

Abstract
Apelin peptides and the apelin receptor represent a relatively new therapeutic axis for the potential treatment of cardiovascular disease. Several reports suggest apelin receptor activation with apelin peptides results in cardioprotection as noted through positive ionotropy, angiogenesis, reduction of mean arterial blood pressure, and apoptosis. Considering the potential therapeutic benefit attainable through modulation of the apelinergic system, research is expanding to develop novel therapies that limit the inherent rapid degradation of endogenous apelin peptides and produce metabolically stable small molecule agonists and antagonists to more rigorously interrogate the apelin receptor system. This review details the structure–activity relationships for chemically modified apelin peptides and recent disclosures of small molecule agonists and antagonists and summarizes the peer reviewed and patented literature. Development of metabolically stable ligands of apelin receptor and their effects in various models over the coming years will hopefully lead to establishment of this receptor as a validated target for cardiovascular indications.
Graphical abstract
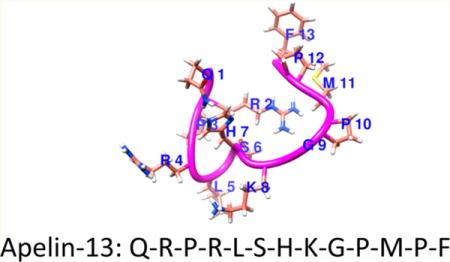
INTRODUCTION
Over the years, G-protein-coupled receptors (GPCRs) have emerged as important targets for medications development. The human genome project in the 1990s and other sequencing initiatives led to the identification of several putative GPCR genes. These genes were typically identified by mining genomic data for sequence identity to known receptors. In 1993, a novel GPCR was cloned that showed some structural homology to the angiotensin 2 receptor 1 (AT1) receptor.1 This gene was predicted to produce a 380 amino acid long class A GPCR with ∼33% sequence identity to AT1. The gene was localized to chromosome 11 and contained consensus sequences for protein kinase A (PKA) phosphorylation. However, the receptor was not activated by angiotensin 2 (Ang 2) and therefore classified as an orphan GPCR and given the name apelin receptor (gene: APLNR or AGTRL1).1 Presence of the apelin receptor gene has been confirmed across many species including rodents, zebrafish, frog, monkey, and pig, indicating that apelin receptor is physiologically important. Rat and mouse orthologues of apelin receptor have been cloned (377 amino acids each), and these genes share ∼90% homology to their human counterpart.
ENDOGENOUS LIGANDS OF THE APELIN RECEPTOR
The apelin receptor remained an orphan until 1998, when Tatemoto and colleagues identified a 36 amino acid peptide from bovine stomach as the native ligand and named it apelin (APLN).2 Studies indicated that apelin-36 was first produced as a 77-residue pre-pro-peptide that consequently underwent cleavage at paired basic residues (Arg-Arg and Arg-Lys) to produce smaller peptides of various lengths.3,4 While the proteolytic process needs further clarification, recent studies by Shin and colleagues have demonstrated that proprotein convertase subtilisin kexin 3 (PCSK3 or furin) preferentially cleaves proapelin to apelin-13.5 Of these peptides, the full length apelin-36 and smaller peptides including apelin-13, apelin-17, apelin-12, and a pyroglutamated version of apelin-13 (pyr-apelin-13) have been the most thoroughly investigated in vitro and in vivo (Table 1).4,6–8 These peptides are sequentially conserved across many species.9 For example, the last 23 residues of apelin are conserved in mammals. Studies with C-terminal synthetic fragments representing the last 36, 17, 13, and 12 residues of pre-pro-apelin indicate that these peptides are potent agonists of apelin receptor with nanomolar affinity and activity at the receptor. Apelin peptides are readily metabolized in vivo with a half-life (t1/2) of ∼5 min.10 Past studies have implicated angiotensin converting enzyme 2 (ACE2) in cleavage of the C-terminal phenylalanine residue of apelin-13.11 However, data also indicate that absence or alteration of this residue does not lead to complete inactivation of apelin peptides in vitro.12 In 2014, two different research groups independently identified a second endogenous peptide ligand of apelin receptor. This peptide called TODDLER/ELABELA (HUGO: apelin receptor early endogenous ligand, APELA) is involved in zebrafish embryo development.13–15 Recent studies indicate this peptide is expressed in humans and has nanomolar activity and affinity at apelin receptor in spite of being structurally different from apelin.16 The possibility of a third endogenous regulator of the apelin receptor cannot be ruled out at this time.
Table 1.
Functional Apelin Receptor Activity and Sequence of Predominate Bioactive Apelin Peptides
| compd | peptide | sequence | EC50 ± SEM (nM) |
|---|---|---|---|
| 1 | apelin-36 | LVQPRGSRNGPGPWQGGRRKFRRQRPRLSHKGPMPF | 2.69 ± 0.2617,a |
| 2 | apelin-17 | KFRRQRPRLSHKGPMPF | 1.56 ± 0.1117,a |
| 3 | apelin-13 | QRPRLSHKGPMPF | 7.82 ± 0.6917,a |
| 4 | pyr-apelin-13 | pERPRLSHKGPMPF | 0.20 ± 0.0217,a |
| 5 | apelin-12 | RPRLSHKGPMPF | 0.18 ± 0.0417,a |
| 6 | ELABELA/TODDLER-32 | QRPVNLTMRRKLRKHNCLQRRCMPLHSRVPFP | 11.116,b |
Intracellular calcium mobilization assay. CHO cells expressing apelin receptor were treated with peptides, and calcium mobilization was monitored using fluorescent dye.
cAMP inhibition assay. CHO cells expressing apelin receptor were treated with forskolin, ELA, and inhibition of cAMP was measured by TR-FRET.
Identification of endogenous peptidergic apelin receptor ligands facilitated molecular characterization of the receptor. Apelin receptor activation is associated with inhibition of forskolin-stimulated cAMP production, and this pathway is sensitive to the effects of pertussis toxin.3 Thus, apelin receptors are coupled to inhibitory Gαi proteins. Additionally, some studies indicate that apelin receptors can also activate PLC mediated calcium flux through Gαq coupling.18,19. It is also understood that activated apelin receptor recruits β-arrestin, which in turn desensitizes the receptor utilizing clathrin mediated endocytosis.12
TISSUE EXPRESSION OF APELIN AND THE APELIN RECEPTOR
Apelin has a wide pattern of expression across tissues in humans and rodents. High levels of apelin mRNA have been noted in the central nervous system (CNS), heart, lung, kidneys, and placenta.20–22 In rat CNS, apelin mRNA is expressed within the hypothalamus where it is coexpressed with vasopressin mRNA in paraventricular (PVN) and supraoptic nuclei (SON).23–25 Accordingly, there is some evidence to suggest the apelinergic system counters the effect of the vasopressin system.26 Elegant studies using a transgenic approach have revealed intriguing data related to expression of apelin within heart, lung, and skeletal muscle (quadriceps) in mice.27 A transgenic mouse line was created expressing the bacterial lacZ gene with a nuclear localization signal in the first apelin exon immediately upstream of the translation start site. No additional deletions were made upstream or to intronic sequences of the gene that could affect transcription. Staining with X-gal and CD31 antibody confirmed that expression of the transgene was restricted to endothelial cells, particularly within capillaries and veins.27 Within the heart and skeletal muscle, postarterial capillaries and venous endothelium strongly expressed the reporter protein, whereas expression was absent from large arteries and arterioles. Within the lung, alveolar septal capillaries and pulmonary veins expressed lacZ but no arterial expression was observed. LacZ expression was also absent in type 1 and 2 pneumocytes. However, immunocytological studies by Kleinz and Davenport in fresh frozen human tissues indicated that apelin-like immunoreactivity was present within vascular endothelial cells lining blood vessels in the human heart, kidney, adrenal gland, and lung and in endothelial cells of large conduit vessels. Apelin-like immunoreactivity could also be detected in endocardial endothelial cells lining recesses of the right atrium. Signal was absent or below the level of detection in cardiomyocytes, Purkinje’s cells, pulmonary or renal epithelial cells, secretory cells of the adrenal gland, vascular smooth muscle cells, adipocytes, nerves, and connective tissue.28 These somewhat contrasting data from mice and humans indicate that there might be species differences related to apelin expression. It is also possible that LacZ staining was not sensitive enough to detect apelin in some tissues examined.
The mRNA of apelin receptor is widely distributed in humans and is detected in several tissues including the CNS, heart, lung, placenta, liver, kidney, spleen, and stomach. A similar expression pattern of the receptor was also noted in rodents.22,29,30 Corroborative studies indicated that apelin receptor mRNA was expressed at high levels in endothelial cells and therefore within vascular compartments of most organs.31–37 As such, highly vascularized organs typically express high levels of apelin receptor mRNA.
Several immunohistochemical studies have been performed to investigate apelin receptor protein expression in various tissues, and these studies have at least partially confirmed high levels of protein expression within vascular compartment of several tissues. Unfortunately, definitive immunohistochemical studies of apelin receptor have been difficult to perform and interpret because of a lack of high quality antibodies. Accordingly, conflicting data have been produced related to both subcellular and tissue localization of this receptor. Perhaps the best characterized antibody to apelin receptor was described by Puffer et al. in 2000 in collaboration with a commercial entity (R&D Systems).38 This particular monoclonal antibody was designated mAb 856, and the authors confirmed the specificity of this reagent by detecting apelin receptor in 293T cells transfected with a cDNA vector of the gene versus control cells harboring the vector alone. The authors also established that localization of apelin receptor was primarily at the cell surface using flow cytometry and immunofluorescence. This pattern of localization was consistent with expected subcellular localization of GPCRs. Immunoblot analysis revealed apelin receptor to be N-glycosylated, and aggregation of the protein upon heating in a reducing environment was also reported in the same study. In studies reported by Pope et al., in situ hybridization histochemistry with APJ riboprobes demonstrated strong hybridization specifically in the paraventricular (PVN) and supraoptic (SON) nuclei of the hypothalamus and in the anterior pituitary, along with low level expression in posterior pituitary in mice. In the periphery, strong hybridization was observed in the lung, heart, adrenal cortex, renal medulla, ovary, and uterus. Autoradiographic binding to apelin receptor with 125I-labeled pyr-apelin-13 exhibited significant binding in the anterior pituitary, while lower levels were observed in the posterior pituitary and PVN and SON. In the periphery, strong receptor binding was observed in tissues exhibiting intense riboprobe hybridization, indicating a good correlation between receptor transcription and translation.39
Some of the controversies related to apelin receptor protein expression at the tissue level were recently addressed through generation of a transgenic mouse model wherein nuclear lacZ was knocked into the endogenous Aplnr locus.40 Both traditional X-gal staining with histochemical observations and double staining of tissues from various organs along with marker proteins neuropilin-1 (Nrp-1) for arteries and nuclear receptor subfamily 2 group F member 2 (NR2f-2) for veins indicated that lacZ expression was largely restricted to cardiomyocytes in heart and venous endothelium of most major organs examined. LacZ expression was completely absent from lymphatic vessels. Taken together, these reporter gene studies provided complementary evidence suggesting that at least in mice, apelin receptor and apelin proteins were highly expressed within the venous capillary beds of various organs.27,40 Further, lacZ staining of the myocardium suggested a possible role of the apelinergic system in cardiovascular regulation. However, these data related to apelin and apelin receptor expression using transgenic animals should be cautiously interpreted because there might be species differences affecting expression. Apelin receptor-like immunoreactivity has been noted in human endothelial cells, vascular smooth muscle cells, and cardiomyocytes.33 This is particularly important within the context of human clinical data discussed in later sections. Further, an altered physiological state could also affect gene and protein expression. For example, hypoxia induces both apelin receptor and apelin peptide expression through HIF1α sensitive transcriptional regulation.35,41–43 Therefore, under hypoxic conditions, the apelinergic system could be upregulated in tissues where it is normally expressed at very low levels.
