

Abstract
Paralysis following spinal cord injury (SCI) is due to failure of axonal regeneration. It is believed that the capacities of neurons to regrow their axons are due partly to their intrinsic characteristics, which in turn are greatly influenced by several types of inhibitory molecules that are present, or even increased in the extracellular environment of the injured spinal cord. Many of these inhibitory molecules have been studied extensively in recent years. It has been suggested that the small GTPase RhoA is an intracellular convergence point for signaling by these extracellular inhibitory molecules, but due to the complexity of the central nervous system (CNS) in mammals, and the limitation of pharmacological tools, the specific roles of RhoA are unclear. By exploiting the anatomical and technical advantages of the lamprey CNS, we recently demonstrated that RhoA knockdown promotes true axon regeneration through the lesion site after SCI. In addition, we found that RhoA knockdown protects the large, identified reticulospinal neurons from apoptosis after their axons were axotomized in spinal cord. Therefore, manipulation of the RhoA signaling pathway may be an important approach in the development of treatments that are both neuroprotective and axon regeneration-promoting, to enhance functional recovery after SCI.
Keywords: RhoA, spinal cord injury, neuronal survival, apoptosis, axon regeneration, morpholino, C3 transferase
Introduction
Recovery of function after spinal cord injury (SCI) has been limited by failure of axons in the central nervous system (CNS) to regenerate and restore neurological connectivity. This has been ascribed to both inhibitory environmental molecular cues and the intrinsic characteristics of CNS neurons. Ras homolog gene family, member A (RhoA) is a small GTPase that is activated and/or upregulated locally at the site of SCI, and is thought to be an important participant in limiting recovery. This increase in RhoA expression can last for 4 weeks (Dubreuil et al., 2003; Conrad et al., 2005; Hu et al., 2017). Evidence suggests that RhoA is a convergence point for intraneuronal signaling of several extracellular molecules implicated in restricting recovery after SCI, e.g., the chondroitin sulfate proteoglycans (CSPGs), and myelin-associated glycoprotein (MAG), oligodendrocyte myelin glycoprotein (OMgp), Nogo and their corresponding receptors. This makes RhoA a promising target to promote axon growth and improve functional recovery after SCI (Lehmann et al., 1999; Dergham et al., 2002). The means by which RhoA limits recovery are complex, and may involve at least two types of neural responses to SCI: 1) neuronal apoptosis; and 2) regenerative failure. How these two phenomena relate to each other is not clear, but it is not simply that dead neurons cannot regenerate. The results of experiments on SCI in vivo are difficult to interpret in mammals for three reasons: 1) It is difficult to distinguish true regeneration of injured axons from collateral sprouting of spared axons. 2) The cause of neuronal death near the lesion site is ambiguous. 3) The time courses of the two effects overlap.
We have used the sea lamprey as a model to address these ambiguities because its large, individually identified reticulospinal (RS) neurons show great heterogeneity in the ability of their axons to regenerate through the same spinal cord environment (Jacobs et al., 1997) (Figure 1), and to survive long term after axotomy (Shifman et al., 2008). We have used these features of the lamprey to analyze the factors underlying axotomy-induced retrograde neuronal death, and also the failure of axons to regenerate. Moreover, the neurons whose axons regenerate poorly at early time points post-SCI, eventually die at a later time (Shifman et al., 2008). This has suggested that the pathways for inhibition of axon regeneration and for survival of the parent neurons may converge, possibly through RhoA. RhoA and its downstream signaling molecules are highly conserved between lamprey and mammals, so the mechanisms revealed by these experiments are likely to be the same. It is the anatomical simplicity of the lamprey CNS and the presence of individually identified RS neurons that make the results of experiments easier to interpret in the lamprey compared to mammals.
Figure 1.
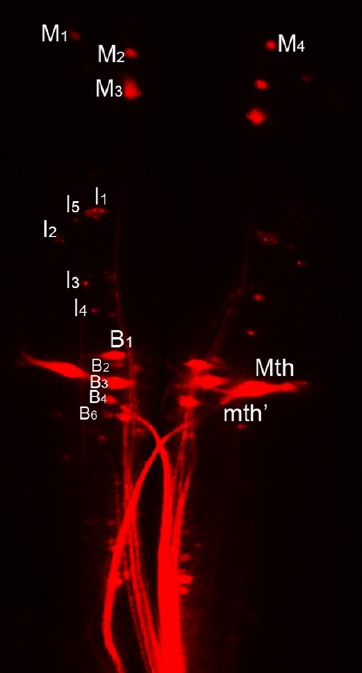
Anatomy of the regenerating spinal-projecting system in lamprey.
