation regions; Chr, chromosome; FDMR, fetus differential methylation regions.
Figure 1.
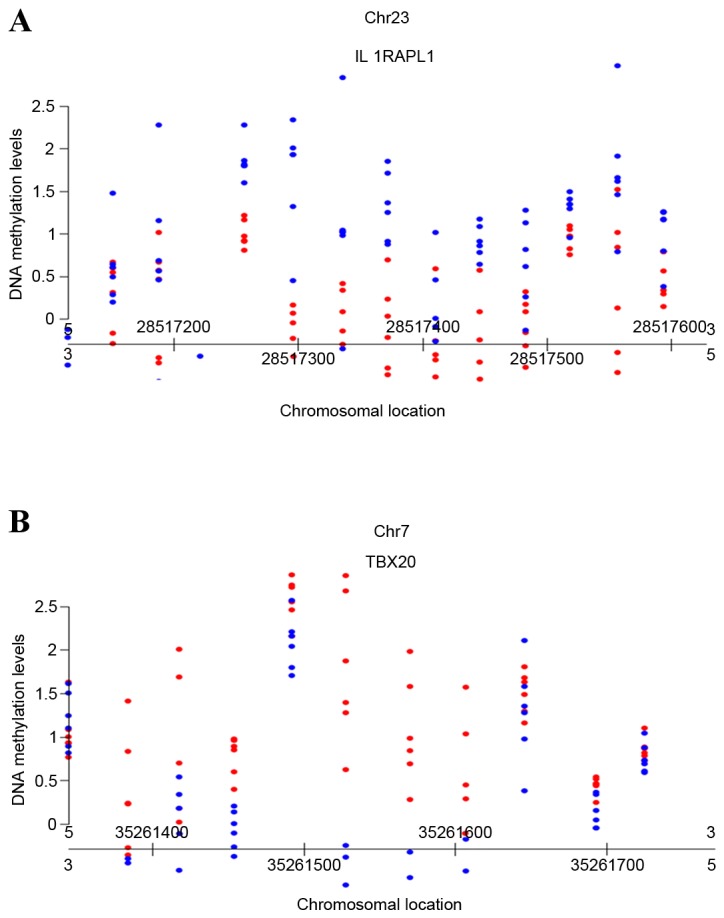
Scatter plots generated using R software packages based on the methylation degree. (A) The fetus hypomethylation region on chromosome X. (B) The fetus hypermethylation region on chromosome 7. The X axis refers to chromosomal location; the Y axis refers to the degree of the DNA methylation. Red dots refer to fetal DNA methylation CpG sites; blue dots refer to maternal DNA methylation CpG sited. CpG, C-phosphate-G.
Table V.
Further information concerning the 5 FHypeMR and 6 FHypoMR.
| Marker ID | Chromosomal location NCBI36/hg18 | Associated gene |
|---|---|---|
| FHypeMR 1 | Chr4:81,219,256-81,219,713 | Anthrax toxin receptor 2 (ANTXR2) |
| FHypeMR 2 | Chr6:50,894,411-50,894,837 | Transcription factor AP-2β (TFAP2B) |
| FHypeMR 3 | Chr21:38,791,187-38,792,106 | Erythroblastosis virus E26 oncogene homolog (ERG) |
| FHypeMR 4 | Chr7:35,261,345-35,261,771 | T-box 20 (TBX20) |
| FHypeMR 5 | Chr22:25,205,384-25,205,989 | Hermansky-Pudlak syndrome 4 (HPS4) |
| FHypoMR 1 | Chr1:41,215,030-41,215,734 | Cytidine triphosphate synthase (CTPS) |
| FHypoMR 2 | ChrX:28,517,115-28,517,628 | Interleukin 1 receptor accessory protein-like 1 (IL1RAPL1) |
| FHypoMR 3 | Chr6:132,312,025-132,312,573 | Connective tissue growth factor (CTGF) |
| FHypoMR 4 | Chr1:10,194,437-10,194,830 | Kinesin family member 1B (KIF1B) |
| FHypoMR 5 | Chr6:6,267,573-6,268,106 | Coagulation factor XIII, A1 polypeptide (F13A1) |
| FHypoMR 6 | Chr13:72,527,950-72,528,544 | Kruppel-like factor 5 (KLF5) |
Chr, chromosome; FHypeMR, fetus hypermethylation regions; FHypoMR, fetus hypomethylation regions.
