

Abstract
Neuroendocrine tumor (NET) occurring in association with other endocrine syndromes forms a distinct entity. The aim was to assess the therapy response profile of the routine peptide receptor radionuclide therapy (PRRT) in this relatively uncommon but clinically challenging subgroup of patients. A retrospective analysis was undertaken from the case records from those who were treated with 177Lu-DOTATATE for metastatic NET. In addition to assessing the therapeutic efficacy, emphasis was also given to study lesional sites and scan pattern. A total of 5 cases were found: In this series of five cases, four belonged to multiple endocrine neoplasia type 1 (MEN1) syndrome; in these four MEN1 syndrome patients, the primary site of NET was thymic region (n = 1), duodenum (n = 1), and pancreas (n = 2). The fifth case was of Cushing's syndrome with the primary site of NET in the thymus. A good symptomatic response was observed in all MEN1 syndrome cases (100%) and progression of symptoms in the patient with Cushing's syndrome. The biochemical response (assessed by measurement of tumor marker serum chromogranin A) demonstrated very good partial response (defined by more than 75% reduction of tumor marker) in 2 MEN1 cases and Cushing's syndrome, good partial response (25–75% reduction of tumor marker) in the remaining 2 MEN1 cases. Scan wise (assessed by technetium [99mTc]-hydrazinonicotinamide [HYNIC]-tektrotyd [TOC]/68Ga-DOTA-NOC/TATE positron emission tomography-computed tomography [PET-CT] and fluorodeoxyglucose [FDG] PET-CT) partial response was observed in 3 MEN1 cases, stable disease was noted in one MEN1 case and disease progression was noted in the patient with Cushing's syndrome. The change in FDG uptake was found to be an important sensitive scan parameter in the treatment evaluation of NETs compared to somatostatin receptor-based imaging in the cases with low MiB1 index. In our series, good palliative response to 177Lu-DOTA-octreotate (DOTATATE) PRRT was observed in most NET patients associated with MEN1 syndrome without any major hematological or renal toxicity.
Keywords: 177Lu-DOTATATE, multiple endocrine neoplasia, neuroendocrine tumor, peptide receptor radionuclide therapy
Introduction
Multiple endocrine neoplasia type 1 (MEN1) syndrome is a very rare, inherited disorder characterized primarily by tumors of the anterior pituitary (15–90% of cases), parathyroid glands (95% of cases), and pancreatic tumors (30–80% of cases).[1] This syndrome also includes the other rare neoplasms such as thymic, bronchial, and gastric carcinoids, meningiomas, adrenocortical and thyroid tumors, facial angiofibromas and collagenomas, visceral and cutaneous lipomas. Approximate prevalence of MEN1 syndrome is 1 in 35000 (1 in 20,000–1 in 40,000).[2] It is a genetic disorder and runs in families with autosomal dominant trait. It is also known by name “Wermer syndrome” and occurs due to mutation of gene MEN1, which encodes Menin, a putative tumor suppressor.
Involvement of parathyroid may manifest with excess of serum calcium levels and rarely with renal calculi. Pancreas involvement may present in the form of islet cell tumor, which leads to excess production of gastrin, which in turn excess production of acid in stomach and leading to various symptoms such as pain abdomen, vomiting, diarrhea, gastrointestinal bleed, gastric and peptic ulcers, and perforations. Involvement of pituitary gland may manifest with tumors which produce excess of prolactin and growth hormones and manifests with various symptoms such as menstrual abnormalities, breast secretions, decreased sexual desire, erectile dysfunction, acromegaly, and gigantism. Diagnosis is made by proper family history, ultrasonography of the involved anatomical region, computed tomography (CT) scan, genetic tests, molecular imaging, and tumor markers/hormone levels of involved glands.[2] There is no definitive treatment available for MEN1 syndrome per se treatment of MEN1 syndrome is done by surgical excision of the involved endocrine gland followed by hormone replacement. Gastroenteropancreatic neuroendocrine tumors (GEP-NET) are also manifested in association with MEN1 syndrome.