Individually identified reticulospinal (RS) neurons in the brain were labeled retrogradely with dextran tetramethylrhodamine (DTMR). These neurons are named according to the nomenclature of Rovainen (1967) as modified by Swain et al. (1993) and Jacobs et al. (1997). M: Mesencephalic; I: isthmic; B: bulbar; Mth: mauthner cell.
Pharmacological Inhibition of RhoA Enhances Axon Growth
C3 transferase (C3) from Clostridium botulinum can block Rho activation by ADP-ribosylation. C3 was first used to inhibit Rho activation in the mouse optic nerve crush (ONC) model (Lehmann et al., 1999). The investigators applied C3 either at the crush site or to the cell bodies of the retinal ganglion cells (RGCs), and found that it promoted regrowth of the optic nerve axons across the lesion site. Soon after, C3 was tested in SCI models, and was found to promote functional recovery and growth of corticospinal tract (CST) fibers beyond a partial SCI in mice (Dergham et al., 2002). These studies in mammalian models suggest that inhibiting Rho produced a complicated and only partial axon regeneration benefit. In these studies, C3 was applied directly to the lesion either by gelform plus Elvax tube (Lehmann et al., 1999) or a fibrin adhesive delivery system (Dergham et al., 2002). In these procedures, the C3 available to the target tissue was relatively limited, and the mechanical procedures themselves might have caused secondary damage. It also is possible that Rho activity affects collateral sprouting of spared CST or other axons, rather than true regeneration of injured ones (Tuszynski and Steward, 2012). Recently, a non-replicating herpes simplex virus (HSV) vector carrying the gene coding for C3 has been generated. Subcutaneous inoculation of the vector into the skin of the forepaw 1 week after a dorsal C5–T1 rhizotomy resulted in expression of C3 in dorsal root ganglion (DRG) neurons. This efficiently inhibited Rho activation, resulting in extensive axonal regeneration into the spinal cord and improved sensory-motor coordination of the forepaw (Zhou et al., 2012). However, the inhibition of RhoA by C3 is not specific. C3 also can activate microglia, which is the immune-competent element of the CNS and believed to protect the neurons against any potentially harmful change of the environment. Activated microglia can trigger the release of nitric oxide and several pro-inflammatory cytokines and chemokines (Hoffmann et al., 2008), thereby affecting axon growth. Thus, any regenerative benefits of C3 might not be limited to its effects on RhoA. To clarify the role of RhoA in axon regeneration after injury, there is a need to increase the specificity and efficiency with which RhoA is inhibited in vivo.
Increasing the Specificity of RhoA Manipulation
Several methods other than C3 application have been developed in the past few years to improve the specificity with which RhoA expression can be manipulated. Adeno-associated viral (AAV) vectors have been generated to downregulate RhoA specifically via shRNA (Koch et al., 2014). In the rat ONC model in vivo, specific RhoA knockdown significantly enhanced axon regeneration. Moreover, survival of RGC transduced with AAV expressing RhoA-shRNA was substantially increased at 2 weeks after ONC (Koch et al., 2014). The cationic, amphiphilic copolymers poly-(lactide-co-glycolide)-g-polyethylenimine (PgP) have been used to deliver siRNA targeting RhoA, and were evaluated in a SCI compression model (Gwak et al., 2017). PgP/siRhoA polyplexes were locally injected into the lesion site and significantly reduced RhoA mRNA and protein expression for up to 4 weeks post-injury (Gwak et al., 2017). Histological analysis showed that RhoA knockdown was accompanied by a reduction in local apoptosis, cavity size, and astrogliosis, and an increase in axonal regeneration within the lesion site at 4 weeks post-injury, suggesting that PgP is an efficient non-viral carrier for delivery of therapeutic siRhoA to the injured spinal cord, and may be a promising platform for the development of combinatorial therapies (Gwak et al., 2017). Specificity of RhoA knockdown has been increased in the lamprey SCI model through the use of morpholino antisense oligonucleotides (MOs) (Hu et al., 2017).