FHypeMR 1 was associated with the anthrax toxin receptor 2 (ANTXR2) gene. This gene encodes a receptor for anthrax toxin, and mutations in this gene result in juvenile hyaline fibromatosis and infantile systemic hyalinosis (30). FHypeMR 2 was associated with the transcription factor AP-2β gene. Mutations in this gene result in autosomal dominant Char syndrome and congenital heart disease (31). FHypeMR 3 was associated with erythroblastosis virus E26 oncogene homolog. This gene encodes a member of the erythroblast transformation-specific family of transcription factors. All members of this family are key regulators of embryonic development, cell proliferation, differentiation, angiogenesis, inflammation and apoptosis. FHypeMR 4 was associated with T-box 20. This gene is essential for heart development, and mutations in this gene are associated with diverse cardiac pathologies, including defects in septation, valvulogenesis and cardiomyopathy (32). FHypeMR 5 was associated with Hermansky-Pudlak syndrome 4, and mutations in this gene result in subtype 4 of Hermansky-Pudlak syndrome (33). FHypoMR 1 was associated with the cytidine triphosphate synthase gene. The protein encoded by this gene is an enzyme responsible for the catalytic conversion of uridine triphosphate to cytidine triphosphate. Activity of this protein is important for the immune system, and loss of function of this gene is associated with immunodeficiency (34). FHypoMR 2 was associated with the interleukin 1 receptor accessory protein-like 1 gene. Deletions and mutations in this gene are present in patients with mental retardation (35). FHypoMR 3 was associated with the connective tissue growth factor gene. The protein encoded by this gene is a mitogen which is involved in cell adhesion and chondrocyte proliferation and differentiation in multiple cell types (36). FHypoMR 4 was associated with the kinesin family member 1B gene. Mutations in this gene result in to Charcot-Marie-Tooth disease, type 2A1 (37). FHypoMR 5 was associated with the coagulation factor XIII, AI polypeptide gene. This gene encodes the coagulation factor XIII A subunit, and a dysfunctional A subunit results in factor XIII deficiency (type II deficiency) (38). FHypoMR 6 was associated with the Kruppel-like factor 5 gene. Expression of this gene is altered in a variety of different cancers and in cardiovascular disease (39).
To verify the 11 selected methylation regions by genome-wide microarray and to detect the differential C-phosphate-G (CpG) sites in these regions, a bisulfate direct sequencing strategy was used. The results of bisulfate direct sequencing were analyzed by QUMA. The methylation status of 12 detected CpG loci in the ANTXR2 gene promotor from a blood sample from one mother and the respective placental tissue was visualized in Fig. 2A and B, respectively. The bisulfate sequencing results of this same promotor were visualized in Fig. 3, revealing that the 12 CpG sites of the ANTXR2 gene promotor were completely methylated in the placental tissue sample (Fig. 3A) but were completely unmethylated in the maternal peripheral blood sample (Fig. 3B).
Figure 2.

Bisulfate clone sequencing results from 2 samples for 12 CpG loci of the anthrax toxin receptor 2 gene promoter (Chr4:81,219,256-81,219,713). (A) The bisulfate direct sequencing result of (A) the mother and (B) the fetus. The X axis refers to 10 different clone sequencing results; the Y axis refers to 12 detected CpG loci. Black circles refer to methylated loci; white circles refer to unmethylated loci. CpG, C-phosphate-G.
Figure 3.

Bisulfate sequencing results of the ANTXR2 gene promoter for 2 samples. (A) The 12 CpG sites of the ANTXR2 gene promoter were completely methylated in the placental tissue sample. (B) The 12 CpG sites of ANTXR2 gene promoter were completely unmethylated in maternal peripheral blood cell. ANTXR2, anthrax toxin receptor 2; CpG, C-phosphate-G.
According to these results, 87 fetal differential methylation CpG sites in the 11 methylation DNA regions were selected (Table VI). In order to clarify whether the differences between mothers and fetuses were due to fetus sex, the bisulfate sequencing results were grouped according to fetus sex. The analysis of variance results revealed that there were no significant differences between male and female fetuses at any of the identified differential methylation CpG site. Therefore, it was concluded that the differences in methylation CpG sites between mothers and fetuses was not associated with the sex of the fetus in the present study.