The apelinergic system is expressed at a fairly high level within the cardiovascular system, and several studies have been undertaken to understand the role of apelin and apelin receptor in regulation of cardiovascular physiology. Several recent reviews have discussed these results.44–46 Briefly, apelin has been described as a positive ionotropic and cardioprotective agent. In a majority of reported in vivo examinations in rodents, peripheral administration of apelin led to reduction of mean arterial blood pressure (MABP) presumably through prostanoids4 and/or nitric oxide47 dependent mechanisms. However, reports to the contrary have also emerged. For example, administration of apelin to normal anesthetized dogs produced no effect on the mean pulmonary artery pressure,48 and vasoconstriction upon administration of apelin-13, pyr-apelin-13, and apelin-36 has been reported in endothelium denuded saphenous vein.4 In another report, administration of apelin-36 did not alter MABP in normal SD rats or in Lewis rats following myocardial infarction (MI). However, both apelin-36 and a PEGylated stable analogue of the peptide with longer circulating half-life improved cardiac ejection fraction by ∼20% and 40%, respectively, in MI rats confirming positive ionotropic effects of apelin peptides in agreement with previous studies.49 Additionally, apelin-17 and mutated apelin-17 fragments have been shown to regulate vasopressin secretion and fluid homeostasis.50 Internalization of apelin receptor is also affected by mutations to apelin-17, and there is a direct correlation between internalization of the receptor and hypotensive action of apelin fragments.12,51 Finally, a apelin independent, mechanical stretch dependent activation pathway has also been described for apelin receptor.52 This activation is pathological in mice under the condition of chronic pressure overload.
APELIN RECEPTOR KNOCKOUT STUDIES
Independent research groups have produced apelin receptor knockout animals and studied their physiology. Charo et al. reported that apelin receptor-deficient mice were not born in the expected Mendelian ratio and manifested developmentally related cardiovascular defects. In addition, both apelin and apelin receptor knockout animals had decreased exercise capacity due to reduced sarcomeric shortening of isolated cardiomycotytes and impaired velocity of contraction.53 Similarly, apelin knockout animals developed progressive impairment of cardiac contractility associated with systolic dysfunction in the absence of histological abnormalities upon aging.54 Ishida et al. also created apelin receptor knockout mice and reported that apelin receptor-deficient animals showed an increased vasopressor response to the vasoconstrictor Ang 2, and the baseline blood pressure of double mutant mice homozygous for both apelin receptor and angiotensin-type 1a receptor was significantly elevated compared with that of angiotensin-type 1a receptor-deficient mice.55 These results demonstrated that apelin receptor exerted hypotensive effects in vivo and counteracted the pressor effect of Ang 2. However, a recent publication by Lathen et al. has challenged some of these earlier reports, which suggested that apelin receptor has effects in the adult rodent arterial circulation.40 As discussed previously, these investigators created a line of lacZ reporter mice from the APLNR locus and noted a lack of receptor gene expression within the arteries whereas strong expression was noted in veins and cardiomyocytes. According to the authors, these results suggested that reported effects of apelin in arteries could be due to interaction with a different receptor and not apelin receptor. Interestingly in the same paper, global or endothelial specific knockout of APLNR led to development of pulmonary veno-occlusive disorder (PVOD) consistent with reporter gene expression from the Aplnr locus.40 The potential of apelin receptor manipulation in small vessel diseases remains largely unexplored and could potentially open up new possibilities for medications development.
HUMAN TRIALS USING APELIN PEPTIDES
Studies in humans with apelin peptides have produced very interesting results. Japp et al. conducted studies in healthy human volunteers to investigate direct vascular effects of apelin-36 and pyr-apelin-13 infusions.47 Intra-arterial infusions of apelin peptides followed by measurement of forearm blood flow using venous occlusion plethysmography indicated that apelin peptides caused reproducible vasodilation of arterial vessels. However, there were subtle differences elicited by each peptide. Pyr-apelin-13 produced a rapid dose-dependent increase of blood flow that reached a plateau, but the effect was reproducible after a saline washout period. Apelin-36 in contrast produced a more durable response that could not be recapitulated after a washout period. These differences could be related to differential internalization and/or desensitization of apelin receptor upon activation by each peptide and requires further elucidation. However, intravenous infusions of each apelin peptide into a nonbranching dorsal hand vein (DHV) preconstricted with norepinephrine did not increase DHV diameter. The vasodilatory effect of pyr-apelin-13 infusion within the arteries was attributable to nitric oxide production but unrelated to prostanoids, as oral aspirin did not inhibit these effects. Interestingly, the lack of effect within the venous bed was in sharp contrast to what could be predicted based upon rodent studies of Lathen et al., discussed above, and possibly points to species differences and the need to test more stable and druglike pharmacological agents. Follow-up studies by the same group in heart disease patients indicated that acute pyr-apelin-13 systemic infusion increased cardiac index and lowered mean arterial pressure and peripheral vascular resistance as well as increased cardiac output in patients suffering from chronic heart failure.56 Additional studies in humans indicated that apelin receptor agonism by pyr-apelin-13 produced sustained beneficial cardiovascular effects that were preserved even in the presence of renin–angiotensin system activation.57 Taken together, these data indicate that the apelinergic system is a promising but controversial new target for cardiovascular indications that will benefit from the discovery and application of stable ligands for both in vitro and in vivo studies.
PURSUIT OF METABOLICALLY STABLE APELIN ANALOGS
In vivo proof of principle studies with endogenous peptides are often challenging because of limited plasma stability. The majority of studies on apelin peptides have so far focused on the smaller bioactive fragments in part because of the ability to rapidly synthesize and test chemical modifications. One of the early discoveries with apelin-13 was that it undergoes rapid cleavage to inactive fragments by a number of purified enzymes including angiotensin converting enzyme type 2 (ACE2).11 In order to further understand the potential sites of backbone cleavage, Murza and colleagues10,58 studied the time course and cleavage patterns of pyr-apelin-13 in plasma, cerebrospinal fluid (CSF), and in vivo. The primary cleavage site in plasma from rat, mouse, and human occurred between Leu5 and Ser6 (Figure 1A). Additional cleavage then occurs to delete Ser6 from the secondary fragment. An interesting finding from this study demonstrated that ACE2 cleavage was not a significant contributor of apelin-13 degradation in isolated plasma. In vivo, however, ACE2 cleavage was observed between Pro11 and Phe12 in rat (Figure 1B). This suggested that either ACE2 activity is degraded during plasma preparation or apelin peptides accumulate in tissues with high ACE2 activity. In light of these data, several structure–activity studies have now been undertaken to understand the importance of each apelin-13 residue on apelin receptor binding and function which provides a template for chemical modification to block proteolytic degradation and enhance metabolic stability.
Figure 1.
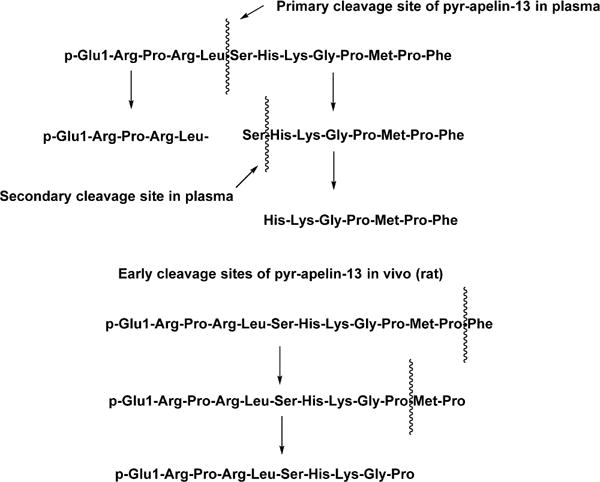
(A, top half) Cleavage sites of pyr-apelin-13 in plasma. (B, bottom half) Early cleavage steps of pyr-apelin-13 in vivo (rat). ACE2 cleavage occurred only in vivo.59 Reproduced with permission from Biopolymers (Murza, A.; Belleville, K.; Longpre, J. M.; Sarret, P.; Marsault, E. Stability and degradation patterns of chemically modified analogs of apelin-13 in plasma and cerebrospinal fluid. 2014, 102, 297–303). Copyright 2014 John Wiley and Sons.
One method of stabilizing metabolically labile peptides is through the use of polyethylene glycol (PEG) linkers that add molecular mass and thus shield the peptides from proteolytic digestion. The first reported study to attempt this approach with apelin peptides was by Jia and colleagues.58,60 The authors utilized PEG branched aldehydes of 10, 30, and 40 kDa to conjugate apelin-36 and found that the larger molecular weight PEG conjugation occurred primarily once and on the N-terminal nitrogen. The 10 kDa PEG coupling was less specific, and levels of mono-PEGylated apelin-36 were <20%. The 40 kDa PEGylated apelin-36 retained functional activity (cAMP) and was only 1.5-fold less potent than apelin-36. Moreover, the Ki of the 40 kDa PEGylated apelin-36 was 0.3 nM compared apelin-36 (Ki = 0.05 nM). Blood levels of both peptides were evaluated at 20 min after iv infusion, and 40 kDa PEG apelin-36 exhibited a 1.6-fold higher concentration suggesting reduced metabolic transformation of PEGylated apelins. Cardiac function was also assessed with apelin-36 and 40 kDa PEG apelin-36. Both peptides exhibited an increase in ejection fraction (EF) in myocardial infarcted rats after iv administration (30% and 40% at 20 min, respectively). Importantly, these changes in EF were observed without any alteration of MABP. Overall these results suggest that structural modifications that stabilize in vivo degradation of apelin related peptides may have value as pharmacotherapies for cardiovascular disease by promoting cardiac contractility through positive ionotropy.
ALANINE SCANNING STUDIES
One of the primary methods of determining a bioactive peptide core and assessing the contributions of individual side chain residues on biological activity is through alanine scanning. In one of the first SAR studies on pyr-apelin-13, Medhurst and coworkers established the importance of an N-terminal RPRL recognition element as key to its interaction with the apelin receptor.22 The importance of this motif was determined through both alanine scanning and N-terminal deletions (Table 2). Substitutions at Arg222 (9), Arg422 (10), and Leu522 (11) with Ala reduce the pIC50’s and pEC50’s substantially indicating the critical role of these residues in promoting a favorable backbone conformation in this region and/or favorable interactions with complementary receptor features.22 Alanine substitutions at positions other than 2, 4, and 5 had only minor or no impact on pIC50, cAMP derived EC50’s, and FLIPR calcium mobilization EC50 values. This early study began to define the N-terminal RPRL region as a critical functional element of the peptide but also suggested that additional residues were important for receptor recognition.
Table 2.
Alanine Scanning Effects on Pyr-apelin-13 Binding and Function22,a
| compd | peptide | binding (pIC50) | cAMP (pEC50) | Ca2+ (pEC50) |
|---|---|---|---|---|
| 4 | pyr-apelin-13 | 8.88 | 10.02 | 7.70 |
| 7 | N-acetyl-apelin-13 (8–13) | <5 | <4.5 | <4.5 |
| 8 | N-acetyl-apelin-13 (5–13) | 5.45 | 5.88 | <4.5 |
| 9 | pGlu1,Ala2-apelin-13 | 7.12 | 8.37 | 7.29 |
| 10 | pGlu1,Ala4-apelin-13 | 8.05 | 9.0 | 7.61 |
| 11 | pGlu1,Ala5-apelin-13 | 7.15 | 7.74 | 6.73 |
| 12 | pGlu1,Ala8-apelin-13 | 8.04 | 9.21 | 7.23 |
| 13 | pGlu1,Ala13-apelin-13 | 7.91 | 9.63 | 7.08 |
Data adapted from Medhurst, A. D.; Jennings, C. A.; Robbins, M. J.; Davis, R. P.; Ellis, C.; Winbornl, K. Y.; Lawrie, K. W.; Hervieu, G.; Riley, G.; Bolaky, J. E.; Herrity, N. C.; Murdock, P.; Darker, J. G. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin, J. Neurochem. 2003, 84, 1162–1172 (Copyright 2003 John Wiley and Sons).
