

Abstract
Defects in DNA repair can result in oncogenic genomic instability. Cancers occurring from DNA repair defects were once thought to be limited to rare inherited mutations (such as BRCA1 or 2). It now appears that a clinically significant fraction of cancers have acquired DNA repair defects. DNA repair pathways operate in related networks, and cancers arising from loss of one DNA repair component typically become addicted to other repair pathways to survive and proliferate. Drug inhibition of the rescue repair pathway prevents the repair-deficient cancer cell from replicating, causing apoptosis (termed synthetic lethality). However, the selective pressure of inhibiting the rescue repair pathway can generate further mutations that confer resistance to the synthetic lethal drugs. Many such drugs currently in clinical use inhibit PARP1, a repair component to which cancers arising from inherited BRCA1 or 2 mutations become addicted. It is now clear that drugs inducing synthetic lethality may also be therapeutic in cancers with acquired DNA repair defects, which would markedly broaden their applicability beyond treatment of cancers with inherited DNA repair defects. Here we review how each DNA repair pathway can be attacked therapeutically and evaluate DNA repair components as potential drug targets to induce synthetic lethality. Clinical use of drugs targeting DNA repair will markedly increase when functional and genetic loss of repair components are consistently identified. In addition, future therapies will exploit artificial synthetic lethality, where complementary DNA repair pathways are targeted simultaneously in cancers without DNA repair defects.
Our DNA is not contained pristine in the nucleus, but rather is subject to assault by endogenous and exogenous genotoxins. Exogenous insults to DNA include hypoxia, lack of nutrients, radiation, dietary carcinogens, and medications (1–3). Endogenous insults include oxygen-free radicals from metabolism, aberrant incision of DNA by immune or repair nucleases, and collision of replication forks with messenger RNA transcription or noncanonical DNA structures (1–4). Almost every element of the DNA structure can be damaged, from base damage to breaks in phosphodiester bonds. Given the precarious existence of DNA and the need to maintain genome stability to prevent cell death or neoplastic transformation, DNA repair is a critical function for all cells.
Defects in DNA repair can lead to an increase in genomic instability, which is one mechanism of oncogenic transformation (5–8). Genomic instability produces the mutations that dysregulate growth and promote tumor cell invasion and metastasis (5,9,10). However, DNA repair defects can be exploited in cancer therapy because excessive genomic instability itself can have lethal consequences by inducing deadly mutations, mitotic catastrophe, or chromothripsis (11,12). The same defects in DNA repair that produced oncogenesis in the first place make replication more stressful for that cell because the continuous DNA replication a cancer cell undergoes requires many DNA repair components (13,14). The cancer cell must find replacements for the original oncogenic loss of the DNA repair component to continue replicating. These replacement DNA repair components can be targeted to prevent the repair and restart of stressed replication forks (15,16).
There are four major types of DNA repair pathways, some with multiple subpathways (Figure 1) (17–19). These repair pathways operate within the DNA damage response (DDR), a complex network of checkpoint signaling and DNA repair pathways that promote cell survival and genome stability or trigger programmed cell death when damage is excessive (20–23). Defects in DDR components predispose to cancer, determine tumor response to chemo- and radiotherapy, and underlie several congenital conditions including multiple types of Seckel syndrome, primordial dwarfism, and premature aging syndromes (24–26). DDR components are often defective in cancer, but the DDR comprises interacting/crosstalking pathways, and defects in one can be compensated by alternative pathways. Such compensatory pathways are formidable obstacles to successful cancer treatment.
Figure 1.

DNA repair pathways in mammalian cells. A) Double-strand breaks (DSBs) activate DNA damage response signaling including checkpoint arrest through ATM, ATR, and DNA-PKcs. DSB repair pathway choice is determined by the amount of 5’ end resection at the DSB, inhibited by 53BP1/RIF1, promoted by BRCA1/CtIP. MRE11 initiates limited end resection, and this is followed by Exo1/EEPD1 and Dna2 nucleases for extensive resection. 53BP1/RIF1 and Ku protect DSB ends from resection, promoting classical nonhomologous end joining (cNHEJ). PARP1 competes with Ku and promotes limited end resection for alternative nonhomologous end joining (aNHEJ). RAD51 catalyzes invasion by the resected 3' end into the sister or other homologous sequences, and Pol δ catalyzes repair synthesis across the DSB. The amount of 3’ end resection regulates DSB pathway choice. cNHEJ requires little or no end resection, aNHEJ requires limited resection, and homologous recombination (HR) and single-strand annealing (SSA) require extensive resection. DNA polymerase θ (Pol θ) promotes a microhomology search by the opposing 3’ single strands after short resection in aNHEJ. The HR subpathway SSA requires extensive end resection to expose large homologous regions, usually direct repeats, with RAD52 promoting annealing, producing large deletions between direct repeats. B) Base excision repair repairs minor lesions (eg, oxidized bases), promoted by PARP1. Repair intermediates include an abasic site and nicking of the DNA backbone, with short gap filling by Pol β. C) Nucleotide excision repair (NER) repairs large helix-distorting lesions and involves excision of about 15 nucleotides on either side of the lesion, followed by gap filling. GG-NER and TC-NER differ in lesion recognition by XPA or CSA/B, respectively. D) Mismatch repair involves recognition of the mismatched nucleotide by MutSα/β, followed by strand nicking with MutL, extensive resection with Exo1, and repair synthesis. DNA ligases catalyze religation, the final step in all pathways. aNHEJ = alternative nonhomologous end joining; cNHEJ = classical nonhomologous end joining; DDR = DNA damage response; DSB = double-strand break; HR = homologous recombination; SSA = single-strand annealing.
Among the most dangerous DNA lesions are double-strand breaks (DSBs), which can trigger apoptosis or lead to oncogenic translocations (1–3,17–19,27). There are two major DSB repair pathways, nonhomologous end joining (NHEJ) and homologous recombination (HR), each with two subpathways (Figure 1A) (17–19,27). In NHEJ, DSB ends are trimmed and ligated, and NHEJ is therefore error prone, while HR uses a homologous sequence (typically the sister chromatid) as a repair template and is generally accurate (27,28).
NHEJ includes classical (cNHEJ) and alternative (aNHEJ) pathways (Figure 1A). The choice between these pathways is regulated by 53BP1/RIF1, which promotes cNHEJ, the dominant pathway (29), and PARP1, which promotes aNHEJ (30). Similar to HR, aNHEJ involves 5’ strand end resection, which reveals microhomologies for annealing. By contrast, cNHEJ directly ligates free DSB ends (29,31). aNHEJ serves as the backup repair pathway for two important HR functions. If HR is not functional, aNHEJ can rescue resected DSBs (31,32), such as might occur in G0/G1 of the cell cycle or abnormally in BRCA1/2-mutant cancers. aNHEJ also provides a backup mechanism to repair broken replication forks when HR is deficient at the cost of genome stability (2,31,32).
HR has accurate (conservative) and inaccurate subpathways that require extensive 5’ end resection to produce 3’ SS DNA tails, a process that initiates when BRCA1/CtIP out-competes 53BP1/RIF1 for DSBs (Figure 1A) (28,29). In the accurate HR subpathway, 3’ SS DNA coated with RAD51 invades a homologous sequence to copy genetic information to effect repair. Inaccurate HR, termed single-strand annealing (SSA), is RAD52 dependent and deletes one repeat and DNA between linked repeats, or it can cause translocations when two DSBs occur in or near repetitive elements on different chromosomes (33,34). RAD51-mediated HR is the dominant pathway for restarting stalled or broken replication forks, which if processed improperly by NHEJ cause genome instability and neoplastic transformation (2,27,28).
Three pathways repair single-strand damage: base excision repair (BER), mismatch repair (MMR), and nucleotide excision repair (NER) (35–37). In these pathways, the undamaged, complementary strand serves as repair template. BER is initiated by glycosylases that remove the damaged base, followed by strand nicking, PARP1-promoted DNA synthesis across the lesion site and strand religation (Figure 1B) (35). NER repairs bulky DNA lesions by excising approximately 30 nt containing the lesion, followed by DNA synthesis and ligation (Figure 1C) (37). MMR recognizes and excises mismatched nucleotides introduced during DNA replication (and HR heteroduplex intermediates), and repair is completed by DNA synthesis and ligation (Figure 1D) (36).
Like DSBs, DNA interstrand crosslinks (ICLs) are dangerous because they present an absolute block to replication. ICL repair in the G1/0 phase involves dual incisions flanking the ICL, excision via NER, and DNA synthesis to fill the gap. ICL repair in the S phase is similar but involves HR to provide an accurate template for repair synthesis across the excised lesion (Figure 2) (38). When replication forks converge on an ICL, BRCA1 contributes to replisome dissociation, and the consequent SS DNA is protected with RAD51. A host of Fanconi anemia proteins activate ATR and recruit XPF and MUS81-EME1 nucleases that make two incisions on one strand flanking the ICL. This creates a substrate for translesion synthesis in one duplex (39) and a DSB on the other. NER then excises the lesion, and HR completes repair (Figure 2).
Figure 2.