Morpholino Knockdown of RhoA in the Lamprey Increases True Axon Regeneration
Compared to mammals, the lamprey CNS is relatively simple, with special advantages for the study of axon regeneration and related mechanisms. In the lamprey, the main supraspinal inputs are the RS neurons, which are responsible for initiating and controlling locomotion, steering, and equilibrium. Of the approximately 2,000 RS neurons, 18 pairs are individually identified. Their axons extend the entire length of spinal cord, and therefore are always severed by a complete spinal cord transection (TX). The regeneration capabilities of these axons after TX have been documented extensively, and are very heterogeneous. Moreover, the severed axons and their neuronal cell bodies can be back-labeled with dextran Alexa Fluor 488 (DAF-488, Green) (Figure 2A). Uninjured axons do not pick up the dye. Thus if a second TX is made 5 mm caudal to the first TX, and dextran tetramethyrhodamine (DTMR, Red) is applied to the rostral cut end, axons that have regenerated ≥ 5 mm will be re-axotomized, and their neurons will be double-labeled with DAF-488 and DTMR (Yellow, Figure 2B). On the other hand, neurons and axons that did not regenerate, or regenerated less than 5 mm, will be labeled only with DAF-488 (Green, Figure 2B). With this design, because RhoA is highly conserved across species, we have evaluated the effects of MO-mediated specific RhoA knockdown on true axon regeneration, as opposed to collateral sprouting, and on neuronal apoptotic signaling after axotomy. In the MO experiments, we used either fluorescently labeled RhoA MO or control MO as the first dye to label the neurons and axons undergoing axotomy, and DTMR as the second dye to label the regenerated neurons and axons (Hu et al., 2017).
Figure 2.
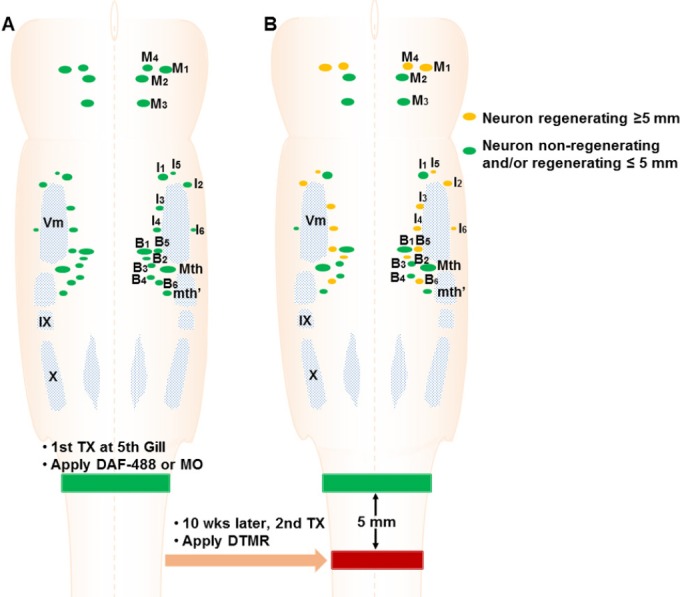
Experimental design.
(A) A first transection (TX) is performed at the level of the 5th gill, and Gelfoam soaked with dextran Alexa Fluor 488 (DAF-488) or oligonucleotides (MO) (Green) is inserted into the TX site. Axons that have been severed by this lesion will take up the dye at the cut tip and the label transported retrogradely to the identified reticulospinal (RS) neurons in brainstem. (B) 10 weeks later, a second TX is performed 5 mm caudal to the first TX and Gelfoam soaked with dextran tetramethyrhodamine (DTMR) (Red) is applied, to label the neurons whose axons have regenerated more than 5 mm beyond the original TX.
MOs are oligomers, usually 25 bases in length, which can bind complementary RNA to knock down gene expression specifically. We designed a MO to target the start codon of RhoA mRNA and its upstream, untranslated region, thus blocking translation of RhoA protein (Hu et al., 2017). In the lamprey, MOs are efficiently transported retrogradely in axotomized RS axons from the TX site to their neurons in brain. With the fluorescently labeled MOs, we could determine which neurons had taken them up. Thus, we used MOs in vivo to specifically knockdown RhoA in RS neurons after TX, and imaged the cut axon tips. During the first 2 weeks post-TX, axons retracted, and this early retraction was inhibited by the RhoA MO. Thereafter, axon forward growth was accelerated. By 10 weeks post-TX, RhoA knockdown greatly increased the number of axons regenerating more than 5 mm (Hu et al., 2017) (Figure 3).
Figure 3.