Cushing's syndrome is described as a group of signs and symptoms due to excessive cortisol levels and also known as hypercortisolism.[3] It can be due to exogenous or endogenous causes [Figure 1]. In majority of the cases, the cause is exogenous such as employment of external medications such as glucocorticoids such as hydrocortisone, prednisolone, and dexamethasone. Endogenous cause includes pituitary tumor that secrets excess amount of adrenocorticotropic hormone (ACTH), also known as Cushing's disease as observed in our case. Cushing's syndrome is manifested by group of symptoms such as moon facies, red cheeks, buffalo hump, truncal obesity, red striation over the abdomen, high blood pressure, poor wound healing, thin arms, headaches, rapid weight gain, and diabetes mellitus. Diagnosis is usually made by dexamethasone suppression test or a 24-h urinary measurement for cortisol, 24 h cortisol levels in saliva, CT scan, magnetic resonance imaging scan, and adrenal scintigraphy. Treatment is undertaken by gradual tapering of exogenous medication in most of the cases. Surgery is usually done for pituitary and adrenal adenomas and other incidentalomas. Medical management is tried with cortisol synthesis inhibitors (such as ketoconazole or metyrapone) in patients who are unwilling for surgery. Peptide receptor radionuclide therapy (PRRT) is relatively new targeted therapeutic approach for patients, who harbor metastatic/inoperable NETs and can be given to those who express somatostatin receptor (SSTR) receptor at the tumor sites on SSTR-based imaging such as 68Ga-DOTA-NOC/TATE positron emission tomography (PET)-CT.[4,5,6] We assessed therapy response to PRRT in this relatively rare group of patients harboring metastatic NETs in the setting of MEN1 syndrome and Cushing's syndrome and present the profile and outcome of this treatment modality in this particular subgroup.
Figure 1.
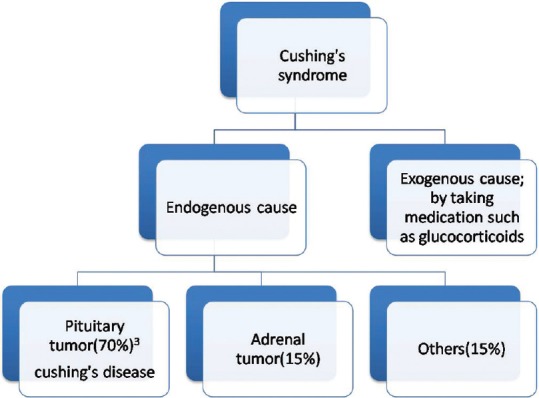
Etiologies of Cushing's syndrome
Materials and Methods
We performed a retrospective analysis of histopathologically proven NETs associated with MEN1 syndrome and Cushing's syndrome from a population of patients who had undergone PRRT with 177Lu-DOTATATE. The patients included in this study demonstrated histopathologically proven NET with raised serum chromogranin A and increased tracer uptake noted on initial diagnostic study (technetium [99mTc]-hydrazinonicotinamide [HYNIC]-tektrotyd [TOC]/68 Ga-DOTA-NOC/TATE PET-CT and fluorodeoxyglucose [FDG] PET-CT).
These patients were analyzed for response evaluation under three broad headings:
Symptomatically
Biochemically (tumor marker with serum chromogranin A) and
Scan (99mTc HYNIC-TOC or 68Ga-DOTA-NOC/TATE PET-CT and fluorodeoxyglucose [FDG] PET-CT scan) wise.
Hematological and renal toxicity were evaluated using National Cancer Institute-Common Terminology Criteria for Adverse Events version 4.0 score.
Observations and Results
On retrospective analysis of around 350 patients, we found five rare syndromic NET cases which included four MEN1 syndromes and one Cushing's syndrome. Most of the cases had a family history and was initially asymptomatic with incidental finding of tumor on routine evaluation of nonspecific symptoms. In our series, the age ranged from 34 to 52 years (for MEN1 cases age ranges from 41 to 52) and had male predominance (male to female ratio of 3:2). The follow-up duration ranged from 7 to 26 months. The lesion site and its histopathological characterization, the details of PRRT administered (with its toxicity profile), the treatment response profile, and the dual tracer imaging features and its correlation with histopathology and prognosis are tabulated in Tables 1–4, respectively [Figures 2–8].
Table 1.