Interstrand crosslinks can be repaired by replication-independent (left) and replication-dependent (right) mechanisms. Both repair pathways involve dual incisions, translesion synthesis across the crosslinked segment, and nucleotide excision repair (NER) to remove the incised crosslink. Replication-dependent interstrand crosslink (ICL) repair also involves homologous recombination (HR). When forks converge on an ICL, BRCA1 and RAD51 protect the stalled fork, and the Fanconi anemia (FA) repair pathway repairs the crosslink. FA repair is initiated by the ubiqutination of FANCD2, which recruits nucleases XPF, MUS81/EME1, and SLX1 to incise the crosslink, followed by translesion synthesis across the lesion, NER to remove the lesion, and HR to repair the replication fork. Both mechanisms are error free, except for mutations that may be introduced by translesion synthesis polymerases. DSB = double-strand break; FA = Fanconi anemia; HR = homologous recombination; ICL = interstrand crosslink; NER = nucleotide excision repair; TC-NER = transcription-coupled NER.
Defects in any of these repair pathways can lead to malignant transformation, and any pathway can also be subverted to assist the cancer cell in resisting therapy. There is considerable crosstalk among the single- and double-strand lesion repair pathways and replication fork restart pathways. This crosstalk reflects the many mechanistic commonalities in the pathways: lesion recognition, SS DNA binding, structure-specific endo- and exonuclease cleavage, strand annealing, polymerase gap filling, and ligation. Repair pathways display several types of crosstalk. There is signaling crosstalk between the HR and cNHEJ pathways through ATR, ATM, and DNA-PK (40,41). There is functional crosstalk, shown by several examples in which overexpression of a DNA repair component in one pathway compensates for a repair defect in another, conferring therapeutic resistance (42). Finally, there is direct crosstalk when specific components are shared among pathways, for example, PARP1 functions in BER and in aNHEJ (Figure 1) (30). Although PARP1 is not required for HR repair of frank DSBs (43), PARP1 promotes MRE11 recruitment to collapsed replication forks prior to HR repair (Figure 3). PARP1 may also promote HR by increasing repair factor accessibility to damage by modifying chromatin (44,45). Also, when PARP1-dependent BER is blocked, unrepaired lesions cause fork collapse, which requires HR for proper restart (13–15,46). Thus, PARP1 plays similar roles in repairing different types of DNA lesions (27,30,35). This promiscuous functionality makes PARP1 a common crutch for malignancies that arise due to defects in DNA repair, and thus an attractive synthetic lethal target. Other shared DNA repair components may similarly prove useful as synthetic lethal targets, including RPA, DNA polymerases, and structure-specific nucleases.
Figure 3.

PARP1 at intersecting repair pathways. PARP1 promotes base excision repair (BER) and cell survival. PARP1 inhibitors (PARP1i) cause unrepaired intermediates such as SS nicks to accumulate. They also trap PARP1 on chromatin, causing replication forks to stall. Double-strand breaks are either from the collision of the fork with a BER SS nick repair intermediate or from nuclease cleavage of a stalled replication fork. Collapsed replication forks are repaired and restarted by homologous recombination (HR) promoted by PARP1 modification of MRE11, which initiates 5’ end resection. In HR-deficient cells (eg, BRCA1/2 mutants), HR repair of stalled forks cannot occur, accounting for the synthetic lethality of PARP1i in HR-deficient cancers. BER = base excision repair; DSB = double-strand break; HR = homologous recombination; SSB = single-strand break.
The Concept of Synthetic Lethality
Original oncogenic events are mutations that promote uncontrolled cell replication, and these will occur more frequently in cells with inherited or acquired DNA repair defects (7,8,20). Cell replication requires several of the DNA repair pathways to be functional (15,16,18,22,27). When there is a systemic and persistent defect in DNA repair in the transformed cell, then that cell becomes dependent on another repair pathway that can back up the defective pathway for replication (27,28,34). Synthetic lethality occurs when the backup pathway is chemically inhibited and the cancer cell can no longer replicate (13–15). Such stalled replication induces apoptosis.
A well-known example of this is the inherited mutations in the HR components BRCA1 and 2 in breast and ovarian cancer (13,14). All the patient’s cells have one mutated allele of BRCA1 or 2, but the cancer has both alleles mutated, and the cancer cell has an unstable genome because it has lost HR capability. Because HR is the most important pathway for repairing and restarting stalled replication forks, these cancers become dependent on PARP1-mediated SSB repair and alternative nonhomologous end joining to repair and restart their replication forks (12–15). When PARP1 is inhibited by a drug, then these cancers cannot repair and restart replication forks (Figure 3) (13,14,19,42,44). The stalled forks collapse and can aberrantly ligate together, and distinct chromosomes can fuse, resulting in mitotic catastrophe and subsequent apoptosis (45,46). Because the nonmalignant cells have normal HR as they retain one normal BRCA1 and 2 allele, only the cancer cells are sensitive to PARP1 inhibition (42–46).
Subversion of Synthetic Lethality: Synthetic Rescue
Synthetic lethality is an important tool in cancer treatment, but it has limitations. For example, PARP1 inhibitors are very effective against BRCA1-defective cancers, but treatment can lead to development of resistance because of second-site mutations in other checkpoint signaling or DNA repair proteins, such as 53BP1, Rif1, PTIP, SFLM11, JMJD1C, and REV7 (47–52). At least three PARP1 inhibitor resistance mechanisms have been identified: restoration of HR, for example, by mutation or downregulation of 53BP1; loss of PARP1 itself; and upregulation of the PgP drug transporter (53). PARP1 inhibitor resistance by 53BP1 loss appears specific to BRCA1-defective tumors, reflecting the interplay between BRCA1, 53BP1, and RIF1 (Figure 1A); in BRCA2-defective tumors, resistance to PARP1 inhibition develops because of secondary BRCA mutations (53).
These findings have clinical relevance. For example, 53BP1 expression is reduced in a fraction of sporadic triple-negative and BRCA1-defective breast cancers (47). Generally, breast cancer patients respond well to PARP1 inhibitors, but eventually resistance develops and disease progresses (54). This has stimulated efforts to identify targets to prevent or overcome resistance to PARP1 inhibition and to identify markers of resistance (55–57).
Drugging Base Excision Repair
The most important rationale for targeting BER is that cancer cells have a higher oxidative status than normal cells and therefore suffer more oxidative DNA damage (4,35,58). Oxidized nucleotides can result in two forms of crosslinks that block replication; the abasic deoxyribose can react with an adenine on the opposing strand or covalently link with DNA polymerase β (Pol β), forming a DNA-protein adduct (59–61). In addition, BER repairs alkylation damage, and thus can mediate chemotherapy resistance (35,62). The rate-limiting step in BER is APE1 phosphodiester cleavage 5’ to an abasic site, following glycosylase removal of the oxidized base (35). Not surprisingly, APE1 is overexpressed in many cancer types, and there have been multiple attempts to target APE1 for cancer therapy (42,63–65). However, APE1 inhibitors have not had much clinical impact because their biochemical activity did not translate well to cell and animal models (63–65).
We and others have described compounds that inhibit Pol β (42,66,67), but these have not been pursued pharmaceutically because of their relatively modest activity. In addition, specificity for Pol β compared with other DNA polymerases has not been defined, so these compounds may have in vivo toxicity that would limit their usefulness (66,67). However, Pol β remains an attractive target in BRCA1/2-defective tumors. Inhibiting Pol β generates the same cleaved, unrepaired SS BER intermediate that PARP1 inhibition does, and also causes replication fork DSBs (35,66–68). Thus, Pol β inhibitors should also be effective in treating HR-deficient cancers, similar to PARP1 inhibitors.
PARP1 is an essential component for BER, and its inhibition in HR-defective cancers has been by far the most effective means of targeting of DNA repair for cancer therapy to date (Figure 3) (69,70). Two groups demonstrated that BRCA1/2-mutant tumors, defective in HR, were sensitive to PARP1 inhibitors (13,14). DSBs generated at replication forks are largely repaired by HR (2,27,28), and HR requires BRCA1/2 (27,28). PARP1 is required for BER, and catalytic inactivation causes accumulation of SSBs. Thus, replication fork collision with SSBs causes fork collapse to DSBs, and repair occurs predominantly (and most accurately) by HR (Figure 3, middle). In BRCA1/2 mutants, or other HR-defective cancers, such forks cannot be repaired appropriately (13,14,69). Replication-associated DSBs are highly toxic DNA lesions that cause mitotic catastrophe and apoptosis if improperly repaired or unrepaired (13,14,69,70). Apart from SSB generation, PARP1 inhibition kills BRCA1/2-mutant cancer in at least two other ways, trapping PARP1 on DNA at the lesion site (71) and increasing ribonalactone-adenine and ribonalactone-Pol β adducts (59–61). Trapped PARP1 and DNA adducts stall replication forks, which are cleaved by structure-specific nucleases (MUS81-EME2, EEPD1, Metnase) (72–75), causing fork collapse (Figure 3), again requiring HR for restart and lacking in BRCA1/2-mutated malignancies (66–68). Thus, the remarkable efficacy of PARP1 inhibitors in HR-deficient malignancies is due in part to PARP1’s multiple roles in DNA repair.
Drugging Mismatch Repair
During DNA replication, incorrect nucleotides are occasionally incorporated into the daughter DNA strand, creating mismatched base pairs corrected by MMR (36); mismatches also form in heteroduplex DNA during HR (76). MMR comprises four key steps: mismatch recognition, excision of the lesion, DNA synthesis across the SS gap, and ligation (Figure 1D) (36,77). Two heterodimeric proteins recognize the lesion: MutSα (MSH2/6 complex) recognizes short mismatches, and MutSβ (MSH2/3 complex) recognizes longer insertion-deletion loops (36,77,78). Binding of either heterodimer recruits the heterodimer MutL (MLH1 and PMS2 complex). MutL recruits Exo1, which excises the mismatched DNA (79) in a reaction enhanced by PARP1 (80). Pol δ fills in the gap, and DNA ligase I seals the nick (81).