In a subsequent study, Fan and co-workers61 also pointed out the importance of positively charged residues such as Arg as well as turn inducing residues such as Pro in apelin-13 using alanine scanning experiments but extended these results to receptor internalization in stable apelin receptor-EGFP-expressing 293 cells. Mutation of Arg4Ala, Ser6Ala, and Gly9Ala all had diminished capacity to induce apelin receptor internalization while reducing receptor binding. Arg2Ala, Pro3Ala, Leu5Ala, Lys8Ala, Pro10Ala, and Met11Ala mutations reduced or eliminated the ability to induce receptor internalization along with reduced binding. In particular, the authors noted one surprise that Arg2Ala had diminished receptor internalization but increased calcium mobilization. However, the authors did not quantitatively assess receptor function and internalization. As such, any aberrations noted in this study could be a result of lack of quantitative information related to apelin receptor function. In spite of these limitations, these data generally indicate that reduced binding of mutated peptides to apelin receptor correlated well with decreased function as assessed by calcium mobilization and receptor internalization using GFP-tagged apelin receptor.
D-SCANNING STUDIES
In order to better determine the effects of chirality on pyr-apelin-13 binding and function, D-scanning studies were conducted by Murza and colleagues.10 As anticipated based on previous alanine scanning data, D-substitution within the RPR region produced significant reductions (>200-fold) in apelin receptor affinity but D-Leu5 substitution resulted only in a 10-fold reduction in affinity suggesting that chirality of the Leu5 side chain is less critical for receptor interaction than previously thought. Overall, D-substitution reduced affinity compared to pyr-apelin-13 for all other residues: D-Ser6 (100-fold decrease), D-His7 (52-fold decrease), D-Lys8 (80-fold decrease), D-Pro10 (288-fold decrease), D-Met11 (2-fold decrease), D-Pro12 (7-fold decrease), D-Phe13 (20-fold decrease).10 In total, these studies corroborate the data from alanine scanning and suggest that the RPRL region most likely plays a critical role in determining an overall architecture of the peptide in addition to those side chains engaging in favorable receptor interactions.
PEPTIDE TRUNCATION STUDIES
In order to better transition toward molecules with improved metabolic stability and druglikeness, the minimal residue content necessary for apelin-13 potency was assessed through both C-terminal and N-terminal truncation studies. On apelin-13 analogs, Glu1 or pyr-Glu1 has been shown to play a limited role in determining potency from both Ala- and D-scanning studies. Therefore, several studies on C-terminal truncated analogs of apelin-12 have demonstrated efficacy in vivo for a number of end points including hypertension, myocardial infarction, and generic cardioprotection.62–64 Medhurst and colleagues22 also determined that apelin-13 N-terminal truncated peptides 5–13 and 8–13 lacking the R-P-R-L region, which is conserved throughout species, had no appreciable activity at the apelin receptor (Table 2). Systematic N-terminal truncations were not undertaken past Glu1 because of the recognized importance of the R-P-R-L region in recognition and binding. In contrast, a comprehensive C-terminal truncation study was able to provide smaller peptides with appreciable activity at the apelin receptor. C-terminal truncations of Phe13 and Pro12 resulted in shorter peptides with moderate activity, but when Met11 was removed, activity fell 6000-fold.22 Zhang and colleagues demonstrated that Ac-R-P-R-L-S-H-K-G-P-M65 (14) has an EC50 of 4.4 nM in calcium mobilization experiments confirming that Glu1, Phe13, and Pro12 play a limited functional role regarding the primary activity of apelin-13. These data suggested that the smallest fragment of apelin-13 that would retain activity is the 10-mer Ac-R-P-R-L-S-H-K-G-P-M-NH2.
Considering that metabolic deactivation of apelin-13 was one of the major obstacles preventing more detailed pharmacological studies, Murza and colleagues focused on blocking the known cleavage sites in plasma and in vivo between Leu5 and Ser6 as well as the ACE2 cleavage site between Pro12 and Phe13.10 In several studies the presence of Phe13 has been shown to impart limited effects on agonist potency; therefore, a number of unnatural amino acids were substituted for Phe13 in peptides having the general formula pGlu-R-P-R-L-S-H-K-G-P-Nle-P-X (Figure 2). Some of the most potent and metabolically resistant analogs were X = 2-naphthyl10 (15) (IC50 = 1.2 nM and 84% remaining at 1 h), X = cyclohexylalanine10 (16) (IC50 = 2.3 nM with 65% parent remaining at 1 h), and X = (4-phenyl)phenylalanine10 (IC50 = 7.8 nM with 67% parent remaining at 1 h). A subsequent study10 focused on replacement of Pro12 to block ACE2 cleavage. Peptides having the formula pGlu-R-P-R-L-S-H-K-G-P-Nle-X-F were prepared where X = a series of unnatural amino acids (Figure 2). The most metabolically resistant substitutions were X = tert-butylleucine10 (17) (IC50 = 15.6 nM with 61% parent remaining at 1 h) and aminoindane10 (18) (IC50 = 20 nM with 57% parent remaining at 1 h) in plasma. Another primary cleavage site in plasma occurs between Leu5 and Ser6. It appears that natural amino acid substitutions do little to improve stability, but substitution of D-leu5 improves plasma half-life (t1/2) from 14.1 min in pyr-apelin-13 to 52 min. The same holds true for D-Ser6 with a t1/2 of 45 min in rat plasma, although D-Ser6 has limited affinity for the apelin receptor (642 nM).
Figure 2.
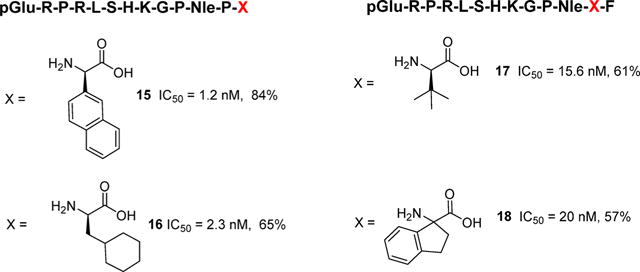
Analogs of apelin-13 with enhanced half-life (t1/2) designed to block ACE2 cleavage. % is amount of parent remaining at 1 h.
Modifications to the apelin-13 template were also conducted by Wang and colleagues with the goal of producing peptides that are resistant to cleavage mediated by ACE2 in vivo.66 Expanding upon the SAR generated by Murza et al.10 led to substitution of residues 11, 12, and 13 to afford two analogues with enhanced proteolytic stability compared to pyr-apelin-13. pGlu-Arg-Pro-Arg-Leu-Ser-His-Lys-Nle-Inp-4-bromoPhe66 (19) and pGlu-Arg-Pro-Arg-Leu-Ser-His-Lys-Nle-Aib-4-bromoPhe66 (20) were both deemed completely resistant to ACE2 cleavage after 48 h, whereas ∼78% of pyr-apelin-13 was degraded within 2 min of incubation. Although these peptides were not evaluated for functional activity at the apelin receptor using in stably transfected cell lines, they were directly tested in a number of assays to determine whether durable agonism of apelin receptor could provide a benefit in myocardial infarction. First, the authors tested the two peptides in a perfused isolated heart model of ischemia reperfusion (IR) injury. In ex vivo Langendorff heart preparations, analog 20 expedited recovery of heart function using a postconditioning protocol wherein the peptide was infused (1.5 μg/mL, 20 min) in the postischemic period to stimulate a clinical situation. This effect was postulated to be mediated by increased phosphorylation of Akt and ERK1/2, two kinases that are protective during IR injury by 20 in these isolated heart preparations. Analog 20 also stimulated lengthening and sprouting of endothelial progenitor cells at a concentration of 100 ng/mL in vitro by activating Akt signaling. The authors went on to conclude that reduced apelin signaling was a contributor to cell loss and organ damage during IR injury, and analog 20 reduced injury by promoting cardiac function, cell survival, and angiogenesis.
A more recent study looking at the specific signaling properties for C-terminal apelin-13 analogs identified the most potent peptides to date (Figure 3).67 Substitution of the Phe13 with aryl and aliphatic analogs indicated that the most beneficial modifications were obtained through substitution at the 4-position of the aromatic Phe. Substitution of Phe13 with P-Tyr(OBn)13 afforded analog 2167 that possessed a roughly 60-fold enhancement in receptor binding compared to pyr-apelin-13 but retained similar inhibition in cAMP production (EC50 = 0.35 nM) and activation of Gαi (EC50 = 1.1 nM). Further modification of the 4-position led to analog 2267 that incorporated a P-Bpa group. This specific analog possessed a slightly distinct functional profile being 35-fold more potent in cAMP accumulation assays (EC50 = 0.04 nM) compared to pyr-apelin-13 but was similar to pyr-apelin-13 in receptor binding assays. The ability to recruit β-arrestin 1 and 2 was also measured for 21. Analog 21 was roughly 2- to 3-fold more potent than pyr-apelin-13 at recruiting arrestins 1 and 2. Considering the very high affinity of 21, Murza and colleagues evaluated it in an anesthetized rodent model of blood pressure regulation. In a dose dependent manner, 21 reduced mean arterial blood pressure to a greater extent than pyr-apelin-13. The authors suggest that this effect could be in part due to the enhanced t1/2 of 21 (66 min) compared to that of pyr-apelin-13 (14 min) and/or the increased affinity at the receptor. Overall these studies indicate that modification of the C-terminal residues of apelin-13 can lead to enhanced potency in a variety of assays and also enhance metabolic stability.67
Figure 3.
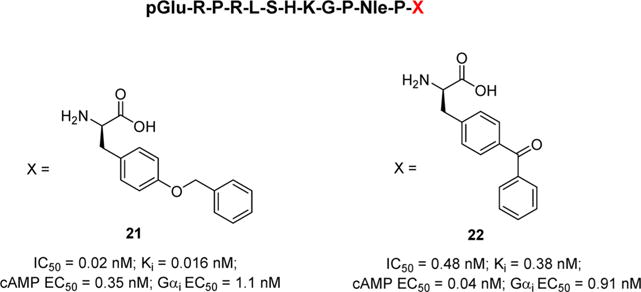
The most potent analogs of apelin-13 analogs with differential signaling properties.
In contrast to concentrating on blocking proteolytic cleavage, Zhang and colleagues65 focused on defining receptor tolerances at specific positions with the goal of driving discovery toward more druglike entities. A very detailed SAR study of pyr-apelin-13 was conducted on the shortest known fragment of apelin that retained potency and then evaluated tolerances to substitution at individual positions (Figure 4). The 10-mer Ac-R-P-R-L-S-H-K-G-P-M-amide65 (14) (EC50 = 4.4 nM) was utilized as the starting scaffold. Replacement of Met11 with Phe, Val, and Leu to improve stability indicated that steric bulk played a role at this position. Phe11 and Leu11 were also tolerated, but Val11 was 157-fold reduced in potency compared to Met11. Adding in Ala substitutions for Ser6 and His7 provided Ac-R-P-R-L-A-A-K-G-P-L-amide65 with an EC50 of 1.1 nM and 100% efficacy compared to pyr-apelin-13. Unfortunately, this analog had a very short t1/2 in rat plasma (<5 min). Other important modifications on this scaffold demonstrated that Arg4 and Lys8 could be substituted with norleucine (Nle) to afford Ac-R-P-Nle-L-A-A-Nle-G-P-L-amide65 with moderate activity (EC50 = 33 nM) and maximal efficacy. Overall this study more broadly defined substituent requirements at positions 4, 8, and 11 on truncated apelin analogs. These conclusions should be of use in defining a better understanding of the most critical receptor interactions with chemically modified peptides and may aid the development of small molecules when suitable apelin receptor models become available.