Site and histopathological characterization of the lesion
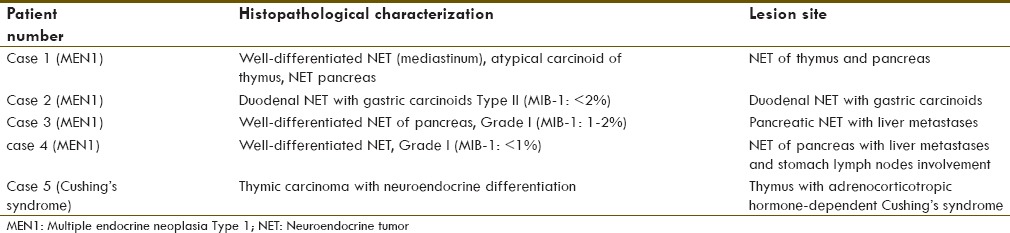
Table 4.
Dual tracer imaging features: Relevance to prognosis
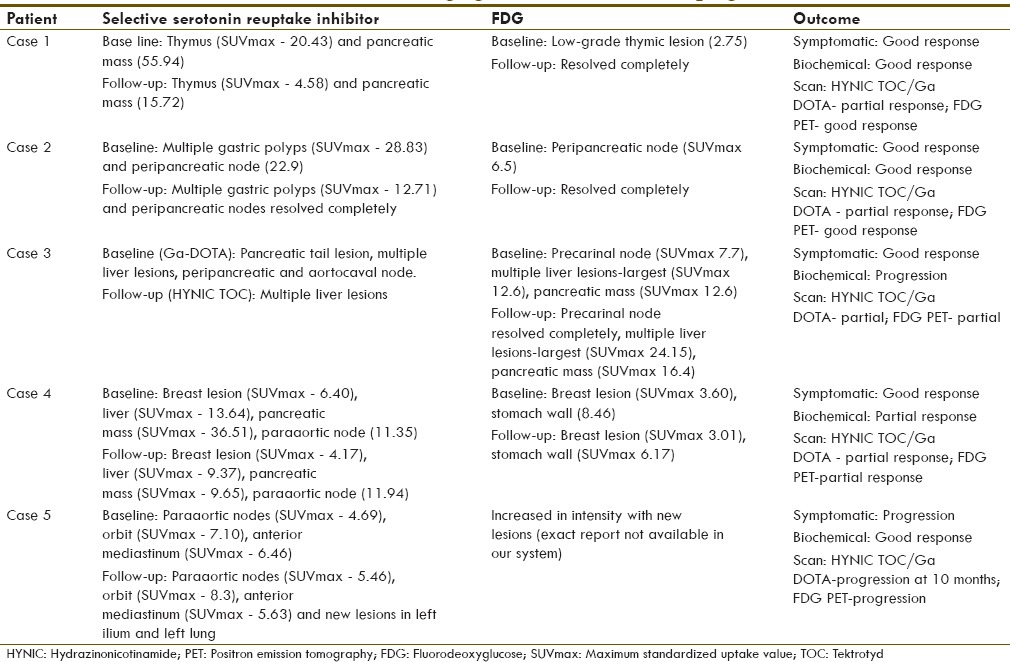
Figure 2.
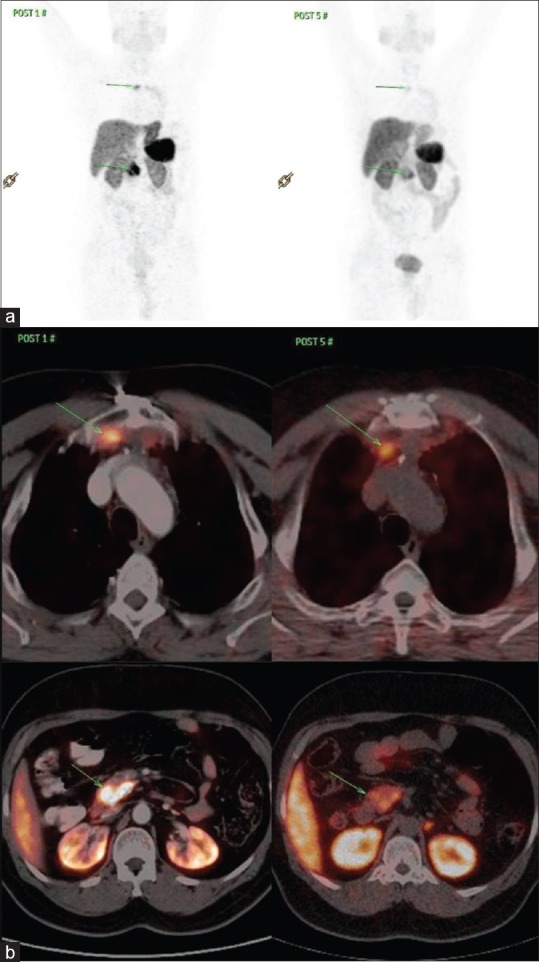
(a) Comparison of 68Ga-DOTATATE positron emission tomography-computed tomography of case 1. (b) Transaxial 68Ga DOTATATE positron emission tomography-computed tomography fused images of case 1
Figure 8.