Hereditary nonpolyposis colorectal cancer (HPNCC or Lynch syndrome) is an inherited autosomal-dominant disease resulting from defects in MMR proteins, with the majority of mutations affecting MLH1, MSH2, and MSH6 (82). Silencing by somatic methylation of MMR gene promoters also decreases MMR (83,84) and confers resistance to platinum-based chemotherapy (85). DNA demethylating agents such as 5-azacytidine induce re-expression of MMR components in these cancers, restoring sensitivity to cisplatin or carboplatin (86,87).
Cancers with MLH1, MSH2, or MSH6 defects display synthetic lethality with therapeutic potential, but it is important to identify the specific MMR deficiency in the tumor as they differ in therapeutic response. For example, MSH2-mutant cancers are sensitive to methotrexate, an antimetabolite that inhibits DNA synthesis, and psoralen, a DNA crosslinking agent, but MLH1-mutant cancers are resistant to both treatments (88,89).
Unrepaired oxidized nucleotides accumulate upon BER repression (eg, Pol β or PINK1 inhibition) or methotrexate treatment, which increases mismatch formation during DNA synthesis, increasing the burden on MMR, and mutagenesis. These effects are strongly exacerbated in MMR-defective cancers, a dynamic that presents synthetic lethal opportunities. For example, Pol β inhibition is synthetically lethal in MSH2- or MLH1-deficient tumors (90) (Figure 4A). Thus, Pol β inhibitors are promising agents for treatment of MMR-deficient cancers, as well as the aforementioned BRCA1/2-mutant cancers (42,66).
Figure 4.

Synthetic lethality with mismatch repair (MMR) defects. A) MSH2 or MLH1 defects are synthetically lethal, with increased oxidative damage caused by inhibition of Pol β or PINK1, or by treatment with methotrexate. PINK1 increases oxidation of nucleotides, burdening base excision repair (BER), and Pol β inhibition decreases BER repair of oxidized nucleotides, which induce DNA mismatches during DNA synthesis. B) MSH3 assists in loading RAD51 onto end-resected SS DNA during homologous recombination (HR) repair. MSH3 defects can behave similarly to HR defects and are synthetically lethal when BER is blocked by inhibition of PARP1. MSH3 deficiency could also increase neoantigen generation within the tumor, which would result in synergy between the PARPi and immune checkpoint inhibitors in these cancers. BER = base excision repair; HR = homologous recombination; i = inhibition; MMR = mismatch repair.
Some MMR-deficient cancers behave like BRCA1/2-mutant cancers, with defects in stressed replication fork repair. MSH3 is critical for loading RAD51 during HR repair, and thus MSH3-deficient cancers are sensitive to PARP1 inhibition (Figure 4B) (91,92). Clinical trials with PARP1 inhibitors in MSH3-mutant colon cancer are warranted, especially in conjunction with an immune checkpoint inhibitor because genomic instability associated with MMR-deficient colon cancer increases neoantigen production and thus increases the chance of immune recognition (93).
Drugging Nucleotide Excision Repair
NER processes DNA lesions resulting from exposure to UV light, environmental toxins, and some chemotherapeutic drugs (37,94,95). NER has two subpathways: global genome NER (GG-NER) and transcription-coupled NER (TC-NER). GG-NER repairs lesions across the whole genome, whereas TC-NER repairs lesions in transcribed DNA, initiated by RNA polymerase II stalling at DNA lesions. These pathways differ in only two respects: how the DNA lesion is recognized and kinetically (TC-NER is faster than GG-NER) (37).
In GG-NER DNA, damage is recognized by XPC-RAD23B, which binds to the undamaged DNA strand opposite the lesion, recruiting downstream NER components. Helicase XPB unwinds the DNA, and XPD recruits the RPA/XPA/XPG complex. This complex recruits the nuclease ERCC1-XPF, which incises 5’ to the lesion, and initiates DNA synthesis across the gap by Pol δ and Pol κ or Pol ε, followed by 3’ incision by XPG to remove the damaged DNA and ligation by DNA ligase III/XRCC1 or DNA ligase I (Figure 1C) (37,94,95). In TC-NER lesions that stall RNA polymerase II are recognized by WD repeat protein CSA, SWI/SNF family member CSB, and XAB2. This complex is exchanged with the TFIIH complex, and repair proceeds as above (96).
Three known autosomal recessive inherited diseases are associated with defects in the NER pathway: xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy (TTD). These inherited mutations result in either an extreme predisposition to cancer (XP) or neurodevelopmental defects associated with rapid aging but without cancer predisposition (CS and TTD) (97).
NER deficiency confers sensitivity to crosslinking agents such as cisplatin, reflecting reduced crosslink repair (98,99). The ERCC1-XPF nuclease complex is essential for repair of platinum-DNA crosslinks, as well as trimming flaps during DSB repair by SSA (98) and aNHEJ (100). Unrepaired crosslinks cause replication stress and eventually apoptosis (2,17,18). Importantly, lower expression of ERCC1 correlated with increased sensitivity to platinum agents in several tumor types (98,99,101). Low expression of ERCC1 is a biomarker for response to cisplatin in non–small cell lung cancers (NSCLCs), and PARP1 inhibitors are synthetically lethal in cancers with low ERRC1 (102), implying that PARP1 inhibitors may be effective in this fraction of NSCLC patients, especially when combined with cisplatin (103). Inhibitors targeting the ERCC1-XPF active site are in development as adjuncts to platinum-based chemotherapy (104,105).
Protein-DNA interactions that mediate NER have also been targeted, and small molecules have been identified that block DNA interaction with RPA and XPA (106,107). RPA is important for both NER and HR, and RPA inhibition causes cell cycle arrest, cell death, and enhances sensitivity to cisplatin and etoposide (106,108). This suggests that RPA inhibitors could be combined with PARP1 inhibitors to mediate synthetic lethality in cancers with nonmutated BRCA1/2.
Perturbation of NER components may be synthetically lethal with PARP1 inhibition. For example, PARP1 inhibition in combination with DDB1 or XAB2 deficiency is synthetically lethal in non-BRCA mutant cells (109), and combined inhibition of PARP1 and topoisomerase I (with camptothecin) has greater cytotoxicity in cancer cells depleted of XPF-ERCC1 (Figure 5) (110).
Figure 5.

Synthetic lethal targeting with PARP1 and ERCC1-XPF deficiencies. Camptothecin traps TopoI covalently onto DNA, blocking replication. Repair can proceed via a PARP-TDP and ERCC1-XPF1 pathway (left) or by fork repair and restart via homologous recombination; PARP1 inhibition, coupled with ERCC1-XPF deficiency, is synthetically lethal (middle). PARP1 inhibition also blocks base excision repair of single-strand lesions that block replication; these lesions are similarly lethal with ERCC1-XPF deficiency (right). BER = base excision repair; CPT = camptothecin; DSB = double-strand break; HR = homologous recombination.
ATR inhibition in ERCC1-depleted cancer cells is synthetically lethal (111). ERCC1 depletion not only increases DNA mismatches, but also single-strand lesions, which stall replication forks. Repair of stalled forks requires ATR, accounting for the synthetic lethality of ATR inhibition and ERCC1 depletion; this effect of ATR inhibition is specific to ERCC1 as no other NER deficiencies are sensitive to ATR inhibition, consistent with ERCC1-XPF functioning in other DNA repair pathways (99). The combination of an ERRC1 inhibitor with a PARP1 or ATR inhibitor may create artificial synthetic lethality, where one DNA repair inhibitor induces dependency on another pathway blocked by a second drug. An example of this, mentioned above, is combining RPA and PARP1 inhibitors, which could generate artificial synthetic lethality in cancers that do not harbor an HR defect. This principle could be widely applied to many clinical scenarios (66–68).
Drugging HR and CrossLink Repair
In addition to defects in BRCA1 or 2, cancer genome sequencing revealed mutations in many other HR pathway components that promote oncogenesis, including PALB2 (112), BRCA1-interacting protein 1 (BRIP1; also termed FANCJ and BACH1) (113), BARD1 (114), BAP1 (115) and RAD51C (116). This expansion of HR driver mutations should broaden the clinical application of PARP1 inhibitors (117). To achieve this goal, oncologists need reliable methods to identify patients carrying mutations in any HR components. Exome sequencing is currently the most common method and is accepted by regulatory bodies that approve PARP1 inhibitor indications (118,119). However, such sequencing misses a fraction of tumors that are functionally deficient in HR but lack mutations in known HR genes. One solution to this problem is to use DNA sequencing to measure genomic abnormalities that are due to functional HR loss, such as loss of heterozygosity, telomeric allelic imbalance, and chromosomal translocations (118,120,121). In this way, tumors with functional HR deficiency but without mutations in known HR genes can be identified and treated using synthetic lethal approaches. Such an approach has been used with great success in recent trials with PARP1 inhibitors (118,120).
Several upstream modulators of HR have been identified as synthetic lethal targets that can sensitize cancer cells to PARP1 inhibitors (Figure 6). For example, several cyclin-dependent kinases (CDKs) are upstream HR regulators, and a recent study showed that the pan-CDK inhibitor dinaciclib impairs HR repair and sensitizes cancer cells to the PARP1 inhibitor veliparib (122). Dinaciclib probably acts by blocking required phosphorylation of the HR components Exo1 and BRCA1 (17,28,29,122). ATR is another upstream HR regulator that promotes restart/repair of stalled replication forks. Not surprisingly, PARP1 inhibition synergizes with ATR blockade (123).
Figure 6.