Figure 4.
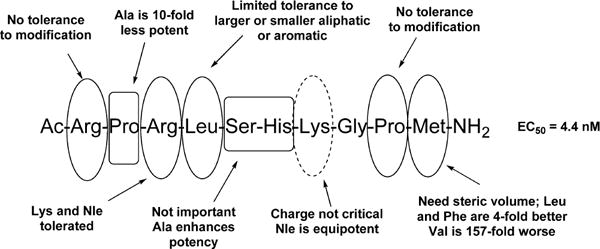
Compilation of SAR results for smallest apelin-13 fragment.
CONFORMATIONAL STUDIES ON APELIN PEPTIDES
Given the lack of an X-ray structure for the apelin receptor, understanding the bioactive conformation of apelin peptides is an important component of computational approaches focused on the development of stable peptides or small molecule therapeutics. Once information about the bioactive conformation is gathered, better predictions of ligand receptor interaction can be ascertained. A solution based conformational study by Langelaan and co-workers utilized temperature dependent NMR and circular dichroism (CD) of apelin-36, apelin-17, apelin-13, pyr-apelin-13, and apelin-12 at 5 and 35 °C.6 Far-UV CD spectra of apelin-17 at both temperatures showed little evidence of α helix or β sheet and were consistent at 5 °C with random coil. The CD spectral characteristics at 5 and 35 °C were found to be quite similar for the five peptides studied on the 210–230 nm wavelength. A crossing at ∼208–211 nm and decreased circular dichroism at 5 °C vs 35 °C were observed below the spectral crossing point. A positive band with a maximum at 217–218 nm was observed for all peptides except apelin-36 and F13A-apelin-13. While some characteristics such as a positive band at 217–230 nm were consistent with a PPII structure, other facets such as a band at 194–195 nm were not consistent. Previous reports indicated the presence of a random coil and slight PPII analogous to that found in this work.61 Characteristic bands of β-turns were also not present in the CD spectra.
A series of apelin-17 NMR investigations were also performed at both 5 and 35 °C consisting of 13C–1H HSQC (heteronuclear single quantum coherence correlation experiment), homonuclear TOCSY, NOESY, and SQF-COSY experiments. A distillation of the structural information obtained show conformational exchange in the C-terminal region for both apelin-17 and the shorter peptides which are indications of proline isomerization (Figure 5). The chemical shifts at 5 and 35 °C for apelin-17 were consistent with formation of a random coil. The global picture of apelin-17 was one in which some regions have considerable structure that undergo conformational transitions relative to each other. No short peptide close to room temperature will have deep potential surface minima but may have regions in configuration space with shallow, sharp, minima that are occupied for short times before undergoing transition to another conformation. This conception is not unlike the results derived by Krivov and Karplus68 who performed an early investigation of the nature of the free-energy surfaces for peptides employing a Monte Carlo exploration of a β-hairpin (Gly-Glu-Trp-Thr-Tyr-Asp-Asp-Ala-Thr-Lys-Thr-Phe-Thr-Val-Thr-Glu).68 In this manner, type IV β turns were present at several residues in the RPRL region as well as in the H-K-G-P C-terminal region6 while others had a greater preference for PPII. Langelaan and colleagues hypothesize, based on differential structuring of apelin-17 at 5 and 35 °C, that the Arg6-Leu9 region may undergo conformation selection in order to bind a recognition element in the N-terminal region of apelin receptor.
Figure 5.
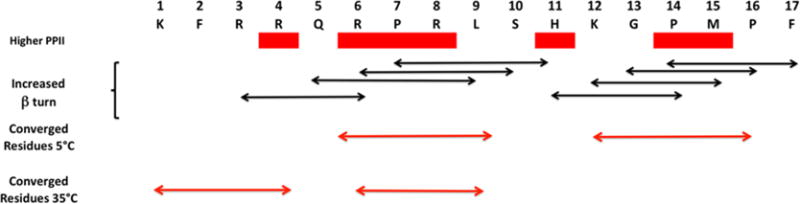
Summary NMR evidence illustrating regions of heightened PPII and type IV β turn character. Bars over four-residue range defining the β-turn are indicated.6 Adapted from Biochemistry (Langelaan et al.6).
CYCLIC PEPTIDES
NMR studies of apelin-17 indicated that significant turn character was energetically accessible in the RPRL region. In order to better understand the overall conformation of apelin peptides and simultaneously enhance metabolic stability, Hamada and co-workers synthesized a number of cyclic peptides based on apelin-13 and utilized inhibition of adenylyl cyclase via PTx-sensitive Gαi/o proteins to validate potency (Figure 6).69 Cyclization between Arg-2 and Phe-12 peptide 169 (23) was reduced in potency 1300-fold compared to pyr-apelin-13 but still possessed a moderate EC50 of 493 nM, confirming that pyr-apelin-13 must possess some degree of turn character for activation (Figure 6A). Building off this concept, Hamada et al. prepared peptide 269 (24) with a cycle within the R-P-R-L region between Arg2 and Lys8 that was 400-fold reduced in potency compared to pyr-apelin-13.69 Finally, peptide 369 (25) employed a cycle in the C-terminal portion between Lys8 and Phe12 and had an EC50 of 79 nM. Examination of these results without undue focus on the magnitude of potency difference between the cyclic compounds raises the question of bioactive conformations that may differ from solution based assessments. It is clear the three peptides depicted in Figure 6A all bind to and activate the apelin receptor. The cyclic structure of 23 (Figure 6B) retains some activity and is suggestive of bioactive conformations that may be accessible in the linear apelin peptides. Figure 6B shows a representative of one highly populated cluster from a 100 ns MD 300 K simulation in water of 23.
Figure 6.
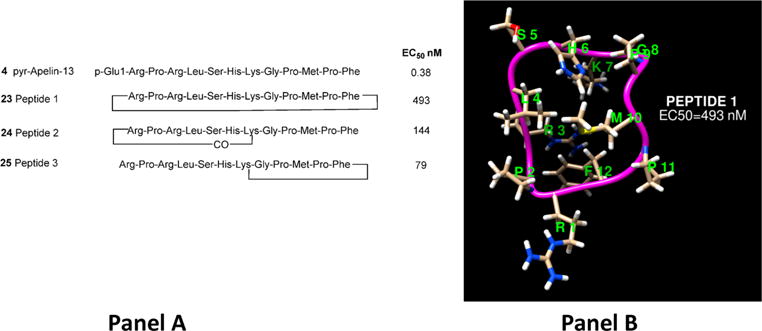
(A) Cyclic apelin-12 peptides. (B) One of the most populated solution structures of 23 from average-linkage clustering of a 100 ns simulation of the peptide in TIPS3P water using an AMBER ff99SB force field.
Depictions of the probable backbone secondary structure from some studies6 have suggested that in solution apelin-17 demonstrates a propensity to possess turn character, and this facet is commensurate with the potency of cyclic 23 that inescapably locks in conformations with high turn character. Figure 7 depicts five of the most populated average linkage clusters for 23 which show extensive turn character throughout the sequence on an average basis over 100 ns (deduced from a 300 K MD simulation employing the ff99SB force field in a box of 3239 waters employing both NPT ensemble for initial density equilibration followed by NVT ensemble production dynamics). It should be noted that none of the secondary structural motifs (turn, α helix, etc.) within the cyclic peptide subsequences persist for extended durations during the 100 ns aqueous simulation but rather undergo rapid fluctuation and interchange conformational states on short time scales. The data generated by Hamada et.al.69 as well as some of the replica exchange studies of Macaluso and Glen70 suggest apelin receptor recognition may involve a peptide conformation with significantly more turn character than was determined by others71,72 for pyr-apelin-13 or the random coil evidence in the CD work of Langelaan for apelin-17.6
Figure 7.
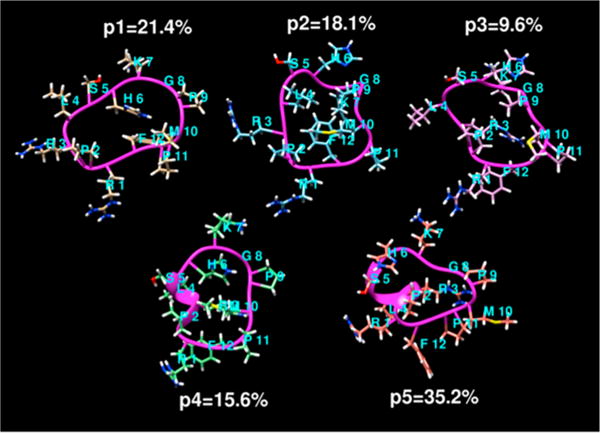
Five rmsd-average-linkage clusters of backbone conformations of 23 sampled from a 100 ns 300 K aqueous (TIP3P water) simulation (the percentage of each cluster of the total sample is indicated).
Macaluso and Glen prepared and evaluated additional cyclic peptides where the import of the RPRL motif and turn character was further examined (Figure 8A).70 They focused on cyclizations of smaller N-terminal segments between Glu1 and Ser6 (peptide A, 26),70 Glu1 and His7 (peptide B, 27),70 Glu1 and Lys8 (peptide C, 28),70 and Glu1 and Gly9 (peptide D, 29).70 They observed roughly 50% inhibition of radiolabeled pyr-apelin-13 binding at 10 μM for the two shortest peptides 26 and 27 but saw no inhibition of binding for the two longer peptides 28 and 29. It should be noted that Ki’s were not reported for these analogs. The authors concluded that turn percentage was important for binding to the apelin receptor, since 26 and 27 both possessed a much higher degree of turn character in the RPRL region than 28 and 29.
Figure 8.
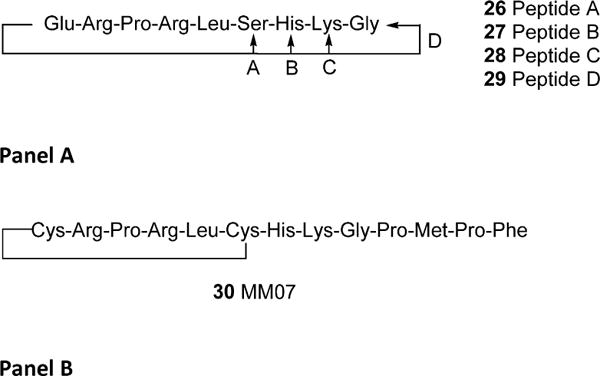
(A) Small cyclic N-terminal apelin based peptides. Peptide cyclization is between Glu1 and peptides A–D. (B) 30, an apelin receptor biased agonist.