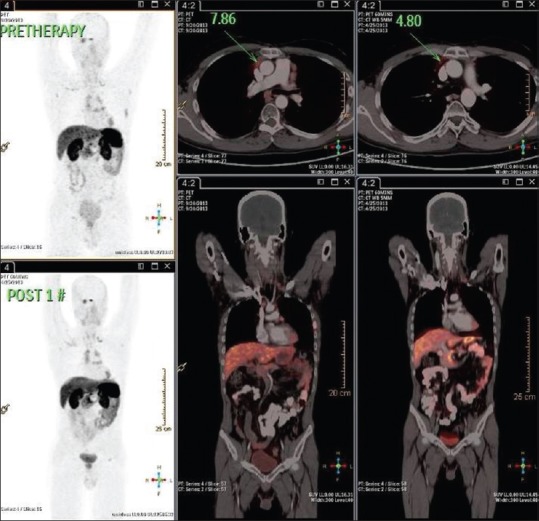
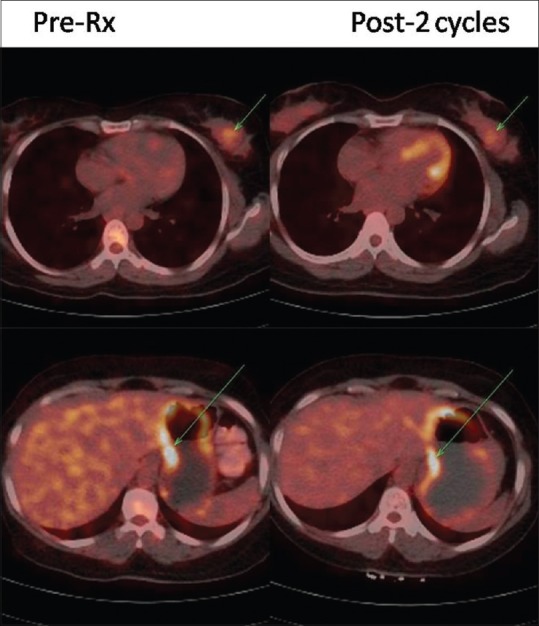
Documentation of initial stable disease on 68GaDOTATATE at post 1 cycle peptide receptor radionuclide therapy at 3 months with some metabolic scan response in case 5
Figure 3.
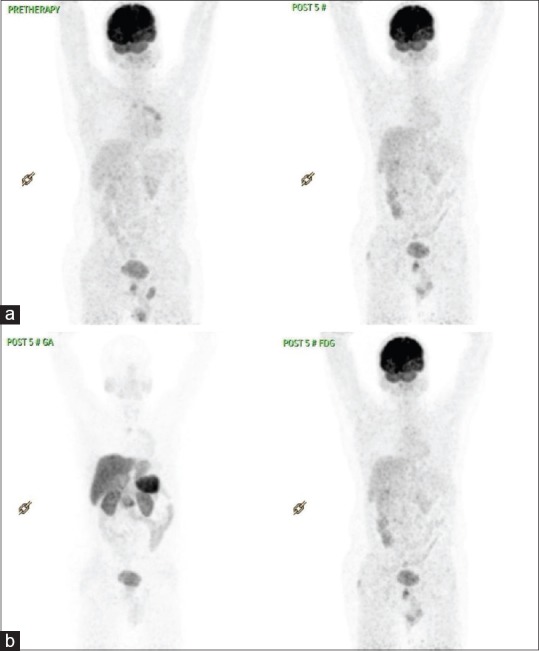
(a) Fluorodeoxyglucose-positron emission tomography comparison – case 1. (b) Ga-DOTATATE and fluorodeoxyglucose post-5# of peptide receptor radionuclide therapy in case 1
Figure 4.