Repression of or mutation in several upstream regulators of homologous recombination (HR) can be synthetically lethal with PARP1 inhibition because they can cause a variety of HR repair defects that prevent fork restart. These upstream regulators include several CDKs, PTEN, USP11, and the cohesins. Inherited or acquired mutations in downstream HR components such as RAD51, MRE11, BLM, and WRN can lead to cancer. Such cancers also demonstrate synthetic lethality with PARP1 inhibition, although there could be biologically significant normal tissue toxicity when the mutation is an inherited autosomal recessive. When HR is defective in any of these cases, Pol θ inhibition would also be synthetically lethal because Pol θ is also required for the alternative nonhomologous end joining backup replication fork repair pathway.
Interestingly, PARP1 is also essential for the survival of malignancies with isocitrate dehydrogenase 1 and 2 mutations (IDH1 or 2, mainly glioblastomas and acute myeloid leukemias). These IDH1/2 mutations generate the oncometabolite 2-hydroxuglutarate (2-HG). 2-HG inhibits the dioxygenase class of enzymes, such as histone demethylases, and inhibits HR repair. These IDH1/2-mutant cancers are exquisitely sensitive to PARP1 inhibition with olaparib or BMN-673 (124). Adding exogenous 2-HG to non-IDH1/2-mutant cancers produced PARP1 sensitivity (124). Thus, PARP1 inhibitors may be effective to treat these malignancies, and 2-HG itself may be another tool to induce artificial synthetic lethality.
The tumor suppressor phosphatase and tensin homolog (PTEN) promotes HR repair during replication stress (125). PTEN is mutated in a fraction of malignancies; this mutation sensitizes cancer cells to PARP1 inhibitors (Figure 6) (125). These results suggest that clinical trials might be warranted to test PARP1 inhibitor effects on cancers with PTEN mutations, including breast cancer and glioma (126). PTEN mutations are also responsible for PTEN hamartomatous syndromes (such as Cowden’s syndrome), and while not true malignancies, these syndromes are debilitating and disfiguring and can progress to cancer. The relatively low toxicities of PARP1 inhibitors suggest these syndromes as attractive targets for such intervention (127).
The clinical success of PARP1 inhibitors stimulated efforts to identify other proteins whose inactivation in cancer might make those cancers responsive to PARP1 inhibition. RNAi screening approaches have identified the deubiquitinating enzyme USP11 (128), CDK12 (129), and cohesins (130) that when silenced or mutated cause synthetic lethality with PARP1 inhibitors (Figure 6). Interestingly, acute myeloid leukemia driven by aberrant transcription factors is also highly sensitive to PARP1 inhibition (131), suggesting that PARP1 inhibition may be synthetically lethal in many types of cancers with defects in various DNA repair genes.
HR is important for the resolution of DNA crosslinks by the Fanconi anemia (FA) DNA repair pathway (Figure 2) (38,132), and inherited mutations in FA components cause cancer predisposition syndromes (133). Although autosomal recessive biallelic mutations of FA genes contribute to leukemogenesis, targeting FA components may be exploited for therapeutic gain in malignancies in patients without FA. For example, cancer cells depleted of FA components are extremely sensitive to crosslinking agents such as cisplatin (134,135), and FA pathway inhibition confers sensitivity to PARP1 inhibition (136). Thus, combining FA and PARP1 inhibitors might expand the use of PARP1 inhibitors beyond BRCA1/2-mutant cancers.
The FA component FANCD2 must be mono-ubiquitinated to activate FA pathway crosslink repair (Figure 2), and most therapeutic efforts have targeted this event (136). For example, proteosome inhibition with bortezomib was reported to decrease FANCD2 mono-ubiquitination and block crosslink repair (137). A PARP1 inhibitor may enhance bortezomib efficacy in mantle cell lymphoma or myeloma (136,137). A small molecule inhibitor of NEDD8 activation, MLN4924, decreases FANCD2 activation, which would sensitize cancer cells to crosslink damage (138). Small molecule inhibitors of USP1-UAF-mediated deubiquitination of FANCD2 prevent FANCD2 recycling and ultimately decrease FA pathway activity (134,139). These selective USP1/UAF1 deubiquitinase inhibitors also enhance sensitivity of cancer cells to crosslinkers such as cisplatin (134–136,139).
RAD52 mediates SSA (Figure 1A) in humans and also serves as a backup for BRCA2 to load RAD51 onto SS DNA during HR (140). BRCA1/2-mutant cancer cells are forced to rely on RAD52 to repair replication forks (140,141). Thus, depleting RAD52 in BRCA1/2-mutant cancer presents another synthetic lethal approach distinct from PARP1 inhibition (141,142). Several groups, including ours, have generated small molecule inhibitors of RAD52 that are cytotoxic to cancer cells with BRCA1/2 defects (143–145). RAD52 and PARP1 inhibitors could be combined to treat HR-deficient cancers to increase the duration or depth of response (66–68).
Drugging NHEJ
There is substantial crosstalk among DSB repair pathways (15,17,28,29), and this presents many opportunities to exploit synthetic lethal interactions to improve cancer therapy (68). HR, cNHEJ, and aNHEJ share the MRE11/RAD50/NBS1 (MRN) complex, an early DSB sensor important for activation of ATM-dependent DNA damage checkpoint signaling (15,28,29,146). HR and aNHEJ share 5’ end resection, which is regulated by 53BP1/RIF1, DNA-PK, and BRCA1/CtIP (28,29,32,146) and mediated by several nucleases (MRE11, Dna2, Exo1, EEPD1) (17,75,147). As discussed above, the extent of 5’ end resection regulates DSB repair pathway choice (Figure 1A) (28,29,147).
In cNHEJ, broken ends are initially bound by the Ku heterodimer that inhibits end resection and recruits DNA-PKcs, which then recruits the end-ligation complex XRCC4/ligase 4/XLF (31,148). PARP1 initiates aNHEJ by out-competing Ku for DSB ends (Figure 1A) (30,148). Thus, Ku and PARP1 competition regulates cNHEJ vs aNHEJ choice, and it is likely that cNHEJ is favored approximately 10:1 because Ku is more abundant and has high affinity for DNA ends (30,148–150).
In aNHEJ, Mre11/CtIP promote limited 5’ end resection, and Pol θ mediates micro-homology-mediated alignment between 3’ SS DNA tails at each end of the break (151,152). This creates 3’ flaps that are trimmed by one or more structure-specific endonucleases such as FEN1; repair is completed by ligase III/XRCC1 (148,150,153). Although cNHEJ is inaccurate, producing small insertions and deletions at the repair junction, inherited defects in cNHEJ confer genome instability and predispose to cancer (18,27,28,148,150), probably because the aNHEJ backup pathway is even more inaccurate, generating larger deletions (148,153,154) and mediating most chromosomal translocations (34,69,155,156).
Targeting cNHEJ to enhance cancer treatment was originally studied in radiation therapy (42,157,158). Radiation therapy typically uses x-rays or protons, whose most deleterious lesions are DSBs repaired by cNHEJ (157). Carbon ion radiotherapy uses high-mass/high-charge particles to create dense ionization tracks that produce clustered DNA damage that is effective against radio-resistant cancers (159). While cNHEJ is the dominant repair pathway for sparsely ionizing x-rays and protons, HR appears to play a more important role in repair of clustered DSBs (160–163). HSP90 inhibitors can block HR by downregulating RAD51, and these agents sensitize cancer cells to carbon ions (163–166).
Other HR inhibitors such as the CDK inhibitor dinaciclib, or ATR inhibitors, may also potentiate the lethal effects of heavy ion DNA damage, but these agents may have limited use in x-ray or proton therapy, where cNHEJ dominates. Thus, it is critical to have a clear understanding of the relevant repair pathways when attempting to augment particular radiotherapy modalities. This concept also extends to chemotherapy. For example, repressing cNHEJ sensitizes tumor cells to etoposide, a TopoIIα inhibitor that generates DSBs, but the same cNHEJ defect confers resistance to camptothecin, a TopoI inhibitor that produces SSBs (167).
PARP1 functions in multiple DNA repair pathways (Figures 1, 3, and 6), and synthetic lethal strategies that exploit this fact are promising (13,14,68–70,93,94). For example, nearly 60% of prostate cancer patients carry a TMPRSS2–ERG translocation event, and this fusion protein interferes with cNHEJ (168). PARP1 functions in aNHEJ and BER, suggesting that TMPRSS2-ERG prostate cancer cells treated with a PARP1 inhibitor would be deficient in BER, cNHEJ, and aNHEJ. This would increase unrepaired DNA damage with lethal consequences (168). Indeed, the PARP1 inhibitor olaparib was effective in relapsed prostate cancers carrying BRCA1/2 mutations, although these patients were not stratified by TMPRSS2-ERG (69,119).
KRAS mutations are common in acute leukemias, and it was recently found that these mutations correlate with overexpression of the aNHEJ factors PARP1, ligase III, and XRCC1 (169). Overexpression of these aNHEJ components produced an increased reliance on aNHEJ to repair DNA damage in these malignancies, and this could mediate treatment resistance. Combining PARP1 inhibitors with chemotherapeutics that induce replication stress may overcome resistance of KRAS-mutant leukemias and improve outcomes (169).
Histone deacetylases (HDACs) play important regulatory roles in chromatin function, and HDAC inhibitors have been developed as antineoplastic agents and radiosensitizing agents (170,171). HDACs are not restricted to histone targets; they are more properly described as protein deacetylases. Acetylation is important for activation of certain NHEJ components. Consistent with this, the pan-HDAC inhibitor trichostatin A enhances acetylation of the critical cNHEJ factor Ku and the aNHEJ initiator PARP1, which inhibits cNHEJ and traps PARP1 on chromatin. This blocks both NHEJ pathways and is synthetically lethal in leukemia cells (172).