One of the most recent examples of cyclic apelin peptide agonists was reported by Brame and colleagues.73 Using the knowledge that small N-terminal cyclic fragments such as 26 (Figure 8A) possessed some affinity for the apelin receptor, the authors prepared MM0773 (30) which is cyclized between two noncritical residues of an apelin-13 analog (Figure 8B). Modification of the natural sequence of apelin-13 to incorporate a Cys at positions 1 and 6 provided the template to generate cyclic 30.73 This is one of the most significant studies with modified apelin analogs because peptide 30 was evaluated in humans. The authors postulated that production of an agonist with bias toward the Gαi pathway and reduced preference for the β-arrestin desensitization pathway would lead to peptides with improved cardiovascular effects. In anesthetized rats, administration of 30 improved cardiac output significantly without altering blood pressure, heart rate, and respiratory rate, although the last two measurements trended higher. These studies were extended into healthy human volunteers using forearm venous occlusion plethysmography and Aellig hand vein techniques. Both pyr-apelin-13 and 30 increased forearm blood flow in these studies, but effects were only noted at higher doses of 100 nmol/min or greater with peptide 30. Repeated doses 30 produced a significant and reproducible response to 100 nmol/min as well.73 Interestingly, 30 also reversed norepinephrine induced venous constriction assessed using the Aellig technique in a dose-dependent fashion. These studies indicated that both pyr-apelin-13 and 30 performed as potent venous dilators. The venous dilation noted in this study is in sharp contrast to what was reported by Japp et al. in humans but would support the findings of Lathen et al. wherein a predominantly venous expression pattern of apelin receptor was predicted based on lacZ expression. In spite of these differences, the human studies with peptide 30 point to the promise of apelin receptor regulation in cardiovascular indications.
A patent application from Novartis (US 2013/0196899 A1) was published in November 2013 that significantly expands the cyclic peptide agonist work summarized above.74 Working off the pyr-apelin-13 sequence, they created a series of bridged peptides using a variety of linkers including disulfides, esters, amides, and short polyether bridges to prepare the cyclic peptides. Several of the peptides are claimed to be as potent as pyr-apelin-13 itself, and many but not all also show improved resistance to proteolytic degradation as demonstrated by the plasma stability t1/2 values. These approaches differ from that of Macaluso70 by focusing primarily on residues outside the apelin-13 RPRL motif. Examples of the strategies employed by Novartis are depicted in Figure 9 where X designates both natural and unnatural amino acids.
Figure 9.
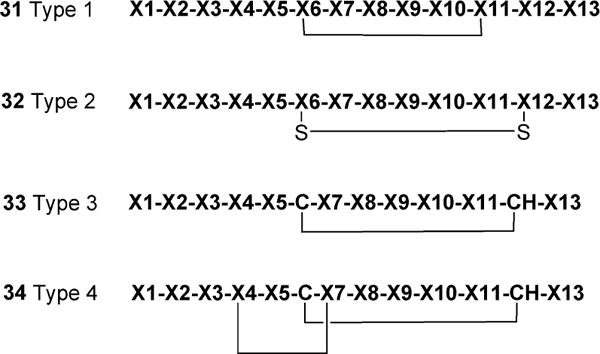
A sampling of the cyclic peptide modifications patented by Novartis. X denotes both natural and unnatural amino acids.
A CYCLIC APELIN PEPTIDE ANTAGONIST
Macaluso and colleagues examined the merits of a bivalent ligand approach utilizing two cyclic (C-R-P-R-L-C) segments separated by two amino acid spacers (Table 3).75 This effort capitalizes on their previous work illustrating that stabilizing particular conformations of the RPRL β-turn can lead to apelin receptor ligands with reasonable activity.70 Table 3 demonstrates that altering the nature of the spacer sequence between the cyclic segments does fine-tune positioning of the bivalent ligand within the apelin receptor binding site. Interestingly, the sequence in 3875 incorporates the natural spacer sequence H-K, but the reverse-sequence K-H in 3975 is the one that leads to the most potent inhibitory peptide with a Ki of 82 nM.75 Although the authors did not specifically identify what may have shifted the compounds’ functional activity from agonist to antagonist, they hypothesize the linkers play a predominate role in anchoring the individual cyclic moieties in the proper orientation. Overall this is a significant finding because little data regarding the development of peptidergic apelin receptor antagonists have been made available.
Table 3.
Cyclic Bivalent Apelin Receptor Antagonists75
| peptide | sequence | % inhibition | IC50 (nm) | Ki (nm) |
|---|---|---|---|---|
| 35 | [CRPRLC]-A-[CRPRLC] | 77 | ||
| 36 | [CRPRLC]-AA-[CRPRLC] | 98 | 950 | 840 |
| 37 | [CRPRLC]-GG-[CRPRLC] | 63 | ||
| 38 | [CRPRLC]-HK-[CRPRLC] | 86 | 1600 | 1400 |
| 39 | [CRPRLC]-KH-[CRPRLC] | 98 | 93 | 82 |
HOMOLOGY MODELS OF APELIN-13 PEPTIDES BOUND TO THE APELIN RECEPTOR
Currently, no X-ray crystal structure of the apelin receptor has been determined; therefore, computational models have been employed to better understand the potential interactions between apelin peptides and the apelin receptor. In one of the recent studies by Gerbier and colleagues, homology models of the rat apelin receptor were constructed using sequence alignments of three crystal structure templates: β2-adrenergic receptor (2RH1), chemokine CXCR4 (3ODU), and a validated model of cholecystokinin-1 receptor (CCK1R) with 20%, 27%, and 23.2% sequence identity to the apelin receptor.71 The pyr-apelin-13 peptide conformations utilized were simulated in aqueous solution, and the conformations thought to best fit the constructed human apelin receptor models were manually docked into the models. The ligand receptor complexes were energy minimized using a CHARMM22 potential function, and then MD equilibration was performed using NAMD. In the CCK1R-based human apelin receptor model, Arg2 and Arg4 of pyr-apelin-13 were proposed to interact with Asp184, Arg2 was proposed to interact with Asp282, and Lys8 of pyr-apelin-13 was proposed to interact with Asp94 and Asp284. In the β2 AR-based human apelin receptor model, Arg2 and Arg4 of pyr-apelin-13 interacted with Asp92, while in the chemokine CXCR4-based human apelin receptor model, Arg2, Arg4, and Lys8 of pyr-apelin-13 interacted with Glu174, Asp284, and Asp94, respectively. On the basis of data obtained from docking pyr-apelin-13 into the three models, the authors proposed Arg2, Arg4, and Lys8 as potentially having important interactions with human apelin receptor polar residues Asp184, Asp282, Asp94, and Asp284.
On the basis of these molecular predictions, the analogous rat apelin receptor (rapelin receptor) was subjected to a series of point mutations. The mutated rapelin receptors were then used to examine the effects on pyr-apelin-13 binding and receptor internalization (Table 4).71 Notably only Ala substitutions for rat Asp92, Glu172, and Asp282 eliminated pyr-apelin-13 binding with concentrations of radioligand up to 5 nM. The authors deduced that these residues must interact with electron deficient charged residues (Lys and Arg) on pyr-apelin-13. Interestingly, mutation of Asp92 and Glu172 with Asn or Gln restored the capacity of the receptor to bind pyr-apelin-13 while replacement of Asn282 with Gln did not restore binding. In principle this effect could be from direct disruption of binding of the acidic residue to the peptide or through an indirect perturbation on the loop structure.
Table 4.
Binding Parameters of Wild-Type and Mutated Rat Apelin Receptor Using [125I] Pyr-apelin-1371
| apelin receptor receptor mutation | Kd (nm) | Bmax (fmol/mg protein) |
|---|---|---|
| WT | 1.51 ± 0.20 | 582 ± 149 |
| E90A | 2.58 ± 1.37 | 217 ± 59 |
| D92A | no binding | no binding |
| D92N | 3.24 ± 1.73 | 140 ± 50 |
| E172A | no binding | no binding |
| E172Q | 2.58 ± 0.79 | 153 ± 49 |
| D182A | 2.94 ± 0.93 | 339 ± 61 |
| D280A | 0.88 ± 0.36 | 620 ± 290 |
| D282A | no binding | no binding |
| D282N | no binding | no binding |
NON-PEPTIDE AGONISTS AND ANTAGONISTS AT THE APELIN RECEPTOR
Because of limited availability of small molecule probes, physiological roles played by apelin receptor are currently being elucidated with apelin peptides having various levels of in vivo stability. In vivo pharmacological studies are thus complicated by the short half-life of the peptides and the need for iv injection. Considering these limitations, potent small molecule agonists or antagonists with improved pharmacological and druglike properties are needed. Such compounds would not only serve as valuable tools to elucidate the physiological role of apelin receptor but also have wider application as oral bioavailable drugs. Herein, we provide a brief summary of non-peptide agonists and antagonists that are reported to date in peer reviewed literature and patents.
NON-PEPTIDE AGONISTS
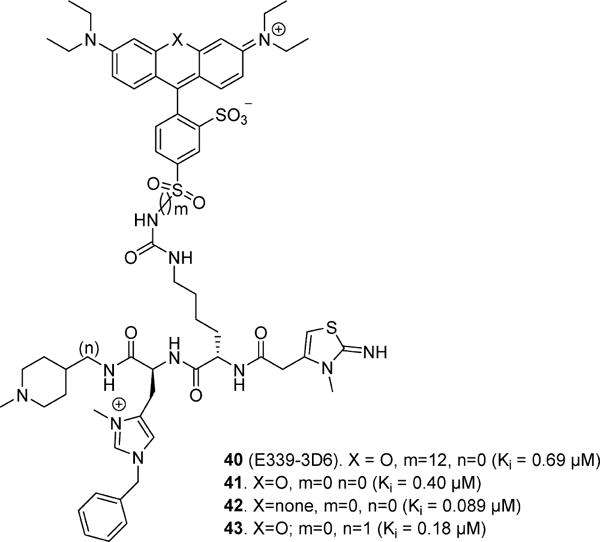
In an attempt to identify non-peptide apelin receptor ligands, Iturrioz and colleagues72 screened a library of GPCR focused fluorescent compounds on human EGFP-tagged apelin receptor using fluorescence resonance energy transfer based assay (FRET). This effort identified the first non-peptide functional apelin receptor agonist. E339-3D672 (40) possessed an EC50 of 90 nM (FRET) and Ki value of ∼400 nM in both rat and human isoforms as confirmed by radioligand binding (Figure 10). Compound 40 behaved as partial agonist inhibiting forskolin-induced cAMP production by 60%; however, it was a full agonist with regard to internalization of the apelin receptor. Compound 40 was selective against a number of GPCRs such as vasopressin V1a, oxytocin OTR, muscarinic m1R, chemokines CXCR4 and CCR5, melanocortin MC3, rat tachykinin NK2 receptors and also demonstrated low affinity for rat AT1A-EGFP receptor (Ki = 8000 nM) and CXCR4 receptor (Kd = 22 000 nM). Ex vivo evaluation of 40 exhibited concentration dependent vasorelaxation of noradrenaline preconstricted aortic rings in normotensive rats. Similar to pyr-apelin-13, when injected icv, 40 potently inhibited AVP release in the blood circulation in water-deprived mice, confirming the significance of apelin receptor agonists for the treatment of water retention.72
Figure 10.
Structural modifications of 40 and effects on activity.