Table VI.
Differential CpG site information.
| Marker ID | Number of CpG sites | Number of differential CpG sites (%) | Location of differential CpG sites |
|---|---|---|---|
| FHypeMR 1 | 12 | 10 (83) | 81,219,476; 81,219,552; 81,219,603; 81,219,612; 81,219,621; 81,219,627; 81,219,638; 81,219,641; 81,219,650; 81,219,653 |
| FHypeMR 2 | 10 | 9 (90) | 50,894,456; 50,894,470; 50,894,526; 50,894,629; 50,894,651; 50,894,741; 50,894,751; 50,894,755; 50,894,806 |
| FHypeMR 3 | 15 | 9 (60) | 38,791,473; 38,791,477; 35,791,498; 38,791,661; 38,791,686; 38,791,698; 38,791,811; 38,791,885; 38,791,979 |
| FHypeMR 4 | 20 | 16 (80) | 35,261,387; 35,261,407; 35,261,449; 35,261,542; 35,261,549; 35,261,582; 35,261,618; 35,261,632; 35,261,645; 35,261,672; 35,261,682; 35,261,684; 35,261,709; 35,261,723; 35,261,745; 35,261,752 |
| FHypeMR 5 | 13 | 7 (54) | 25,205,499; 25,205,552; 25,205,563; 25,205,588; 25,205,652; 25,205,858; 25,205,967 |
| FHypoMR 1 | 7 | 5 (71) | 41,215,318; 41,215,359; 41,215,520; 41,215,533; 41,215,588 |
| FHypoMR 2 | 8 | 5 (63) | 28,517,272; 28,517,282; 28,517,310; 28,517,324; 28,517,574 |
| FHypoMR 3 | 13 | 8 (62) | 132,312,111; 132,312,156; 132,312,241; 132,312,275; 132,312,279; 132,312,289; 132,312,308; 132,312,437 |
| FHypoMR 4 | 13 | 8 (62) | 10,194,426; 10,194,449; 10,194,458; 10,194,502; 10,194,589; 10,194,644; 10,194,659; 10,194,685 |
| FHypoMR 5 | 6 | 4 (67) | 6,267,635; 6,267,782; 6,267,854; 6,267,909 |
| FHypoMR 6 | 6 | 6 (100) | 72,528,064; 72,528,126; 72,528,131; 72,528,261; 72,528,273; 72,528,385 |
CpG, C-phosphate-G; FHypeMR, fetus hypermethylation regions; FHypoMR, fetus hypomethylation regions.
Discussion
Analysis of circulating cff DNA in maternal plasma is a promising, non-invasive strategy to determine fetal sex when diagnosing sex-linked disorders, fetal RhD blood group typing, fetal chromosomal aneuploidy detection and fetal monogenic diseases (4,6–13). However, there remain common diseases of the fetus that it is not possible to diagnose using NIPD methods. There are a variety of technological bottlenecks that remain to be resolved, including cff DNA extraction and detection. These problems will take time to solve. This means that follow-on improvements to the existing technology must be conducted to overcome the complication of the presence of background maternal DNA and to expand the scope of cff DNA application in clinical practice. Therefore, it is necessary to identify novel universal fetal-specific DNA markers to enhance the widespread clinical application of cff DNA.