Recent evidence implicates the aNHEJ component Pol θ as a key target for cancer therapy. High Pol θ levels are associated with poor breast cancer patient survival (173); Pol θ is overexpressed in greater than 80% of NSCLCs, and expression levels correlate with poor outcomes (174). One reason that cancer cells are addicted to DNA repair is that they are programmed to proliferate, regardless of whether their environment is impoverished or the extent of genome damage. Many (but not all) cancers must divide to survive, and replication fork arrest is fatal (15,16,32,75). The common mechanism of action of many chemotherapeutic agents is replication stress (2,19,28). Thus, cancers with HR deficiency become addicted to aNHEJ, which is the backup repair pathway for stalled replication forks (Figure 7) (31,32). It is not surprising then that there appears to be a synthetic lethal relationship between aNHEJ and HR mediated by Pol θ. HR-deficient cancers need all the components of aNHEJ, not just PARP1, to continue replication in the face of an oxidized genome. Consistent with this model, Pol θ depletion in BRCA1/2-deficient ovarian cancer cells was synthetically lethal (175,176). Thus, Pol θ is a very exciting target for HR-deficient cancers (177).
Figure 7.
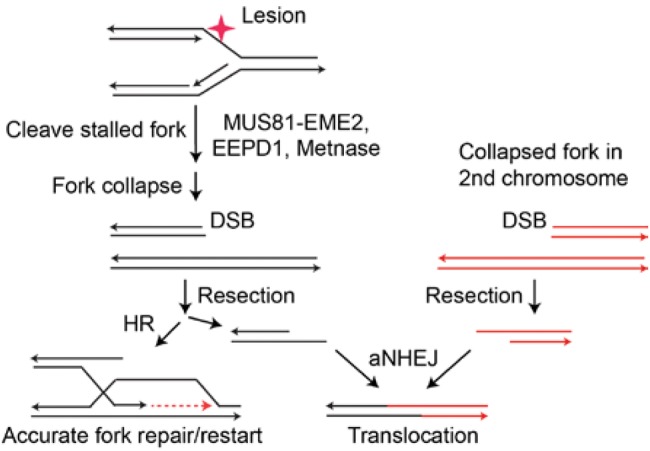
Collapsed forks are normally repaired by homologous recombination (HR). If HR fails, double-strand breaks on different chromosomes may be repaired by the alternative nonhomologous end joining (aNEHJ) backup pathway. The aNHEJ pathway can mediate chromosomal translocations, however, and thus is a riskier repair mechanism for the cell. aNHEJ = alternative nonhomologous end joining; DSB = double-strand break; HR = homologous recombination.
Clinical Effectiveness of Targeting DNA Repair
The most effective clinical drugging of the addiction of cancer to DNA repair has been with PARP1 inhibitors (13,14,68–70,118–121). Many of these compounds also inhibit PARP2 to some extent, although their activity is thought to be due to their PARP1 inhibition (13,14,68–70). The first PARP1 inhibitor to complete clinical evaluation was olaparib (AZD-2281, Lynparza, AstraZeneca). The first phase I trial examined single-agent olaparib in relapsed breast, ovarian, and prostate cancer (178). In that trial, 60 patients were treated with various doses, but objective antitumor activity was observed only in eight ovarian cancer patients who had documented BRCA1/2 mutations. Based on a phase II trial in BRCA1/2-mutant ovarian cancer showing a 34% objective response rate (95% confidence interval = 26% to 42%) and a median response duration of 7.9 months, olaparib was granted accelerated approval by the US Food and Drug Admnistration (179–181). Olaparib was similarly effective against BRCA1/2-mutant ovarian cancers that were sensitive or resistant to platinum-based chemotherapy. Randomized and nonrandomized phase II trials in patients with previously treated ovarian cancer confirmed that approximately one-third of patients with BRCA1/2-mutant ovarian cancer experience objective tumor regression with olaparib monotherapy (182,183).
Modest gastrointestinal toxicity and hematopoietic toxicity seem to be the most common side effects of olaparib, and these are manageable (180,181,183). More worrisome is the 2% rate of myelodysplasia and acute myeloid leukemia seen in olaparib-treated patients (182,183). However, these are known risks of chemotherapeutics used in ovarian cancer, and it is possible that these adverse events were not due to olaparib but rather to prior chemotherapy. Olaparib does not appear to produce objective responses in tumors lacking BRCA1/2 function, although there was some benefit in terms of progression-free survival (182,183).
Ovarian cancers with BRCA1/2 mutations are not the only HR-deficient cancers that respond to olaparib. A recent phase II trial of olaparib in relapsed and refractory metastatic prostate cancer found that 16 of 49 patients had an objective response, and 14 of the responders had mutations in HR components such as BRCA1/2 or ATM (119). This indicates that a clinically significant fraction of prostate cancers have HR defects treatable with PARP1 inhibitors, perhaps as frontline agents to attack metastatic disease given their minimal toxicity.
Responses to olaparib in other BRCA1/2-mutant cancers have not been as clinically significant as in ovarian cancer. BRCA1/2-mutant breast cancers often have a worse outcome than comparable nonmutated breast cancer, with a response rate of 12.9% in one trial (179). In addition, BRCA1/2-mutant pancreatic cancers did not respond as well to olaparib as a single agent as did the ovarian cancers (179). Multiple studies are exploring whether combining olaparib with crosslinking agents such as carboplatin or cisplatin in triple-negative breast cancer will enhance responses in these cancers. However, a randomized study of adding the PARP1 inhibitor veliparib to cyclophosphamide in relapsed triple-negative breast cancer did not show any benefit compared with veliparib alone (184). Veliparib did show single-agent activity in ovarian cancer though, with a 26% response rate (185).
Niraparib is a highly selective PARP1/2 inhibitor soon to be approved in the United States and Europe. Similar to olaparib, niraparib was evaluated in patients with platinum-sensitive ovarian cancer in maintenance therapy. This trial was a randomized, placebo-controlled, phase III study that enrolled 553 patients over 35 months (120). Patients were not required to have BRCA1/2-mutated cancer but were stratified into BRCA1/2-mutated and wild-type cohorts. Patients with wild-type BRCA1/2 were analyzed to determine if tumors were functionally HR deficient by assaying for increased genomic and telomeric rearrangements (120,121). Importantly, niraparib monotherapy showed statistically significant improvement in the primary end point, progression-free survival (PFS), in all three groups. PFS improved from 5.5 to 21.0 months in the BRCA1/2-mutant cohort, from 3.9 to 12.9 months in the BRCA1/2 wild-type/HR-deficient cohort, and from 3.8 to 6.9 months in the BRCA1/2 wild-type/HR-proficient cohort (120). The improvement in PFS with niraparib in the HR-proficient cohort was surprising and could be due to several factors. It could indicate that many HR-proficient ovarian cancers depend on active PARP1 for survival, perhaps to repair oxidative DNA damage. It is also possible that the responsive BRCA1/2 wild-type tumors could have expressed ERCC1 at low levels (102). As PARP1 inhibitors with improved specific activity are developed, they may prove effective against a far broader range of malignancies than originally considered, similar to niraparib.
At least 16 additional PARP1 inhibitors are under clinical development (70). PARP1 inhibitors are being evaluated in combination regimens with DNA-damaging agents, such as platinum analogs or ionizing radiation, or with agents that interfere with other steps in DNA repair or replication. For example, olaparib potentiates the activity of the TopoI inhibitor SN-38 (the active metabolite of the chemotherapeutic agent irinotecan) by blocking RAD51-dependent DNA repair (186). However, a phase I trial combining olaparib with the TopoI inhibitor topotecan was terminated early because of hematologic toxicity at doses below the known effective single-agent dosing levels of each drug (187). Notably, adequate doses of veliparib and topotecan were safely administered together in another phase I study, supporting additional studies of this combination in cervical cancers, where activity was seen in patients with cancers having low PARP1 expression at baseline (188). Veliparib was also examined in multiple types of relapsed hematologic malignancies in combination with carboplatin and topotecan, with some activity seen in chronic myelomonocytic leukemia; interestingly, leukemias with FA pathway deficiency showed the best responses (189).
Summary
There is not a one-to-one correspondence between loss of a repair process and a druggable addiction, and not every cancer has a systemic defect in a DNA repair pathway. While all cancers have many DNA mutations, this is by no means equivalent to the systemic loss of a DNA repair pathway. Such mutations could be random events, with all DNA repair pathways intact. However, the systemic loss of a DNA repair pathway is more common than previously appreciated, and therefore offers a wealth of opportunities to exploit in therapy.
Given the complexity of DSB repair pathways and crosstalk with DNA damage signaling networks, many more synthetic lethal strategies are likely to be revealed through continued study of the regulators of cellular responses to genotoxic cancer therapies. As more data are reported on targeting DNA repair for cancer therapy, several principles are evident that will extend DNA repair targeting beyond those cancers with BRCA1 or 2 defects. First, cancer cells must overcome more endogenous DNA damage than normal cells because of increased oxidative damage and replication stress from forced cell division (2,28,29,58,190). Because specific DNA repair pathways are backed up by other repair pathways, synthetic lethality only occurs when the primary pathway is defective and the backup rescue pathway is repressed (66–68).