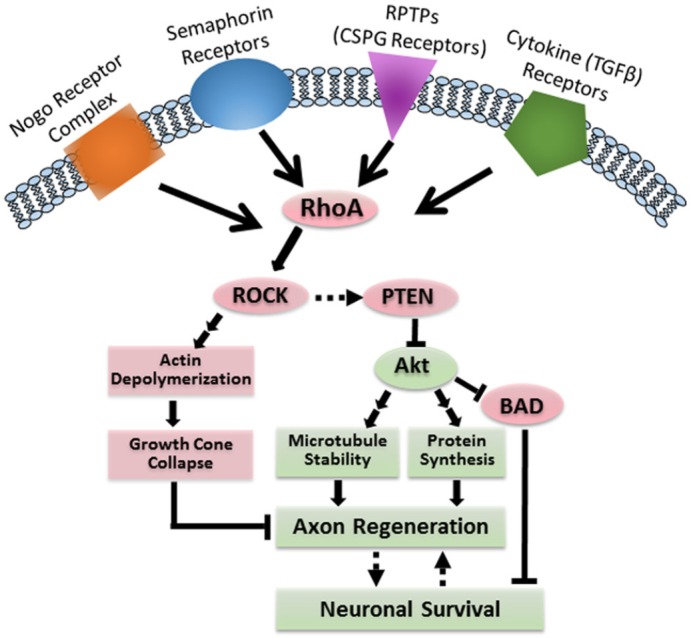
RhoA mediates both apoptosis and failure of axon regeneration after SCI.
Activation of PTEN by RhoA/ROCK has been reported in cell lines but not yet in neurons or in CNS in vivo. BAD: Bcl-2-associated death promoter; CNS: central nervous system; CSPG: chondroitin sulfate proteoglycans; PTEN: phosphatase and tensin homolog; RhoA: ras homolog gene family, member A; ROCK: rho-associated protein kinase; RPTPs: receptor protein tyrosine phosphatases; SCI: spinal cord injury; TGFβ: transforming growth factor β.
RhoA Inhibits Retrograde Apoptotic Signaling after SCI
Near the site of a SCI, neurons and other cells undergo apoptosis, and in mammals, inhibition of RhoA with C3 reduced the apoptotic signaling (Dubreuil et al., 2003; Wu et al., 2016; Gwak et al., 2017). Whether this effect is due to a role for RhoA in axotomy-induced neuronal death is not clear, since non-neuronal cells were also affected, and the neuronal death could have been due to other mechanisms such as inflammation, reactive oxidation and excitotoxicity.
In order to address this question, we used MOs in vivo to retrogradely knockdown RhoA in RS neurons after SCI. The “bad-regenerating” RS neurons often experience very delayed retrograde apoptosis, signaled by late TUNEL staining (Shifman et al., 2008) and earlier caspases activation (Hu et al., 2017). Because these neurons are located far from their cut axon tips, local mechanisms of secondary injury are not responsible for the death of these neurons. Caspase activation was significantly reduced in individual RS neurons, as indicated by fluorochrome-labeled inhibitors of caspases (FLICA). The reduction of retrograde apoptosis signaling began at 2 weeks post-TX and lasted until at least 8 weeks post-TX (Hu et al., 2017). Thus, in vivo RhoA knockdown can both promote true axon regeneration and reduce retrograde apoptosis signaling after SCI (Figure 3). These findings have clarified ambiguities in the results of previous SCI studies using mammalian partial injury or contusion injury models.
Discussion
It is clear that the small GTPase RhoA has effects on axonal outgrowth and neuronal vulnerability to apoptosis after SCI (Figure 3). However, the usefulness of targeting RhoA to promote axon regeneration and neuronal survival depends on our understanding of the molecular mechanisms of RhoA in axon regeneration and apoptosis and the possible toxicities of the therapeutic interventions. In addition to increasing the levels of extracellular growth-inhibitory molecules, SCI also induces neuronal changes in the expression of the corresponding receptors. For instance, p75 has been shown to induce local apoptosis after SCI via RhoA (Dubreuil et al., 2003). LAR and PTPσ also act on RhoA to mediate CSPG inhibitory effects on axon regeneration (Ohtake et al., 2016). Studies also suggest that in addition to its traditional downstream cytoskeletal effects, RhoA activates cPLA2 (Wu et al., 2016) to execute its functions. Interestingly, our preliminary data indicate that RhoA might reduce the phosphorylation of Akt (unpublished), thus inducing retrograde neuronal death and inhibiting axon regeneration.