Epigenetic differences between maternal and placental DNA have previously been explored as a promising strategy to distinguish fetal DNA from maternal DNA for NIPT, by analysing the fetal DNA in maternal plasma (2). In 2005, the first universal epigenetic marker of fetal DNA, serpin family B member 5 (SERPINB5), was described, and fetal SERPINB5 was distinguished from maternal SERPINB5 in maternal plasma DNA using the MeDIP quantitative polymerase chain reaction (qPCR) approach to quantify the fetal sex chromosome copy number in maternal plasma for NIPD (40). Based on methylated DNA immunoprecipitation with high-resolution tiling oligonucleotide array analysis, differentially methylated regions between placenta and maternal blood were identified. Based on the disease-specific methylated regions, by using the MeDIP qPCR approach, it is possible to discriminate normal from abnormal cases and diagnose trisomy 18 and 21 (15–17). This suggests that disease-specific epigenetic markers may be helpful in the development of NIPT for more diseases. Furthermore, based on the differentially methylated regions, by using sequence capture approaches, methylation-sensitive restriction enzymes, methylated DNA immunoprecipitation and qPCR-based approach and adaptor-mediated PCR amplification, it is possible to enrich and quantify the fragment fetal cff DNA from maternal cff DNA simultaneously with parallel sequencing platforms. Therefore, differentially methylated regions may be helpful in the diagnosis of more chromosomal diseases. At present, the lack of a fetal DNA marker that is universally detected in maternal plasma has limited the clinical application of this technology. The aim of the present study was the identification and selection of fetal-specific markers that have the potential to develop NIPD. Compared with previous reports, the present methylation study of differentially methylated regions provides high resolution. The results of the present study agree with previous reports that the array is a high-throughput detecting tool that provides efficient and robust genome-wide methylation profiling (41). However, in order to develop methylation-based NIPT, the technology requires further improvements and further research needs to be conducted.
The present study, concerning genome-wide methylation screening between placenta and maternal peripheral blood lymphocytes, was limited by the relatively small sample size. In addition, the study population was Han Chinese, which may differ from other populations. Therefore, further genome-wide methylation studies should be conducted in larger and different populations. The selected fetal differential methylation DNA regions and fetal differential methylation CpG sites should also be verified in a full clinical trial. In the future more clinical samples will be collected, including maternal plasma, and the selected fetal differential DNA markers will be applied in clinical practice. In the present study, methylation analysis of maternal blood and placental DNA samples was conducted across the whole genome and 5 fetus hypermethylation regions and 6 fetus hypomethylation regions were detected between maternal blood and placental DNA samples. In total, 87 fetal differential methylation CpG sites were identified in the 5 fetus hypermethylation regions and 6 fetus hypomethylation regions. The detection of the fetal differential methylation DNA regions and fetal differential methylation CpG sites may be instrumental in the development of an efficient NIPD, and in the expansion of the application fields in other disorders including Crohn's disease and factor XIII deficiency.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant nos. 81450018, 81471485, 81501336), Shanghai Joint Research Project for Important Disease (grant no. 2013ZYJB0015) and the Science and Technology Research Project of Henan Province (grant. nos. 162102310294 and 172102310295). The authors would also like to thank Dr. Zhou Zaiwei (WuXi App Tec Co., Ltd., Shanghai, China) for the technical assistance.
References
- 1.Xiang Y, Zhang J, Li Q, Zhou X, Wang T, Xu M, Xia S, Xing Q, Wang L, He L, Zhao X. DNA methylome profiling of maternal peripheral blood and placentas reveal potential fetal DNA markers for non-invasive prenatal testing. Mol Hum Reprod. 2014;20:875–884. doi: 10.1093/molehr/gau048. [DOI] [PubMed] [Google Scholar]