Interestingly, it appears that cancer cell death is less due to the lack of repair, but rather the persistence of toxic repair intermediates (75). Incomplete repair can be more toxic than if a repair pathway is never initiated (191). Therefore, defining repair pathway relationships will reveal additional synthetic lethal targets. For example, the understanding that aNHEJ backs up HR in replication fork restart (32) leads to the concept that inhibiting other aNHEJ components besides PARP1, such as Pol θ, will also confer synthetic lethality in HR-defective cancers (177). Such investigations will increase the number of DNA repair components that can be targeted.
A second principle is that cancers can be responsive to drugs that target DNA repair even if they do not carry mutations in known repair proteins such as BRCA1 or 2. Thus, cancers may be functionally defective in repair but not genetically deficient in any known repair pathway (120,121). It will be important to reach a consensus on methods for identifying cancers that are functionally defective in specific repair pathways, such as the genomic instability assay used in the niraparib trial (120), because this will expand the patient base that could benefit from DNA repair–targeted therapies.
Finally, many cancers do not have an identifiable functional or genetic deficiency in a DNA repair pathway that would lend itself to synthetic lethality. Such cancers may be best treated with drug combinations that induce artificial synthetic lethality by blocking primary and backup repair pathways. In these cases, the therapeutic index is not due to differential repair capacity in normal vs cancer cells, but rather to the heavier load of endogenous DNA damage characteristic of many cancers. Such approaches would permit DNA repair targeting to be much more widely applied in cancer therapy. It is also important to identify synthetic rescue pathways and develop strategies to block these before therapeutic resistance develops. It is clear that while drugging DNA repair is still in its infancy, there is enormous potential to this approach because it will be applicable to many other malignancies besides those with BRCA1 or 2 mutations.
Funding
JAN was supported by National Institutes of Health (NIH) GM084020, SHL by NIH CA151367, and RH by NIH GM109645 and NIH CA205224.
Note
The funders had no role in the writing of the review or decision to submit it for publication.
References
- 1. Williams AB, Schumacher B.. DNA damage responses and stress resistance: Concepts from bacterial SOS to metazoan immunity. Mech Ageing Dev. 2016; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mazouzi A, Velimezi G, Loizou JI.. DNA replication stress: Causes, resolution and disease. Exp Cell Res. 2014;329(1):85–93. [DOI] [PubMed] [Google Scholar]
- 3. Mehta A, Haber JE.. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014;6(9):a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res. 2015;763:212–245. [DOI] [PubMed] [Google Scholar]
- 5. Hanks S, Coleman K, Reid S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–1161. [DOI] [PubMed] [Google Scholar]
- 6. Weaver BA, Silk AD, Montagna C, et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11(1):25–36. [DOI] [PubMed] [Google Scholar]
- 7. Shih IM, Zhou W, Goodman SN, et al. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61(3):818–822. [PubMed] [Google Scholar]
- 8. Lengauer C, Kinzler KW, Vogelstein B.. Genetic instabilities in human cancers. Nature. 1998;396(6712):643–649. [DOI] [PubMed] [Google Scholar]
- 9. Hanahan D, Weinberg RA.. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- 10. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12(12):801–817. [DOI] [PubMed] [Google Scholar]
- 11. Forment JV, Kaidi A, Jackson SP.. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat Rev Cancer. 2012;12(10):663–670. [DOI] [PubMed] [Google Scholar]
- 12. Rode A, Maass KK, Willmund KV, et al. Chromothripsis in cancer cells: An update. Int J Cancer. 2016;138(10):2322–2333. [DOI] [PubMed] [Google Scholar]
- 13. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. [DOI] [PubMed] [Google Scholar]
- 14. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. [DOI] [PubMed] [Google Scholar]
- 15. Allen C, Ashley AK, Hromas R, et al. More forks on the road to replication stress recovery. J Moll Cell Biol. 2011;3:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Budzowska M, Kanaar R.. Mechanisms of dealing with DNA damage-induced replication problems. Cell Biochem Biophys. 2009;53(1):17–31. [DOI] [PubMed] [Google Scholar]
- 17. Ciccia A, Elledge SJ.. The DNA damage response: Making it safe to play with knives. Mol Cell. 2010;40(2):179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kennedy RD, D'Andrea AD.. DNA repair pathways in clinical practice: Lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24(23):3799–3808. [DOI] [PubMed] [Google Scholar]
- 19. Gavande NS, VanderVere-Carozza PS, Hinshaw HD, et al. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther. 2016;160:65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson SP, Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roos WP, Kaina B.. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332(2):237–248. [DOI] [PubMed] [Google Scholar]
- 22. Goldstein M, Kastan MB.. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–143. [DOI] [PubMed] [Google Scholar]
- 23. Sirbu BM, Cortez D.. DNA damage response: Three levels of DNA repair regulation. Cold Spring Harb Perspect Biol. 2013;5(8):a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shaheen R, Faqeih E, Ansari S, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24(2):291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Croteau DL, Popuri V, Opresko PL, et al. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Casper AM, Durkin SG, Arlt MF, et al. Chromosomal instability at common fragile sites in Seckel syndrome. Am J Hum Genet. 2004;75(4):654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mladenov E, Magin S, Soni A, et al. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin Cancer Biol. 2016;37–38:51–64. [DOI] [PubMed] [Google Scholar]
- 28. Ceccaldi R, Rondinelli B, D'Andrea AD.. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daley JM, Sung P.. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Moll Cell Biol. 2014;34(8):1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang M, Wu W, Wu W, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iliakis G, Murmann T, Soni A.. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen. 2015;793:166–175. [DOI] [PubMed] [Google Scholar]
- 32. Truong LN, Li Y, Shi LZ, et al. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A. 2013;110(19):7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nimonkar AV, Sica RA, Kowalczykowski SC.. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc Natl Acad Sci U S A. 2009;106(9):3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weinstock DM, Richardson CA, Elliott B, et al. Modeling oncogenic translocations: Distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair. 2006;5(9–10):1065–1074. [DOI] [PubMed] [Google Scholar]
- 35. Bauer NC, Corbett AH, Doetsch PW.. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015;43(21):10083–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reyes GX, Schmidt TT, Kolodner RD, et al. New insights into the mechanism of DNA mismatch repair. Chromosoma. 2015;124(4):443–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marteijn JA, Lans H, Vermeulen W, et al. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15(7):465–481. [DOI] [PubMed] [Google Scholar]
- 38. Michl J, Zimmer J, Tarsounas M.. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35(9):909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Budzowska M, Graham TG, Sobeck A, et al. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;34(14):1971–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ashley AK, Shrivastav M, Nie J, et al. DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair. 2014;21:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu S, Opiyo SO, Manthey K, et al. Distinct roles for DNA-PK, ATM, and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012;40:10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley MR, Logsdon D, Fishel ML.. Targeting DNA repair pathways for cancer treatment: What's new? Future Oncol. 2014;10(7):1215–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schultz N, Lopez E, Saleh-Gohari N, et al. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003;31(17):4959–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Vos M, Schreiber V, Dantzer F.. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem Pharmacol. 2012;84(2):137–146. [DOI] [PubMed] [Google Scholar]
- 45. Luo X, Kraus WL.. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26(5):417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Helleday T, Bryant HE, Schultz N.. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle. 2005;4(9):1176–1178. [DOI] [PubMed] [Google Scholar]
- 47. Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339(6120):700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49(5):858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang J, Aroumougame A, Lobrich M, et al. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014;28(24):2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Watanabe S, Watanabe K, Akimov V, et al. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol. 2013;20(12):1425–1433. [DOI] [PubMed] [Google Scholar]
- 52. Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521(7553):541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lord CJ, Ashworth A.. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. [DOI] [PubMed] [Google Scholar]
- 54. Livraghi L, Garber JE.. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015;13:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murai J, Feng Y, Yu GK, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jiang J, Lu Y, Li Z, et al. Ganetespib overcomes resistance to PARP inhibitors in breast cancer by targeting core proteins in the DNA repair machinery. Invest New Drugs. 2017; in press. [DOI] [PubMed] [Google Scholar]
- 57. Choi YE, Meghani K, Brault ME, et al. Platinum and PARP inhibitor resistance due to overexpression of microRNA-622 in BRCA1-mutant ovarian cancer. Cell Rep. 2016;14(3):429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Helleday T. Poisoning cancer cells with oxidized nucleosides. N Engl J Med. 2015;373(16):1570–1571. [DOI] [PubMed] [Google Scholar]
- 59. Catalano MJ, Liu S, Andersen N, et al. Chemical structure and properties of interstrand cross-links formed by reaction of guanine residues with abasic sites in duplex DNA. J Am Chem Soc. 2015;137(11):3933–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Greenberg MM. Abasic and oxidized abasic site reactivity in DNA: Enzyme inhibition, cross-linking, and nucleosome catalyzed reactions. Acc Chem Res. 2014;47(2):646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinones JL, Thapar U, Yu K, et al. Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase β in vivo. Proc Natl Acad Sci U S A. 2015;112(28):8602–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bobola MS, Kolstoe DD, Blank A, et al. Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to temozolomide. Front Oncol. 2012;2:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kaur G, Cholia RP, Mantha AK, et al. DNA repair and redox activities and inhibitors of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/Ref-1): A comparative analysis and their scope and limitations toward anticancer drug development. J Med Chem. 2014;57(24):10241–10256. [DOI] [PubMed] [Google Scholar]