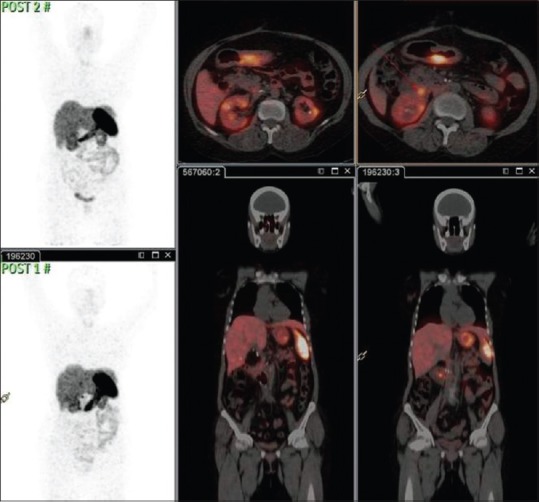
Comparison of 68Ga-DOTATE positron emission tomography-computed tomography in case 2
Figure 5.
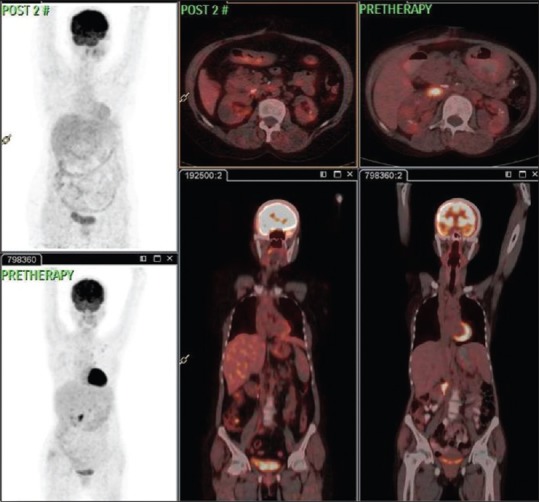
Comparison of fluorodeoxyglucose positron emission tomography-computed tomography in case 2
Figure 6.
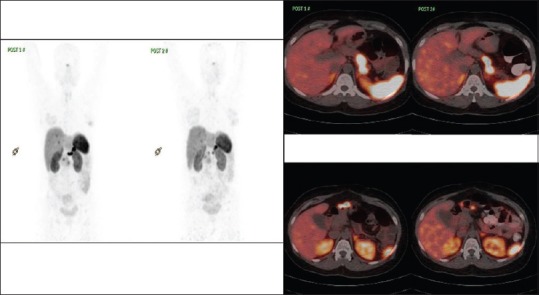
Comparison of 68Ga-DOTATE positron emission tomography-computed tomography in case 4
Figure 7.
Comparison of 18F-fluorodeoxyglucose positron emission tomography-computed tomography in case 4
Primary lesion site and its characteristics
In most of the syndromic NET cases, the primary lesion was from GEP region followed by thymic region. Most of the MEN1 cases were found to have grade 1 well-differentiated NETs, which means MIB-1 index < 2%. In the patient with Cushing's syndrome, the primary lesion involved thymus with histopathology as thymic carcinoma with neuroendocrine differentiation. These characteristics are tabulated in Table 1.
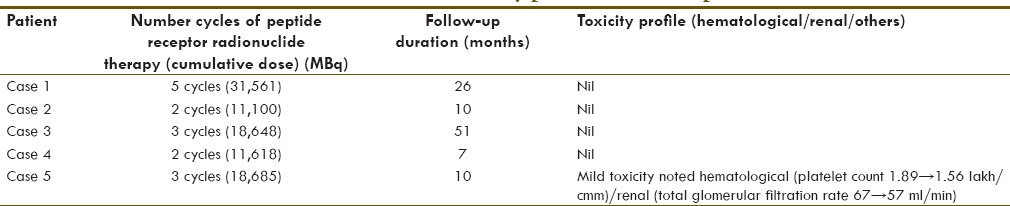
Cumulative dose and toxicity
In most MEN1 cases, there was no specific toxicity noted to PRRT and overall well tolerated with the cumulative activity administered till date without any significant side effects. In Cushing's syndrome case, there was a mild hematological and renal toxicity noted; and that toxicity could have been accentuated with other modes of treatment such as chemotherapy and radiotherapy which were tried earlier in the case. The cumulative dose administered and toxicity profile in each case studied in this series is tabulated in Table 2.
Table 2.