In a subsequent effort, careful characterization of 40 using ES-MS indicated that the original compound was not pure and contained polymethylated compound impurities. By utilization of a novel solid phase approach on DFPE-PS resin, pure 40 was synthesized that exhibited Ki = 0.69 μM.76 Structure–activity relationships on compound 40 were undertaken to evaluate the influence of various components, including length of the spacers (m and n) and heterocyclic substituent (X) on the binding affinity at the apelin receptor (Figure 10). Compounds 41–4376 exhibited improved potency compared to 40 having Ki’s of 0.40, 0.089, and 0.18 μM, respectively. Compound 41 showed >2500-fold selectivity over AT1 receptor and functioned as a full agonist with regard to cAMP production and internalization of the apelin receptor. In addition, compounds 41–43 displayed high plasma stability in mouse (t1/2 > 10 h) in comparison to apelin-17 (t1/2 < 4 min).76
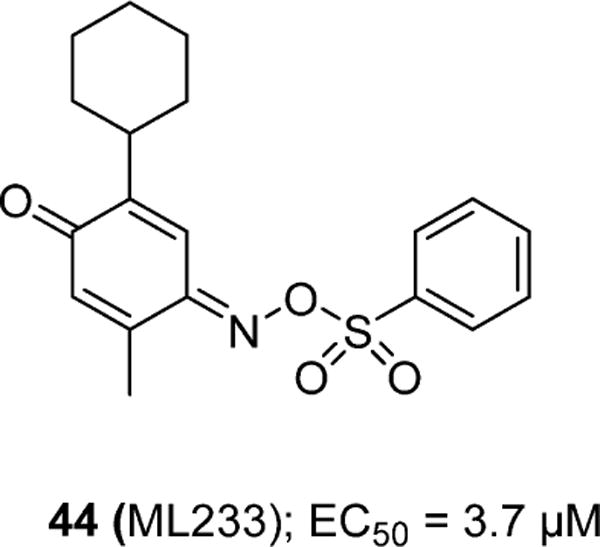
A high throughput screen of ∼330 600 compounds from the NIH small molecule library produced a full agonist, ML23377 (44), with an EC50 of 3.7 μM (Figure 11).77 Compound 44 exhibited >21-fold selectivity over the AT1 receptor and had >50% inhibition at 10 μM against 5-HT1A, adrenergic α2c, benzylpiperazine receptors, and norepinephrine transporter (NET). However, no further characterization or in vitro and in vivo studies utilizing 44 have been reported.
Figure 11.
A low potency apelin receptor agonist (44).
A recent patent disclosure from Sanofi-Aventis detailed a series of promising druglike small molecule apelin receptor agonists based on a substituted benzimidazole core scaffold (Figure 12) using calcium mobilization assays (FLIPR).78 A series of comprehensive scaffold modifications were evaluated on the core as depicted in Figure 12. In general, it appears that modification of R1 with aliphatic groups of moderate size are favored and substitution of R2 with small heterocycles is also preferred. Coupling of the appropriately substituted benzimidazole carboxylic acid with aliphatic amino acids such as Leu provided analogs (45)78 with moderate potency. Extension of the aliphatic amino acid by one methylene (β-amino acids) enhanced potency by roughly 4-fold (46, EC50 = 47 nM).78 Alteration of R2 with thiophene, furan, and thiazole appeared to have little impact on potency (47–49)78 even though small modifications were made to the amino acid side chain. In an apparent effort to improve druglike properties, bioisoteric replacement of the terminal carboxylic acid on 46,78 to tetrazole (50),78 did not alter agonist potency. However, removal of the acidic proton through substitution of oxadiazole (51)78 significantly reduced agonist potency, suggesting an important role of carboxylic acid moiety in determining activity. Disclosure of this class of apelin receptor agonists is likely to have a significant impact on the field of apelin related research considering the need for molecules that have improved oral bioavailability and reasonable t1/2.
Figure 12.
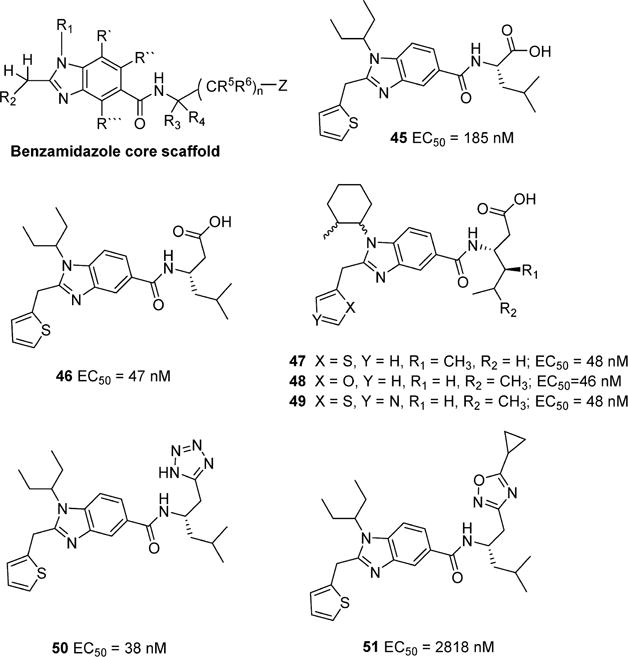
Examples of potent benzimidazole apelin receptor agonists patented by Sanofi-Aventis.
NON-PEPTIDE ANTAGONISTS
Currently, apelin receptor antagonist scaffolds are significantly more limited than those for agonists. In addition to the bicyclic peptides described by Macaluso75 (Table 3) the only other reported antagonists are those reported by Maloney and co-workers.79 A potent small molecule apelin receptor functional antagonist ML22179 (53) that exhibited an IC50 of 0.70 μM in a cAMP assay and 1.75 μM in a β-arrestin assay was discovered through a HTS of ∼330 600 compounds from NIH’s small molecule collection (Figure 13). The initial hit, MLS-022416479 (52) exhibited an IC50 of 4.8 μM and was selective over the AT1 receptor. SAR focused on the terminal phenyl group generated the most potent 4-nitro analog 53; however, the SAR appeared somewhat flat. Compound 53 was selective over range of GPCR receptors except κ opioid and the benzodiazepine receptors. Compound 53 also had poor aqueous solubility (pH 7.4) and exhibited moderate permeability (271 × 10−6 cm/s) in a PAMPA assay at pH 7.4. As expected, because of the presence of a hydrolyzable ester linkage, 53 has poor microsomal stability in both human and mouse liver homogenates which currently limits its utility for vivo studies.79
Figure 13.
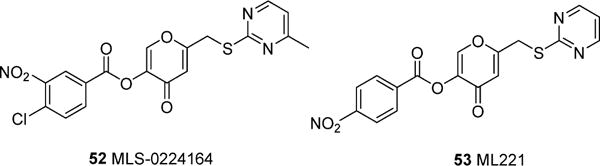
Apelin receptor small molecule antagonists.
FUTURE DIRECTIONS
The apelinergic system appears to be important within the context of both health and disease. While cardiovascular indications have emerged as an area of particular emphasis, regulation of apelin receptor could be beneficial in the treatment of a host of other disorders including diabetes, obesity, stroke, preeclampsia, pulmonary hypertension, and others. However, like many emerging targets, there are important questions that must be answered before apelin receptor is established as an attractive target for medications development. One of the most pressing questions relates to expression of the receptor in various tissues. While immunohistochemical studies and studies in transgenic animals have provided some answers, development of high-quality antibodies coupled with additional well-controlled studies beyond the few that have been reported to date in human tissue samples is urgently needed to address controversies in the field. A second aspect relates to the relative importance of apelin versus TODDLER/ELABELA peptides and their actions on apelin receptor under various physiological and pathophysiological conditions. In spite of these unanswered questions, studies in humans and data from knockout animal models suggest that apelin receptor is poised to become an important target for medications development in the years ahead.
Apelin receptor as a target for cardiovascular disease is being validated with the endogenous agonists apelin-13 and apelin-36 but also with a number of chemically modified analogs. SAR and computational studies on apelin peptides have now been performed to indicate turn characteristics critical for recognition of apelin peptides by the apelin receptor. Mounting evidence from studies with cyclic apelin peptides point to hairpin-like shapes as the most likely bioactive conformations. The importance of individual amino acid side chains on both full length and truncated peptides has been investigated, and strides toward developing more metabolically resistant analogs have been taken. The recent emergence of small molecule agonists and low potency antagonists now provides new molecular templates to expand our understanding of requirements for functional activity at the apelin receptor. What remains lacking is a thorough understanding of the overlap between small molecule and peptide SAR with explicit reference to the probable modes of receptor interaction and the degree to which there may be complementarity between the two. Continued research and resolution of some of these questions will likely enhance our ability to more rapidly bridge the gap from peptides to additional druglike entities for the potential treatment of a number of apelin receptor related disorders.
Acknowledgments
R.M. and S.P.R. are partially supported under Grant R01HD079547-01A1 from the National Institutes of Health.
ABBREVIATIONS
- AT1
angiotensin 2 receptor 1
- PKA
protein kinase A
- PCSK3
proprotein convertase subtilisin kexin 3
- ACE2
angiotensin converting enzyme 2
- CHO
Chinese hamster ovary
- cAMP
cyclic adenosine monophosphate
- ELA
ELABELA
- TR-FRET
time-resolved fluorescence resonance energy transfer
- CNS
central nervous system
- mRNA
messenger ribonucleic acid
- PVN
paraventricular nuclei
- SON
supraoptic nuclei
- CD31
cluster of differentiation 31
- SD
Sprague Dawley
- PVOD
pulmonary veno-occlusive disorder
- DHV
dorsal hand vein
- CSF
cerebrospinal fluid
- cDNA
complementary deoxyribonucleic acid
- HIF1α
hypoxia-inducible factor 1α
- PEG
polyethylene glycol
- MI
myocardial infarction
- Ang 2
angiotensin 2
- MABP
mean arterial blood pressure
- FLIPR
fluorescence imaging plate reader
- GFP
green fluorescent protein
- SAR
structure–activity relationship
- NMR
nuclear magnetic resonance
- HSQC
heteronuclear single quantum coherence correlation
- TOCSY
total correlation spectroscopy
- NOESY
nuclear Overhauser effect spectroscopy
- MD
molecular dynamics
- CD
circular dichroism
- rmsd
root-mean-square deviation
- NAMD
nanoscale molecular dynamics
- FRET
fluorescence resonance energy transfer based assay
- NIH
National Institutes of Health
- NET
norepinephrine transporter
- IR
ischemia/reperfusion
- K
kelvin
Biographies
Sanju Narayanan is a medicinal chemist in the Center for Drug Discovery at RTI International. He received his Ph.D. in 2009 from University of Mississippi under the supervision of Professor Christopher McCurdy and held postdoctoral positions working in lead optimization of GPCRs at Arena Pharmaceuticals Inc., San Diego, and RTI International. Prior to obtaining his Ph.D., he worked as a chemist in Medicinal Chemistry Center at Dr. Reddy’s Laboratories Ltd., India (2001–2004) and received his Masters in Medicinal Chemistry from National Institute of Pharmaceutical Education and Research (NIPER), India, in 2000.
Danni L. Harris received his Ph.D. in Physical Chemistry from Purdue University in 1983, working on computational (Monte Carlo/QM) and spectroscopic studies of solvent mediation of ionic association and reactions in aprotic solvents. He did postdoctoral study at the University of Oregon, probing the effects of mutations on active-site dynamics via picosecond time-resolved fluorescence spectroscopy and molecular dynamics. His work at the Molecular Research Institute and Moltech Corporation involved molecular dynamics/QM and QM/MM predictions of structure/spectra/function relationships, developing new structure based approaches to predict P450 metabolism employing configuration sampling approaches and transition state metrics. His work at RTI includes molecular design of GPCR and ion-channel ligands, dynamics studies probing biased signaling, and development of palliatives for autism spectrum disorder and pain.
Rangan Maitra, Ph.D., conducts preclinical drug discovery research related to metabolic syndrome and liver disease. His work in pharmacology has included studying the mechanisms of chemotherapy-induced differential regulation of ABC transporters CFTR and P-glycoprotein as well as preclinical drug discovery research related to cystic fibrosis, cannabinoid receptors, apelin receptor, and alcohol-induced liver disease. Previous to his work at RTI, Dr. Maitra was employed at Paradigm Genetics as Group Leader of Experimental Biology. He received a B.S. in Chemistry from Angelo State University with honors and Ph.D. in Pharmacology and Toxicology from Dartmouth Medical School.