- 2.Lapaire O, Holzgreve W, Oosterwijk JC, Brinkhaus R, Bianchi DW. Georg Schmorl on trophoblasts in the maternal circulation. Placenta. 2007;28:1–5. doi: 10.1016/j.placenta.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–487. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 4.Lo YM, Hjelm NM, Fidler C, Sargent IL, Murphy MF, Chamberlain PF, Poon PM, Redman CW, Wainscoat JS. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N Engl J Med. 1998;339:1734–1738. doi: 10.1056/NEJM199812103392402. [DOI] [PubMed] [Google Scholar]
- 5.Lun FM, Chiu RW, Chan KC, Leung TY, Lau TK, Lo YM. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem. 2008;54:1664–1672. doi: 10.1373/clinchem.2008.111385. [DOI] [PubMed] [Google Scholar]
- 6.Costa JM, Benachi A, Gautier E, Jouannic JM, Ernault P, Dumez Y. First-trimester fetal sex determination in maternal serum using real-time PCR. Prenat Diagn. 2001;21:1070–1074. doi: 10.1002/pd.219. [DOI] [PubMed] [Google Scholar]
- 7.Khorshid HR Khorram, Zargari M, Sadeghi MR, Edallatkhah H, Shahhosseiny MH, Kamali K. Early fetal gender determination using real-time PCR analysis of cell-free fetal DNA during 6th-10th weeks of gestation. Acta Med Iran. 2013;51:209–214. [PubMed] [Google Scholar]
- 8.Rijnders RJ, van der Schoot CE, Bossers B, de Vroede MA, Christiaens GC. Fetal sex determination from maternal plasma in pregnancies at risk for congenital adrenal hyperplasia. Obstet Gynecol. 2001;98:374–378. doi: 10.1097/00006250-200109000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, Foo CH, Xie B, Tsui NB, Lun FM, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma; Proc Natl Acad Sci USA; 2008; pp. 20458–20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porreco RP, Garite TJ, Maurel K, Marusiak B, Obstetrix Collaborative Research Network. Ehrich M, van den Boom D, Deciu C, Bombard A. Noninvasive prenatal screening for fetal trisomies 21, 18, 13 and the common sex chromosome aneuploidies from maternal blood using massively parallel genomic sequencing of DNA. Am J Obstet Gynecol. 2014;211:365.e1–e12. doi: 10.1016/j.ajog.2014.03.042. [DOI] [PubMed] [Google Scholar]
- 11.Norton ME, Rose NC, Benn P. Noninvasive prenatal testing for fetal aneuploidy: Clinical assessment and a plea for restraint. Obstet Gynecol. 2013;121:847–850. doi: 10.1097/AOG.0b013e31828642c6. [DOI] [PubMed] [Google Scholar]
- 12.Lun FM, Tsui NB, Chan KC, Leung TY, Lau TK, Charoenkwan P, Chow KC, Lo WY, Wanapirak C, Sanguansermsri T, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma; Proc Natl Acad Sci USA; 2008; pp. 19920–19925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lam KW, Jiang P, Liao GJ, Chan KC, Leung TY, Chiu RW, Lo YM. Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: Application to β-thalassemia. Clin Chem. 2012;58:1467–1475. doi: 10.1373/clinchem.2012.189589. [DOI] [PubMed] [Google Scholar]
- 14.Papageorgiou EA, Patsalis PC. Non-invasive prenatal diagnosis of aneuploidies: New technologies and clinical applications. Genome Med. 2012;4:46. doi: 10.1186/gm345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim JH, Lee da E, Park SY, Kim DJ, Ahn HK, Han YJ, Kim MY, Ryu HM. Disease specific characteristics of fetal epigenetic markers for non-invasive prenatal testing of trisomy 21. BMC Med Genomics. 2014;7:1. doi: 10.1186/1755-8794-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DE, Kim SY, Lim JH, Park SY, Ryu HM. Non-invasive prenatal testing of trisomy 18 by an epigenetic marker in first trimester maternal plasma. PLoS One. 2013;8:e78136. doi: 10.1371/journal.pone.0078136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong YK, Jin S, Chiu RW, Ding C, Chan KC, Leung TY, Yu L, Lau TK, Lo YM. Noninvasive prenatal detection of trisomy 21 by an epigenetic-genetic chromosome-dosage approach. Clin Chem. 2010;56:90–98. doi: 10.1373/clinchem.2009.134114. [DOI] [PubMed] [Google Scholar]
- 18.Papageorgiou EA, Koumbaris G, Kypri E, Hadjidaniel M, Patsalis PC. The epigenome view: An effort towards non-invasive prenatal diagnosis. Genes (Basel) 2014;5:310–329. doi: 10.3390/genes5020310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianchi DW. Circulating fetal DNA: Its origin and diagnostic potential-a review. Placenta. 2004;25(Suppl A):S93–S101. doi: 10.1016/j.placenta.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Alberry M, Maddocks D, Jones M, Hadi M Abdel, Abdel-Fattah S, Avent N, Soothill PW. Free fetal DNA in maternal plasma in anembryonic pregnancies: Confirmation that the origin is the trophoblast. Prenat Diagn. 2007;27:415–418. doi: 10.1002/pd.1700. [DOI] [PubMed] [Google Scholar]