- 64. Abbotts R, Jewell R, Nsengimana J, et al. Targeting human apurinic/apyrimidinic endonuclease 1 (APE1) in phosphatase and tensin homolog (PTEN) deficient melanoma cells for personalized therapy. Oncotarget. 2014;5(10):3273–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Al-Safi RI, Odde S, Shabaik Y, et al. Small-molecule inhibitors of APE1 DNA repair function: An overview. Curr Mol Pharmacol. 2012;5(1):14–35. [PubMed] [Google Scholar]
- 66. Jaiswal AS, Panda H, Law BK, et al. NSC666715 and its analogs inhibit strand-displacement activity of DNA polymerase β and potentiate temozolomide-induced DNA damage, senescence and apoptosis in colorectal cancer cells. PLoS One. 2015;10(5):e0123808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arian D, Hedayati M, Zhou H, et al. Irreversible inhibition of DNA polymerase beta by small-molecule mimics of a DNA lesion. J Am Chem Soc. 2014;136(8):3176–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shaheen M, Allen C, Nickoloff JA, et al. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood. 2011;117:6074–6082. [DOI] [PubMed] [Google Scholar]
- 69. Palmbos PL, Hussain MH.. Targeting PARP in prostate cancer: Novelty, pitfalls, and promise. Oncology. 2016;30(5):377–385. [PubMed] [Google Scholar]
- 70. Papa A, Caruso D, Strudel M, et al. Update on poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J Transl Med. 2016;14:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Murai J, Zhang Y, Morris J, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349(3):408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pepe A, West SC.. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014;7(4):1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. De Haro LP, Wray J, Williamson EA, et al. Metnase promotes restart and repair of stalled and collapsed replication forks. Nucleic Acids Res. 2010;38:5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim H-S, Chen Q, Kim S-K, et al. The DDN catalytic motif is required for Metnase functions in NHEJ repair and replication restart. J Biol Chem. 2014;289:10930–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wu Y, Lee SH, Williamson EA, et al. EEPD1 rescues stressed replication forks and maintains genome stability by promoting end resection and homologous recombination repair. PLoS Genet. 2015;11(12):e1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Heyer WD, Ehmsen KT, Liu J.. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hsieh P, Yamane K.. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129(7–8):391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Iyama T, Wilson DM 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair. 2013;12(8):620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guillotin D, Martin SA.. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp Cell Res. 2014;329(1):110–115. [DOI] [PubMed] [Google Scholar]
- 80. Liu Y, Kadyrov FA, Modrich P.. PARP-1 enhances the mismatch-dependence of 5'-directed excision in human mismatch repair in vitro. DNA Repair. 2011;10(11):1145–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Larrea AA, Lujan SA, Nick McElhinny SA, et al. Genome-wide model for the normal eukaryotic DNA replication fork. Proc Natl Acad Sci U S A. 2010;107(41):17674–17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fishel R, Kolodner RD.. Identification of mismatch repair genes and their role in the development of cancer. Curr Opin Genet Dev. 1995;5(3):382–395. [DOI] [PubMed] [Google Scholar]
- 83. Moreira L, Munoz J, Cuatrecasas M, et al. Prevalence of somatic mutl homolog 1 promoter hypermethylation in Lynch syndrome colorectal cancer. Cancer. 2015;121(9):1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nagasaka T, Rhees J, Kloor M, et al. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 2010;70(8):3098–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fink D, Aebi S, Howell SB.. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998;4(1):1–6. [PubMed] [Google Scholar]
- 86. Appleton K, Mackay HJ, Judson I, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol. 2007;25(29):4603–4609. [DOI] [PubMed] [Google Scholar]
- 87. Nie J, Liu L, Li X, et al. Decitabine, a new star in epigenetic therapy: The clinical application and biological mechanism in solid tumors. Cancer Lett. 2014;354(1):12–20. [DOI] [PubMed] [Google Scholar]
- 88. Wu Q, Vasquez KM.. Human MLH1 protein participates in genomic damage checkpoint signaling in response to DNA interstrand crosslinks, while MSH2 functions in DNA repair. PLoS Genet. 2008;4(9):e1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Martin SA, McCarthy A, Barber LJ, et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med. 2009;1(6–7):323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Martin SA, McCabe N, Mullarkey M, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17(3):235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hemann MT. From breaking bad to worse: Exploiting homologous DNA repair deficiency in cancer. Cancer Discov. 2014;4(5):516–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Takahashi M, Koi M, Balaguer F, et al. MSH3 mediates sensitization of colorectal cancer cells to cisplatin, oxaliplatin, and a poly(ADP-ribose) polymerase inhibitor. J Biol Chem. 2011;286(14):12157–12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nouspikel T. DNA repair in mammalian cells: Nucleotide excision repair: Variations on versatility. Cell Mol Life Sci. 2009;66(6):994–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5(10):a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vermeulen W, Fousteri M.. Mammalian transcription-coupled excision repair. Cold Spring Harb Perspect Biol. 2013;5(8):a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Reid-Bayliss KS, Arron ST, Loeb LA, et al. Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc Natl Acad Sci U S A. 2016;113(36):10151–10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Usanova S, Piee-Staffa A, Sied U, et al. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer. 2010;9:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kirschner K, Melton DW.. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res. 2010;30(9):3223–3232. [PubMed] [Google Scholar]
- 100. Ahmad A, Robinson AR, Duensing A, et al. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Moll Cell Biol. 2008;28(16):5082–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jordheim LP, Barakat KH, Heinrich-Balard L, et al. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol Pharmacol. 2013;84(1):12–24. [DOI] [PubMed] [Google Scholar]
- 102. Postel-Vinay S, Bajrami I, Friboulet L, et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene. 2013;32(47):5377–5387. [DOI] [PubMed] [Google Scholar]
- 103. Cheng H, Zhang Z, Borczuk A, et al. PARP inhibition selectively increases sensitivity to cisplatin in ERCC1-low non-small cell lung cancer cells. Carcinogenesis. 2013;34(4):739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. McNeil EM, Astell KR, Ritchie AM, et al. Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance. DNA Repair. 2015;31:19–28. [DOI] [PubMed] [Google Scholar]
- 105. Arora S, Heyza J, Zhang H, et al. Identification of small molecule inhibitors of ERCC1-XPF that inhibit DNA repair and potentiate cisplatin efficacy in cancer cells. Oncotarget .2016; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Neher TM, Bodenmiller D, Fitch RW, et al. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol Cancer Ther. 2011;10(10):1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Neher TM, Shuck SC, Liu JY, et al. Identification of novel small molecule inhibitors of the XPA protein using in silico based screening. ACS Chem Biol. 2010;5(10):953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mishra AK, Dormi SS, Turchi AM, et al. Chemical inhibitor targeting the replication protein A-DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem Pharmacol. 2015;93(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lord CJ, McDonald S, Swift S, et al. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair. 2008;7(12):2010–2019. [DOI] [PubMed] [Google Scholar]
- 110. Zhang YW, Regairaz M, Seiler JA, et al. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011;39(9):3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mohni KN, Kavanaugh GM, Cortez D.. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014;74(10):2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pauty J, Rodrigue A, Couturier A, et al. Exploring the roles of PALB2 at the crossroads of DNA repair and cancer. Biochem J. 2014;460(3):331–342. [DOI] [PubMed] [Google Scholar]
- 113. Filippini SE, Vega A.. Breast cancer genes: Beyond BRCA1 and BRCA2. Front Biosci. 2013;18:1358–1372. [DOI] [PubMed] [Google Scholar]
- 114. Irminger-Finger I, Ratajska M, Pilyugin M.. New concepts on BARD1: Regulator of BRCA pathways and beyond. Int J BioChem Cell Biol. 2016;72:1–17. [DOI] [PubMed] [Google Scholar]
- 115. Yu H, Pak H, Hammond-Martel I, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A. 2014;111(1):285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sopik V, Akbari MR, Narod SA.. Genetic testing for RAD51C mutations: In the clinic and community. Clin Genet. 2015;88(4):303–312. [DOI] [PubMed] [Google Scholar]
- 117. Economopoulou P, Dimitriadis G, Psyrri A.. Beyond BRCA: New hereditary breast cancer susceptibility genes. Cancer Treat Rev. 2015;41(1):1–8. [DOI] [PubMed] [Google Scholar]
- 118. Jenner ZB, Sood AK, Coleman RL.. Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol. 2016;12(12):1439–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016; in press. [DOI] [PubMed] [Google Scholar]
- 121. Telli ML, Timms KM, Reid J, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Alagpulinsa DA, Ayyadevara S, Yaccoby S, et al. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol Cancer Ther. 2016;15(2):241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mohni KN, Thompson PS, Luzwick JW, et al. A synthetic lethal screen identifies DNA repair pathways that sensitize cancer cells to combined ATR inhibition and cisplatin treatments. PLoS One. 2015;10(5):e0125482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med. 2017;9(375):eaal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Majuelos-Melguizo J, Rodriguez MI, Lopez-Jimenez L, et al. PARP targeting counteracts gliomagenesis through induction of mitotic catastrophe and aggravation of deficiency in homologous recombination in PTEN-mutant glioma. Oncotarget. 2015;6(7):4790–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Leslie NR, Longy M.. Inherited PTEN mutations and the prediction of phenotype. Semin Cell Dev Biol. 2016;52:30–38. [DOI] [PubMed] [Google Scholar]