It is not clear at what point the effects of RhoA are most acutely felt by the neurons. Axon retraction was reduced in the first 2 weeks after MO application, but immunohistochemical findings suggested that RhoA levels remained reduced for at least 10 weeks (Hu et al., 2017). Since axon regeneration continues for at least that long, it is possible that the influence of RhoA knockdown on regeneration reflected sustained actions. Many questions remain about the role of RhoA in neuronal responses to SCI, and there is a need for more research to elucidate the specific signaling pathways involved in RhoA's role in the pathogenesis of SCI, and on the mechanisms by which RhoA inhibition enhances functional recovery.
Footnotes
Funding: This study was supported by R01-NS092876 (NIH, MES, PI); SHC-85400 (Shriners Research Foundation, MES, PI); SHC-85220 (Shriners Research Foundation, MES, PI) and SHC-84293 (Shriners Research Foundation, JH, PI).
Conflicts of interest: None declared.
References
- Conrad S, Schluesener HJ, Trautmann K, Joannin N, Meyermann R, Schwab JM. Prolonged lesional expression of RhoA and RhoB following spinal cord injury. J Comp Neurol. 2005;487:166–175. doi: 10.1002/cne.20561. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak SJ, Macks C, Jeong DU, Kindy M, Lynn M, Webb K, Lee JS. RhoA knockdown by cationic amphiphilic copolymer/siRhoA polyplexes enhances axonal regeneration in rat spinal cord injury model. Biomaterials. 2017;121:155–166. doi: 10.1016/j.biomaterials.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Hofmann F, Just I, Lehnardt S, Hanisch UK, Bruck W, Kettenmann H, Ahnert-Hilger G, Holtje M. Inhibition of Rho-dependent pathways by Clostridium botulinum C3 protein induces a proinflammatory profile in microglia. Glia. 2008;56:1162–1175. doi: 10.1002/glia.20687. [DOI] [PubMed] [Google Scholar]
- Hu J, Zhang G, Rodemer W, Jin LQ, Shifman M, Selzer ME. The role of RhoA in retrograde neuronal death and axon regeneration after spinal cord injury. Neurobiol Dis. 2017;98:25–35. doi: 10.1016/j.nbd.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AJ, Swain GP, Snedeker JA, Pijak DS, Gladstone LJ, Selzer ME. Recovery of neurofilament expression selectively in regenerating reticulospinal neurons. J Neurosci. 1997;17:5206–5220. doi: 10.1523/JNEUROSCI.17-13-05206.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch JC, Tonges L, Michel U, Bahr M, Lingor P. Viral vector-mediated downregulation of RhoA increases survival and axonal regeneration of retinal ganglion cells. Front Cell Neurosci. 2014;8:273. doi: 10.3389/fncel.2014.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19:7537–7547. doi: 10.1523/JNEUROSCI.19-17-07537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake Y, Wong D, Abdul-Muneer PM, Selzer ME, Li S. Two PTP receptors mediate CSPG inhibition by convergent and divergent signaling pathways in neurons. Sci Rep. 2016;6:37152. doi: 10.1038/srep37152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovainen CM. Physiological and anatomical studies on large neurons of central nervous system of the sea lamprey (Petromyzon marinus). I. Muller and Mauthner cells. J Neurophysiol. 1967;30:1000–1023. doi: 10.1152/jn.1967.30.5.1000. [DOI] [PubMed] [Google Scholar]
- Shifman MI, Zhang G, Selzer ME. Delayed death of identified reticulospinal neurons after spinal cord injury in lampreys. J Comp Neurol. 2008;510:269–282. doi: 10.1002/cne.21789. [DOI] [PubMed] [Google Scholar]
- Swain GP, Snedeker JA, Ayers J, Selzer ME. Cytoarchitecture of spinal-projecting neurons in the brain of the larval sea lamprey. J Comp Neurol. 1993;336:194–210. doi: 10.1002/cne.903360204. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Steward O. Concepts and methods for the study of axonal regeneration in the CNS. Neuron. 2012;74:777–791. doi: 10.1016/j.neuron.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Walker CL, Lu Q, Wu W, Eddelman DB, Parish JM, Xu XM. RhoA/Rho kinase mediates neuronal death through regulating cPLA2 activation. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-0187-6. doi:10.1007/s12035-016-0187-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Peng X, Chiang P, Kim J, Sun X, Fink DJ, Mata M. HSV-mediated gene transfer of C3 transferase inhibits Rho to promote axonal regeneration. Exp Neurol. 2012;237:126–133. doi: 10.1016/j.expneurol.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]