- 21.Keller EF. From gene action to reactive genomes. J Physiol. 2014;592:2423–2429. doi: 10.1113/jphysiol.2014.270991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shenderov BA, Midtvedt T. Epigenomic programing: A future way to health? Microb Ecol Health Dis. 2014;25 doi: 10.3402/mehd.v25.24145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao HW, Li J. Epigenetic modifications in fibrotic diseases: Implications for pathogenesis and pharmacological targets. J Pharmacol Exp Ther. 2015;352:2–13. doi: 10.1124/jpet.114.219816. [DOI] [PubMed] [Google Scholar]
- 24.Geiman TM, Muegge K. DNA methylation in early development. Mol Reprod Dev. 2010;77:105–113. doi: 10.1002/mrd.21118. [DOI] [PubMed] [Google Scholar]
- 25.Patil V, Ward RL, Hesson LB. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics. 2014;9:823–828. doi: 10.4161/epi.28741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak W, Kim JN, Kim D, Hong JS, Jeong JH, Kim H, Cho S, Kim YY. Genome-wide DNA methylation profiles of small intestine and liver in fast-growing and slow-growing weaning piglets. Asian-Australas J Anim Sci. 2014;27:1532–1539. doi: 10.5713/ajas.2014.14309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumaki Y, Oda M, Okano M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res. 2008;36(Web Server Issue):W170–W175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.R Core Team, corp-author. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- 30.Denadai R, Raposo-Amaral CE, Bertola D, Kim C, Alonso N, Hart T, Han S, Stelini RF, Buzzo CL, Raposo-Amaral CA, Hart PS. Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system. Am J Med Genet A. 2012;158A:732–742. doi: 10.1002/ajmg.a.35228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lingaiah K, Sosalagere DM, Mysore SR, Krishnamurthy B, Narayanappa D, Nallur RB. Mutations of TFAP2B in congenital heart disease patients in Mysore, South India. Indian J Med Res. 2011;134:621–626. doi: 10.4103/0971-5916.90986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Posch MG, Gramlich M, Sunde M, Schmitt KR, Lee SH, Richter S, Kersten A, Perrot A, Panek AN, Al Khatib IH, et al. A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects. J Med Genet. 2010;47:230–235. doi: 10.1136/jmg.2009.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saito A, Kuratomi G, Ito C, Matsuoka H, Suzuki T, Ozeki Y, Watanabe T, Fujii K, Shimoda K, Fukushima Y, et al. An association study of the Hermansky-Pudlak syndrome type 4 gene in schizophrenic patients. Psychiatr Genet. 2013;23:163–173. doi: 10.1097/YPG.0b013e32836130a9. [DOI] [PubMed] [Google Scholar]
- 34.Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, Fabrega S, Nitschké P, Esposti MD, Schwartzentruber J, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature. 2014;510:288–292. doi: 10.1038/nature13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Youngs EL, Henkhaus R, Hellings JA, Butler MG. IL1RAPL1 gene deletion as a cause of X-linked intellectual disability and dysmorphic features. Eur J Med Genet. 2012;55:32–36. doi: 10.1016/j.ejmg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burke JP, O'Connell RM, Lennon G, Doherty GA, Keegan D, O'Donoghue D, Mulcahy H, Hyland J, Winter DC, Sheahan K, O'Connell PR. The influence of CTGF single-nucleotide polymorphisms on outcomes in Crohn's disease. Ann Surg. 2013;258:767–774. doi: 10.1097/SLA.0000000000000247. [DOI] [PubMed] [Google Scholar]
- 37.Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/S0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- 38.Schroeder V, Kohler HP. Factor XIII deficiency: An update. Semin Thromb Hemost. 2013;39:632–641. doi: 10.1055/s-0033-1353392. [DOI] [PubMed] [Google Scholar]
- 39.Dong Z, Yang L, Lai D. KLF5 strengthens drug resistance of ovarian cancer stem-like cells by regulating survivin expression. Cell Prolif. 2013;46:425–435. doi: 10.1111/cpr.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chim SS, Tong YK, Chiu RW, Lau TK, Leung TN, Chan LY, Oudejans CB, Ding C, Lo YM. Detection of the placental epigenetic signature of the maspin gene in maternal plasma; Proc Natl Acad Sci USA; 2005; pp. 14753–14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heyn H, Carmona FJ, Gomez A, Ferreira HJ, Bell JT, Sayols S, Ward K, Stefansson OA, Moran S, Sandoval J, et al. DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis. 2013;34:102–108. doi: 10.1093/carcin/bgs321. [DOI] [PMC free article] [PubMed] [Google Scholar]