- 128. Wiltshire TD, Lovejoy CA, Wang T, et al. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J Biol Chem. 2010;285(19):14565–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74(1):287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. McLellan JL, O'Neil NJ, Barrett I, et al. Synthetic lethality of cohesins with PARPs and replication fork mediators. PLoS Genet. 2012;8(3):e1002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Esposito MT, Zhao L, Fung TK, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21(12):1481–1490. [DOI] [PubMed] [Google Scholar]
- 132. Lopez-Martinez D, Liang CC, Cohn MA.. Cellular response to DNA interstrand crosslinks: The Fanconi anemia pathway. Cell Mol Life Sci. 2016;73(16):3097–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Longerich S, Li J, Xiong Y, et al. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124(18):2812–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chen J, Dexheimer TS, Ai Y, et al. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18(11):1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Chirnomas D, Taniguchi T, de la Vega M, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5(4):952–961. [DOI] [PubMed] [Google Scholar]
- 136. Jo U, Kim H.. Exploiting the Fanconi anemia pathway for targeted anti-cancer therapy. Mol Cell. 2015;38(8):669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Jacquemont C, Taniguchi T.. Proteasome function is required for DNA damage response and Fanconi anemia pathway activation. Cancer Res. 2007;67(15):7395–7405. [DOI] [PubMed] [Google Scholar]
- 138. Kee Y, Huang M, Chang S, et al. Inhibition of the Nedd8 system sensitizes cells to DNA interstrand cross-linking agents. Mol Cancer Res. 2012;10(3):369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Liang Q, Dexheimer TS, Zhang P, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10(4):298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hanamshet K, Mazina OM, Mazin AV.. Reappearance from obscurity: Mammalian Rad52 in homologous recombination. Genes. 2016;7(9):E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lok BH, Powell SN.. Molecular pathways: Understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clin Cancer Res. 2012;18(23):6400–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lok BH, Carley AC, Tchang B, et al. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32(30):3552–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chandramouly G, McDevitt S, Sullivan K, et al. Small-molecule disruption of RAD52 rings as a mechanism for precision medicine in BRCA-deficient cancers. Chem Biol. 2015;22(11):1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hengel SR, Malacaria E, Folly da Silva Constantino L, et al. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Sullivan K, Cramer-Morales K, McElroy DL, et al. Identification of a small molecule inhibitor of RAD52 by structure-based selection. PLoS One. 2016;11(1):e0147230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhou Y, Paull TT.. DNA-dependent protein kinase regulates DNA end resection in concert with Mre11-Rad50-Nbs1 (MRN) and ataxia telangiectasia-mutated (ATM). J Biol Chem. 2013;288(52):37112–37125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Symington LS. Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol. 2016;51(3):195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Deriano L, Roth DB.. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–455. [DOI] [PubMed] [Google Scholar]
- 149. Downs JA, Jackson SP.. A means to a DNA end: The many roles of Ku. Nat Rev Mol Cell Biol. 2004;5(5):367–378. [DOI] [PubMed] [Google Scholar]
- 150. Schipler A, Iliakis G.. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013;41(16):7589–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wyatt DW, Feng W, Conlin MP, et al. Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks. Mol Cell. 2016;63(4):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wood RD, Doublie S.. DNA polymerase theta (POLQ), double-strand break repair, and cancer. DNA Repair. 2016;44:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Nussenzweig A, Nussenzweig MC.. A backup DNA repair pathway moves to the forefront. Cell. 2007;131(2):223–225. [DOI] [PubMed] [Google Scholar]
- 154. Corneo B, Wendland RL, Deriano L, et al. Rag mutations reveal robust alternative end joining. Nature. 2007;449(7161):483–486. [DOI] [PubMed] [Google Scholar]
- 155. Simsek D, Jasin M.. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17(4):410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Weinstock DM, Brunet E, Jasin M.. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat Cell Biol. 2007;9(8):978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Jeggo P, Lavin MF.. Cellular radiosensitivity: How much better do we understand it? Int J Radiat Biol. 2009;85(12):1061–1081. [DOI] [PubMed] [Google Scholar]
- 158. Mladenov E, Magin S, Soni A, et al. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol. 2013;3:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Allen CP, Borak TB, Tsujii H, et al. Heavy charged particle radiobiology: Using enhanced biological effectiveness and improved beam focusing to advance cancer therapy. Mutat Res. 2011;711:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Okayasu R, Okada M, Okabe A, et al. Repair of DNA damage induced by accelerated heavy ions in mammalian cells proficient and deficient in the non-homologous end-joining pathway. Radiat Res. 2006;165(1):59–67. [DOI] [PubMed] [Google Scholar]
- 161. Okayasu R. Repair of DNA damage induced by accelerated heavy ions—a mini review. Int J Cancer. 2012;130(5):991–1000. [DOI] [PubMed] [Google Scholar]
- 162. Wang H, Liu S, Zhang P, et al. S-phase cells are more sensitive to high-linear energy transfer radiation. Int J Radiat Oncol Biol Phys. 2009;74(4):1236–1241. [DOI] [PubMed] [Google Scholar]
- 163. Segawa T, Fujii Y, Tanaka A, et al. Radiosensitization of human lung cancer cells by the novel purine-scaffold Hsp90 inhibitor, PU-H71. Int J Mol Med. 2014;33(3):559–564. [DOI] [PubMed] [Google Scholar]
- 164. Hirakawa H, Fujisawa H, Masaoka A, et al. The combination of Hsp90 inhibitor 17AAG and heavy-ion irradiation provides effective tumor control in human lung cancer cells. Cancer Med. 2015;4(3):426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lee Y, Li HK, Masaoka A, et al. The purine scaffold Hsp90 inhibitor PU-H71 sensitizes cancer cells to heavy ion radiation by inhibiting DNA repair by homologous recombination and non-homologous end joining. Radiother Oncol. 2016;121(1):162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lee Y, Sunada S, Hirakawa H, et al. TAS-116, a novel Hsp90 inhibitor, selectively enhances radiosensitivity of human cancer cells to X-rays and carbon ion radiation. Mol Cancer Ther. 2017;16(1):16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Adachi N, So S, Koyama H.. Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J Biol Chem. 2004;279(36):37343–37348. [DOI] [PubMed] [Google Scholar]
- 168. Chatterjee P, Choudhary GS, Alswillah T, et al. The TMPRSS2-ERG gene fusion blocks XRCC4-mediated nonhomologous end-joining repair and radiosensitizes prostate cancer cells to PARP inhibition. Mol Cancer Ther. 2015;14(8):1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Hähnel PS, Enders B, Sasca D, et al. Targeting components of the alternative NHEJ pathway sensitizes KRAS mutant leukemic cells to chemotherapy. Blood. 2014;23(15):2355–2366. [DOI] [PubMed] [Google Scholar]
- 170. Konsoula Z, Velena A, Lee R, et al. Histone deacetylase inhibitor: Antineoplastic agent and radiation modulator. Adv Exp Med Biol. 2011;720:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Li Z, Zhu WG.. Targeting histone deacetylases for cancer therapy: From molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10(7):757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Robert C, Nagaria PK, Pawar N, et al. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. 2016;45:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lemee F, Bergoglio V, Fernandez-Vidal A, et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc Natl Acad Sci U S A. 2010;107(30):13390–13395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Allera-Moreau C, Rouquette I, Lepage B, et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis. 2012;1:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ceccaldi R, Liu JC, Amunugama R, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518(7538):258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Mateos-Gomez PA, Gong F, Nair N, et al. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015;518(7538):254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Killock D. Targeted therapies: DNA polymerase theta-a new target for synthetic lethality? Nat Rev Clin Oncol. 2015;12(3):125. [DOI] [PubMed] [Google Scholar]
- 178. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. [DOI] [PubMed] [Google Scholar]
- 179. Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Matulonis UA, Penson RT, Domchek SM, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann Oncol. 2016;27(6):1013–1019. [DOI] [PubMed] [Google Scholar]
- 181. Kim G, Ison G, McKee AE, et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res. 2015;21(19):4257–4261. [DOI] [PubMed] [Google Scholar]
- 182. Liu FW, Tewari KS.. New targeted agents in gynecologic cancers: Synthetic lethality, homologous recombination deficiency, and PARP inhibitors. Curr Treat Options Oncol. 2016;17(3):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Bixel K, Hays JL.. Olaparib in the management of ovarian cancer. Pharmgenomics Pers Med. 2015;8:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Kummar S, Wade JL, Oza AM, et al. Randomized phase II trial of cyclophosphamide and the oral poly (ADP-ribose) polymerase inhibitor veliparib in patients with recurrent, advanced triple-negative breast cancer. Invest New Drugs. 2016;34(3):355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Coleman RL, Sill MW, Bell-McGuinn K, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation—An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015;137(3):386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Tahara M, Inoue T, Sato F, et al. The use of olaparib (AZD2281) potentiates SN-38 cytotoxicity in colon cancer cells by indirect inhibition of Rad51-mediated repair of DNA double-strand breaks. Mol Cancer Ther. 2014;13(5):1170–1180. [DOI] [PubMed] [Google Scholar]
- 187. Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Invest New Drugs. 2012;30(4):1493–1500. [DOI] [PubMed] [Google Scholar]
- 188. Kunos C, Deng W, Dawson D, et al. A phase I-II evaluation of veliparib (NSC #737664), topotecan, and filgrastim or pegfilgrastim in the treatment of persistent or recurrent carcinoma of the uterine cervix: An NRG Oncology/Gynecologic Oncology Group study. Int J Gynecol Cancer. 2015;25(3):484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Pratz KW, Rudek MA, Gojo I, et al. A Phase I study of topotecan, carboplatin and the PARP inhibitor veliparib in acute leukemias, aggressive myeloproliferative neoplasms and chronic myelomonocytic leukemia. Clin Cancer Res. 2016; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Hills SA, Diffley JF.. DNA replication and oncogene-induced replicative stress. Curr Biol. 2014;24(10):R435–R444. [DOI] [PubMed] [Google Scholar]
- 191. Schlacher K, Christ N, Siaud N, et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]