Scott P. Runyon is the Director of the Center for Drug Discovery at RTI International. He earned his B.S. in Chemistry from Muhlenberg College in 1994 and his Ph.D. in Medicinal Chemistry from the Medical College of Virginia under Drs. Richard Westkaemper and Richard Glennon in 2001. He did his postdoctoral training at RTI under the direction of Dr. Ivy Carroll and pursued the development of κ opioid antagonists and dopamine transporter inhibitors. His current focus is on the development of small molecule agonists for peptidergic GPCRs including the APJ and NPS receptors.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, Shi X, Petronis A, George SR, Nguyen T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–360. doi: 10.1016/0378-1119(93)90495-o. [DOI] [PubMed] [Google Scholar]
- 2.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–476. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 3.Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, Kitada C, Honda S, Kurokawa T, Onda H, Nishimura O, Fujino M. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem. 2000;275:21061–21067. doi: 10.1074/jbc.M908417199. [DOI] [PubMed] [Google Scholar]
- 4.Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP. [Pyr(1)]Apelin-13 identified as the predominant apelin isoform in the human heart vasoactive mechanisms and inotropic action in disease. Hypertension. 2009;54:598–604. doi: 10.1161/HYPERTENSIONAHA.109.134619. [DOI] [PubMed] [Google Scholar]
- 5.Shin K, Pandey A, Liu XQ, Anini Y, Rainey JK. Preferential apelin-13 production by the proprotein convertase PCSK3 is implicated in obesity. FEBS Open Bio. 2013;3:328–333. doi: 10.1016/j.fob.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langelaan DN, Bebbington EM, Reddy T, Rainey JK. Structural insight into G-protein coupled receptor binding by apelin. Biochemistry. 2009;48:537–548. doi: 10.1021/bi801864b. [DOI] [PubMed] [Google Scholar]
- 7.Azizi M, Iturrioz X, Blanchard A, Peyrard S, De Mota N, Chartrel N, Vaudry H, Corvol P, Llorens-Cortes C. Reciprocal regulation of plasma apelin and vasopressin by osmotic stimuli. J Am Soc Nephrol. 2008;19:1015–1024. doi: 10.1681/ASN.2007070816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Mota N, Goazigo ARL, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos FO, Llorens-Cortes C. Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci USA. 2004;101:10464–10469. doi: 10.1073/pnas.0403518101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habata Y, Fujii R, Hosoya M, Fukusumi S, Kawamata Y, Hinuma S, Kitada C, Nishizawa N, Murosaki S, Kurokawa T, Onda H, Tatemoto K, Fujino M. Apelin, the natural ligand of the orphan receptor APJ, is abundantly secreted in the colostrum. Biochim Biophys Acta. 1999;1452:25–35. doi: 10.1016/s0167-4889(99)00114-7. [DOI] [PubMed] [Google Scholar]
- 10.Murza A, Parent A, Besserer-Offroy E, Tremblay H, Karadereye F, Beaudet N, Leduc R, Sarret P, Marsault E. Elucidation of the structure-activity relationships of apelin: influence of unnatural amino acids on binding, signaling, and plasma stability. ChemMedChem. 2012;7:318–325. doi: 10.1002/cmdc.201100492. [DOI] [PubMed] [Google Scholar]
- 11.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 12.El Messari S, Iturrioz X, Fassot C, De Mota N, Roesch D, Llorens-Cortes C. Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J Neurochem. 2004;90:1290–1301. doi: 10.1111/j.1471-4159.2004.02591.x. [DOI] [PubMed] [Google Scholar]
- 13.Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF. Toddler: An embryonic signal that promotes cell movement via apelin receptors. Science. 2014;343:1248636. doi: 10.1126/science.1248636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chng SC, Ho L, Tian J, Reversade B. ELABELA: A hormone essential for heart development signals via the apelin receptor. Dev Cell. 2013;27:672–680. doi: 10.1016/j.devcel.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Laflamme B. ELABELA, a peptide hormone for heart development. Nat Genet. 2014;46:7. [Google Scholar]
- 16.Wang Z, Yu DZ, Wang MQ, Wang QL, Kouznetsova J, Yang RZ, Qian K, Wu WJ, Shuldiner A, Sztalryd C, Zou MH, Zheng W, Gong DW. Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci Rep. 2015;5:8170. doi: 10.1038/srep08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giddings A, Runyon S, Thomas J, Tajuba J, Bortoff K, Maitra R. Development of a functional HTS assay for the APJ receptor. Int J High Throughput Screening. 2010;1:39–47. [Google Scholar]
- 18.Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H, Pikkarainen S, Piuhola J, Rysa J, Toth M, Ruskoaho H. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91:434–440. doi: 10.1161/01.res.0000033522.37861.69. [DOI] [PubMed] [Google Scholar]
- 19.Lee DK, Jeong JH, Oh S, Jo YH. Apelin-13 enhances arcuate POMC neuron activity via inhibiting M-current. PLoS One. 2015;10:e0119457. doi: 10.1371/journal.pone.0119457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brailoiu GC, Dun SL, Yang J, Ohsawa M, Chang JK, Dun NJ. Apelin-immunoreactivity in the rat hypothalamus and pituitary. Neurosci Lett. 2002;327:193–197. doi: 10.1016/s0304-3940(02)00411-1. [DOI] [PubMed] [Google Scholar]
- 21.Reaux A, Gallatz K, Palkovits M, Llorens-Cortes C. Distribution of apelin-synthesizing neurons in the adult rat brain. Neuroscience. 2002;113:653–662. doi: 10.1016/s0306-4522(02)00192-6. [DOI] [PubMed] [Google Scholar]
- 22.Medhurst AD, Jennings CA, Robbins MJ, Davis RP, Ellis C, Winborn KY, Lawrie KW, Hervieu G, Riley G, Bolaky JE, Herrity NC, Murdock P, Darker JG. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem. 2003;84:1162–1172. doi: 10.1046/j.1471-4159.2003.01587.x. [DOI] [PubMed] [Google Scholar]
- 23.De Mota N, Lenkei Z, Llorens-Cortes C. Cloning, pharmacological characterization and brain distribution of the rat apelin receptor. Neuroendocrinology. 2000;72:400–407. doi: 10.1159/000054609. [DOI] [PubMed] [Google Scholar]
- 24.Lee DK, Cheng R, Nguyen T, Fan T, Kariyawasam AP, Liu Y, Osmond DH, George SR, O’Dowd BF. Characterization of apelin, the ligand for the APJ receptor. J Neurochem. 2000;74:34–41. doi: 10.1046/j.1471-4159.2000.0740034.x. [DOI] [PubMed] [Google Scholar]
- 25.Reaux-Le Goazigo A, Morinville A, Burlet A, Llorens-Cortes C, Beaudet A. Dehydration-induced cross-regulation of apelin and vasopressin immunoreactivity levels in magnocellular hypothalamic neurons. Endocrinology. 2004;145:4392–4400. doi: 10.1210/en.2004-0384. [DOI] [PubMed] [Google Scholar]
- 26.De Mota N, Reaux-Le Goazigo A, El Messari S, Chartrel N, Roesch D, Dujardin C, Kordon C, Vaudry H, Moos F, Llorens-Cortes C. Apelin, a potent diuretic neuropeptide counteracting vasopressin actions through inhibition of vasopressin neuron activity and vasopressin release. Proc Natl Acad Sci USA. 2004;101:10464–10469. doi: 10.1073/pnas.0403518101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheikh AY, Chun HJ, Glassford AJ, Kundu RK, Kutschka I, Ardigo D, Hendry SL, Wagner RA, Chen MM, Ali ZA, Yue P, Huynh DT, Connolly AJ, Pelletier MP, Tsao PS, Robbins RC, Quertermous T. In vivo genetic profiling and cellular localization of apelin reveals a hypoxia-sensitive, endothelial-centered pathway activated in ischemic heart failure. Am J Physiol: Heart Circ Physiol. 2008;294:H88–98. doi: 10.1152/ajpheart.00935.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinz MJ, Davenport AP. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul Pept. 2004;118:119–125. doi: 10.1016/j.regpep.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Reaux-Le Goazigo A, Alvear-Perez R, Zizzari P, Epelbaum J, Bluet-Pajot MT, Llorens-Cortes C. Cellular localization of apelin and its receptor in the anterior pituitary: evidence for a direct stimulatory action of apelin on ACTH release. Am J Physiol: Endocrinol Metab. 2007;292:E7–E15. doi: 10.1152/ajpendo.00521.2005. [DOI] [PubMed] [Google Scholar]
- 30.Yokomori H, Oda M, Yoshimura K, Machida S, Kaneko F, Hibi T. Overexpression of apelin receptor (APJ/AGTRL1) on hepatic stellate cells and sinusoidal angiogenesis in human cirrhotic liver. J Gastroenterol. 2011;46:222–231. doi: 10.1007/s00535-010-0296-3. [DOI] [PubMed] [Google Scholar]
- 31.Saint-Geniez M, Argence CB, Knibiehler B, Audigier Y. The msr/apj gene encoding the apelin receptor is an early and specific marker of the venous phenotype in the retinal vasculature. Gene Expression Patterns. 2003;3:467–472. doi: 10.1016/s1567-133x(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 32.Kasai A, Shintani N, Oda M, Kakuda M, Hashimoto H, Matsuda T, Hinuma S, Baba A. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem Biophys Res Commun. 2004;325:395–400. doi: 10.1016/j.bbrc.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 33.Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Ther. 2005;107:198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Cobellis L, De Falco M, Mastrogiacomo A, Giraldi D, Dattilo D, Scaffa C, Colacurci N, De Luca A. Modulation of apelin and APJ receptor in normal and preeclampsia-complicated placentas. Histol Histopathol. 2007;22:1–8. doi: 10.14670/HH-22.1. [DOI] [PubMed] [Google Scholar]
- 35.Eyries M, Siegfried G, Ciumas M, Montagne K, Agrapart M, Lebrin F, Soubrier F. Hypoxia-induced apelin expression regulates endothelial cell proliferation and regenerative angiogenesis. Circ Res. 2008;103:432–440. doi: 10.1161/CIRCRESAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 36.Cox CM, D’Agostino SL, Miller MK, Heimark RL, Krieg PA. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev Biol. 2006;296:177–189. doi: 10.1016/j.ydbio.2006.04.452. [DOI] [PubMed] [Google Scholar]
- 37.Del Toro R, Prahst C, Mathivet T, Siegfried G, Kaminker JS, Larrivee B, Breant C, Duarte A, Takakura N, Fukamizu A, Penninger J, Eichmann A. Identification and functional analysis of endothelial tip cell-enriched genes. Blood. 2010;116(19):4025–4033. doi: 10.1182/blood-2010-02-270819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puffer BA, Sharron M, Coughlan CM, Baribaud F, McManus CM, Lee B, David J, Price K, Horuk R, Tsang M, Doms RW. Expression and coreceptor function of APJ for primate immunodeficiency viruses. Virology. 2000;276:435–444. doi: 10.1006/viro.2000.0557. [DOI] [PubMed] [Google Scholar]