Number of cycles of peptide receptor radionuclide therapy with 177Lu-DOTA-octreotate therapy administered and toxicity profile on follow-up
Treatment response assessment
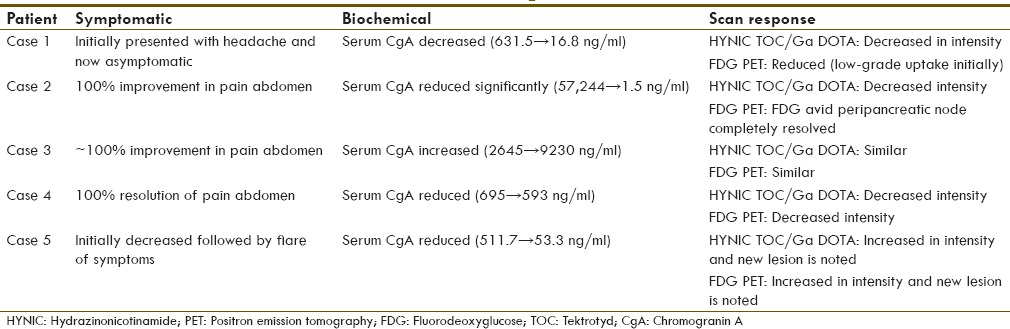
Response assessment was done by three parameters (1) symptomatically (2) Biochemically (tumor marker: Serum chromogranin A) and (3) Scan assessment (99mTc HYNIC-TOC or 68Ga-DOTA-NOC/TATE PET-CT and FDG PET-CT scan).
Scan-wise, in most of the MEN1 cases, there was partial response observed (75%; three out of four MEN2 cases) and one case showed stable disease (25%). There was a disease progression noted in Cushing's syndrome case with initial phase of partial response followed by new lesions observed during follow-up period.
The details of response assessment are tabulated in Table 3. Overall, there was a partial response noted in 60% of the syndromic NET cases and stable disease observed in 20% and disease progression in 20%.
Table 3.
Treatment response assessment
Dual tracer imaging features with somatostatin receptor-based imaging and fluorodeoxyglucose-positron emission tomography-computed tomography
In all MEN1 syndrome cases, there was low to moderate grade FDG uptake when compared to significant HYNIC TOC/DOTATATE uptake at the corresponding lesions; the outcome with respect to scan features in each case has been detailed in Table 4.
Discussion
NETs occurring in the context of hereditary neoplastic syndromes are relatively rare and need a high index of suspicion to diagnose them as early as possible. A slow growing tumor promptly detected and treated in early stages can show a fair clinical outcome. When diagnosed in the late stages, the treatment is primarily palliative in intent. In our series, first four cases were of MEN1 syndrome presenting with different organ involvement. Pain abdomen was a common symptom present in all four cases. The fifth case was an ACTH-dependent Cushing's syndrome due to thymic carcinoma with neuroendocrine differentiation involving multiple organs.
The first case presented with pituitary adenoma and 10 years later was diagnosed to have parathyroid tumor. Subsequently, mediastinal mass pathology was suggestive of neuroendocrine carcinoma with increased SSTR expression on 68Ga-DOTATATE scan. He responded well to 5 cycles of PRRT, showing symptomatic, biochemical, and 68Ga-DOTA/HYNIC TOC and 18F-FDG PET-CT scan response.
The second case was a diagnosed case of MEN1 syndrome found to have SSTR expression on 68Ga-DOTATATE PET-CT scan in thickened proximal two-third of stomach, the first part of duodenum, peripancreatic lymph node. The patient was treated with 2 cycles of 177Lu-DOTATATE therapy and good symptomatic, biochemical, and scan response without any toxicity.
The third case presented with pain abdomen and had a previous history of pituitary adenoma. Subsequently, he was diagnosed as pancreatic NET with liver and multiple abdominal metastases, for which patient was treated with 3 cycles of 177Lu-DOTATATE. The patient responded well symptomatically and had stable disease scan wise, but there was increase in tumor marker which was likely due to intake of proton pump inhibitor during serum chromogranin An estimation. Till date, the patient is asymptomatic with stable disease.
The fourth case also presented with chronic recurrent pain abdomen, initially diagnosed as Zollinger–Ellison syndrome and 6 years later diagnosed as pancreatic NET and medically managed. Now, after 10 years of initial presentation, she received 2 cycles 177Lu-DOTATATE therapy, following which patient shown significant symptomatic, biochemical, and scan response.
The 5th case was a known case of ACTH-dependent Cushing's syndrome, postsurgery, postradiotherapy, and chemotherapy with progressive disease received 3 cycles of 177Lu-DOTATATE therapy. Initially, the patient responded clinically with reduced levels of tumor marker but later, symptoms reappeared and scan showing progressive disease with mild hematological/renal toxicity.
Dual tracer imaging with 68Ga-DOTATATE PET-CT and 18F-FDG PET-CT have a significant role in treatment selection and disease prognosis.[4] Well-differentiated NET with somatostatin expression in the primary lesion as well as metastatic lesions greater than physiological hepatic tracer uptake is a prerequisite for PRRT.[5,6] FDG uptake in primary and metastatic lesions has been equated with metabolic aggressiveness of the tumor and which is more observed in poorly differentiated NET.[7] An inverse relation is also reported with FDG and DOTA with aggressiveness/differentiation of tumor (i.e., well-differentiated tumor showing somatostatin expression with nil/low FDG expression, whereas poorly differentiated tumor shows poor somatostatin expression with high FDG expression).[4] In the present case series, this feature was clearly observed as the cases were of well-differentiated NET with low FDG uptake. On follow-up studies, the FDG uptake reduced earlier than SSTR-based imaging, suggesting the change in FDG uptake to be an important parameter in the treatment evaluation of NETs.
Conclusion
The present retrospective evaluation in our case series showed an overall good response either decrease in metabolic aggressiveness of lesions or stabilization of disease process to PRRT in NETs in association with MEN1 syndrome. There was also good symptomatic and biochemical improvement without any hematological/renal toxicity. Thus, prolongation of symptom-free survival can be expected with 177Lu-DOTATATE therapy in these patients. The fifth case which was a resistant ACTH-dependent tumor presented in the late stage and PRRT could not halt the disease process. Thus, we can conclude that 177Lu-DOTATATE therapy for NETs in association with MEN1 syndrome has a significant role in disease stabilization and good symptomatic response with better health-related quality of life.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Marini F, Falchetti A, Luzi E, Tonelli F, Maria Luisa B. Multiple Endocrine Neoplasia Type 1 (MEN1) Syndrome. In: Riegert-Johnson DL, Boardman LA, Hefferon T, Roberts M, editors. Cancer Syndromes [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2009.-2008. Jul 18, [Last updated on 2008 Aug 09]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21249756 . [PubMed] [Google Scholar]
- 2.Jiang L, Schipper ML, Li P, Cheng Z. 123I labeled metaiodobenzylguanidine for diagnosis of neuroendocrine tumors. Rep Med Imaging. 2009;2:79–89. [Google Scholar]
- 3.Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing's syndrome. Lancet. 2006;367:1605–17. doi: 10.1016/S0140-6736(06)68699-6. [DOI] [PubMed] [Google Scholar]
- 4.Basu S, Sirohi B, Shrikhande SV. Dual tracer imaging approach in assessing tumor biology and heterogeneity in neuroendocrine tumors: Its correlation with tumor proliferation index and possible multifaceted implications for personalized clinical management decisions, with focus on PRRT. Eur J Nucl Med Mol Imaging. 2014;41:1492–6. doi: 10.1007/s00259-014-2805-8. [DOI] [PubMed] [Google Scholar]
- 5.Krenning EP, de Jong M, Kooij PP, Breeman WA, Bakker WH, de Herder WW, et al. Radiolabelled somatostatin analogue(s) for peptide receptor scintigraphy and radionuclide therapy. Ann Oncol. 1999;10(Suppl 2):S23–9. doi: 10.1093/annonc/10.suppl_2.s23. [DOI] [PubMed] [Google Scholar]
- 6.Kwekkeboom DJ, Krenning EP, Lebtahi R, Komminoth P, Kos-Kudla B, de Herder WW, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Peptide receptor radionuclide therapy with radiolabeled somatostatin analogs. Neuroendocrinology. 2009;90:220–6. doi: 10.1159/000225951. [DOI] [PubMed] [Google Scholar]
- 7.Kwee TC, Basu S, Saboury B, Ambrosini V, Torigian DA, Alavi A. A new dimension of FDG-PET interpretation: Assessment of tumor biology. Eur J Nucl Med Mol Imaging. 2011;38:1158–70. doi: 10.1007/s00259-010-1713-9. [DOI] [PubMed] [Google Scholar]