- 39.Pope GR, Roberts EM, Lolait SJ, O’Carroll AM. Central and peripheral apelin receptor distribution in the mouse: species differences with rat. Peptides. 2012;33:139–148. doi: 10.1016/j.peptides.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lathen C, Zhang Y, Chow J, Singh M, Lin G, Nigam V, Ashraf YA, Yuan JX, Robbins IM, Thistlethwaite PA. ERG-APLNR axis controls pulmonary venule endothelial proliferation in pulmonary veno-occlusive disease. Circulation. 2014;130:1179–1191. doi: 10.1161/CIRCULATIONAHA.113.007822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V, Zamanian RT, Quertermous T, Chun HJ. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler, Thromb, Vasc Biol. 2011;31:814–820. doi: 10.1161/ATVBAHA.110.219980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melgar-Lesmes P, Pauta M, Reichenbach V, Casals G, Ros J, Bataller R, Morales-Ruiz M, Jimenez W. Hypoxia and proinflammatory factors upregulate apelin receptor expression in human stellate cells and hepatocytes. Gut. 2011;60:1404–1411. doi: 10.1136/gut.2010.234690. [DOI] [PubMed] [Google Scholar]
- 43.Ronkainen VP, Ronkainen JJ, Hanninen SL, Leskinen H, Ruas JL, Pereira T, Poellinger L, Vuolteenaho O, Tavi P. Hypoxia inducible factor regulates the cardiac expression and secretion of apelin. FASEB J. 2007;21:1821–1830. doi: 10.1096/fj.06-7294com. [DOI] [PubMed] [Google Scholar]
- 44.Kalea AZ, Batlle D. Apelin and ACE2 in cardiovascular disease. Curr Opin Invest Drugs. 2010;11:273–282. [PubMed] [Google Scholar]
- 45.Falcao-Pires I, Goncalves N, Gavina C, Pinho S, Teixeira T, Moura C, Amorim MJ, Pinho P, Areias JC, Leite-Moreira A. Correlation between plasma levels of apelin and myocardial hypertrophy in rats and humans: possible target for treatment? Expert Opin Ther Targets. 2010;14:231–241. doi: 10.1517/14728220903485685. [DOI] [PubMed] [Google Scholar]
- 46.Barnes G, Japp AG, Newby DE. Translational promise of the apelin—APJ system. Heart. 2010;96:1011–1016. doi: 10.1136/hrt.2009.191122. [DOI] [PubMed] [Google Scholar]
- 47.Japp AG, Cruden NL, Amer DA, Li VK, Goudie EB, Johnston NR, Sharma S, Neilson I, Webb DJ, Megson IL, Flapan AD, Newby DE. Vascular effects of apelin in vivo in man. J Am Coll Cardiol. 2008;52:908–913. doi: 10.1016/j.jacc.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 48.Feng JH, Li WM, Wu XP, Tan XY, Gao YH, Han CL, Li SQ, Xie HN. Hemodynamic effect of apelin in a canine model of acute pulmonary thromboembolism. Peptides. 2010;31:1772–1778. doi: 10.1016/j.peptides.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Jia ZQ, Hou L, Leger A, Wu I, Kudej AB, Stefano J, Jiang C, Pan CQ, Akita GY. Cardiovascular effects of a PEGylated apelin. Peptides. 2012;38:181–188. doi: 10.1016/j.peptides.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Reaux A, De Mota N, Skultetyova I, Lenkei Z, El Messari S, Gallatz K, Corvol P, Palkovits M, Llorens-Cortes C. Physiological role of a novel neuropeptide, apelin, and its receptor in the rat brain. J Neurochem. 2001;77:1085–1096. doi: 10.1046/j.1471-4159.2001.00320.x. [DOI] [PubMed] [Google Scholar]
- 51.Llorens-Cortes C, Kordon C. Jacques Benoit lecture: the neuroendocrine view of the angiotensin and apelin systems. J Neuroendocrinol. 2008;20:279–289. doi: 10.1111/j.1365-2826.2007.01642.x. [DOI] [PubMed] [Google Scholar]
- 52.Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang JM, Woods CE, Purcell NH, Catalucci D, Akasaka T, Bueno OF, Vlasuk GP, Kaliman P, Bodmer R, Smith LH, Ashley E, Mercola M, Brown JH, Ruiz-Lozano P. APJ acts as a dual receptor in cardiac hypertrophy. Nature. 2012;488:394–398. doi: 10.1038/nature11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY, Finsterbach TP, Leeper NJ, Ernst KV, Chen MM, Ho YD, Chun HJ, Bernstein D, Ashley EA, Quertermous T. Endogenous regulation of cardiovascular function by apelin-APJ. Am J Physiol: Heart Circ Physiol. 2009;297:H1904–H1913. doi: 10.1152/ajpheart.00686.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y, Leschnik M, Leibbrandt A, Makovic M, Schwaighofer J, Beetz N, Musialek R, Neely GG, Komnenovic V, Kolm U, Metzler B, Ricci R, Hara H, Meixner A, Nghiem M, Chen X, Dawood F, Wong KM, Sarao R, Cukerman E, Kimura A, Hein L, Thalhammer J, Liu PP, Penninger JM. Impaired heart contractility in apelin gene-deficient mice associated with aging and pressure overload. Circ Res. 2007;101:E32–E42. doi: 10.1161/CIRCRESAHA.107.158659. [DOI] [PubMed] [Google Scholar]
- 55.Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, Sugaya T, Matsuzaki H, Yamamoto R, Shiota N, Okunishi H, Kihara M, Umemura S, Sugiyama F, Yagami K, Kasuya Y, Mochizuki N, Fukamizu A. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem. 2004;279:26274–26279. doi: 10.1074/jbc.M404149200. [DOI] [PubMed] [Google Scholar]
- 56.Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, Johnston NR, Denvir MA, Megson IL, Flapan AD, Newby DE. Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation. 2010;121:1818–1827. doi: 10.1161/CIRCULATIONAHA.109.911339. [DOI] [PubMed] [Google Scholar]
- 57.Barnes GD, Alam S, Carter G, Pedersen CM, Lee KM, Hubbard TJ, Veitch S, Jeong H, White A, Cruden NL, Huson L, Japp AG, Newby DE. Sustained cardiovascular actions of APJ agonism during renin-angiotensin system activation and in patients with heart failure. Circ: Heart Failure. 2013;6:482–491. doi: 10.1161/CIRCHEARTFAILURE.111.000077. [DOI] [PubMed] [Google Scholar]
- 58.Holder JR, Swaminath G, Miranda LP, Aral J, Doherty EM, Nixey T. APJ receptor agonist analogs having increased stability relative to wild type apelin-13 and therapeutic uses thereof. WO2014099984. 2014;A1:1–245. [Google Scholar]
- 59.Murza A, Belleville K, Longpre JM, Sarret P, Marsault E. Stability and degradation patterns of chemically modified analogs of apelin-13 in plasma and cerebrospinal fluid. Biopolymers. 2014;102:297–303. doi: 10.1002/bip.22498. [DOI] [PubMed] [Google Scholar]
- 60.Jia ZQ, Hou L, Leger A, Wu I, Kudej AB, Stefano J, Jiang C, Pan CQ, Akita GY. Cardiovascular effects of a PEGylated apelin. Peptides. 2012;38:181–188. doi: 10.1016/j.peptides.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 61.Fan X, Zhou N, Zhang X, Mukhtar M, Lu Z, Fang J, DuBois GC, Pomerantz RJ. Structural and functional study of the apelin-13 peptide, an endogenous ligand of the HIV-1 coreceptor, APJ. Biochemistry. 2003;42:10163–10168. doi: 10.1021/bi030049s. [DOI] [PubMed] [Google Scholar]
- 62.Pisarenko OI, Serebryakova LI, Studneva IM, Pelogeykina YA, Tskitishvili OV, Bespalova ZD, Sidorova MV, Az’muko AA, Khatri DN, Pal’keeva ME, Molokoedov AS. Effects of structural analogues of apelin-12 in acute myocardial infarction in rats. J Pharmacol Pharmacother. 2013;4:198–203. doi: 10.4103/0976-500X.114600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pisarenko OI, Lankin VZ, Konovalova GG, Serebryakova LI, Shulzhenko VS, Timoshin AA, Tskitishvili OV, Pelogeykina YA, Studneva IM. Apelin-12 and its structural analog enhance antioxidant defense in experimental myocardial ischemia and reperfusion. Mol Cell Biochem. 2014;391:241–250. doi: 10.1007/s11010-014-2008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sidorova MV, Az’muko AA, Pal’keeva ME, Molokoedov AS, Bushuev VN, Dvoryantsev SN, Shulzhenko VS, Pelogeykina YA, Pisarenko OI, Bespalova ZD. Synthesis and cardioprotective properties of apelin-12 and its structural analogues. Russ J Bioorg Chem. 2012;38:30–40. doi: 10.1134/s1068162012010177. [DOI] [PubMed] [Google Scholar]
- 65.Zhang YY, Maitra R, Harris DL, Dhungana S, Snyder R, Runyon SP. Identifying structural determinants of potency for analogs of apelin-13: Integration of C-terminal truncation with structure-activity. Bioorg Med Chem. 2014;22:2992–2997. doi: 10.1016/j.bmc.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Wang W, McKinnie SMK, Patel VB, Haddad G, Wang ZC, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, Penninger JM, Kassiri Z, Vederas JC, Murray AG, Oudit GY. Loss of apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic apelin analogues. J Am Heart Assoc. 2013;2:1–17. doi: 10.1161/JAHA.113.000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murza A, Besserer-Offroy E, Cote J, Berube P, Longpre JM, Dumaine R, Lesur O, Auger-Messier M, Leduc R, Sarret P, Marsault E. C-terminal modifications of apelin-13 significantly change ligand binding, receptor signaling, and hypotensive action. J Med Chem. 2015;58:2431–2440. doi: 10.1021/jm501916k. [DOI] [PubMed] [Google Scholar]
- 68.Krivov SV, Karplus M. Hidden complexity of free energy surfaces for peptide (protein) folding. Proc Natl Acad Sci USA. 2004;101:14766–14770. doi: 10.1073/pnas.0406234101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamada J, Kimura J, Ishida J, Kohda T, Morishita S, Ichihara S, Fukamizu A. Evaluation of novel cyclic analogues of apelin. Int J Mol Med. 2008;22:547–552. [PubMed] [Google Scholar]
- 70.Macaluso NJ, Glen RC. Exploring the “RPRL” motif of apelin-13 through molecular simulation and biological evaluation of cyclic peptide analogues. ChemMedChem. 2010;5:1247–1253. doi: 10.1002/cmdc.201000061. [DOI] [PubMed] [Google Scholar]
- 71.Gerbier R, Leroux V, Couvineau P, Alvear-Perez R, Maigret B, Llorens-Cortes C, Iturrioz X. New structural insights into the apelin receptor: identification of key residues for apelin binding. FASEB J. 2015;29:314–322. doi: 10.1096/fj.14-256339. [DOI] [PubMed] [Google Scholar]
- 72.Iturrioz X, Alvear-Perez R, De Mota N, Franchet C, Guillier F, Leroux V, Dabire H, Le Jouan M, Chabane H, Gerbier R, Bonnet D, Berdeaux A, Maigret B, Galzi JL, Hibert M, Llorens-Cortes C. Identification and pharmacological properties of E339-3D6, the first nonpeptidic apelin receptor agonist. FASEB J. 2010;24:1506–1517. doi: 10.1096/fj.09-140715. [DOI] [PubMed] [Google Scholar]
- 73.Brame AL, Maguire JJ, Yang P, Dyson A, Torella R, Cheriyan J, Singer M, Glen RC, Wilkinson IB, Davenport AP. Design, characterization, and first-in-human study of the vascular actions of a novel biased apelin receptor agonist. Hypertension. 2015;65:834–840. doi: 10.1161/HYPERTENSIONAHA.114.05099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zecri F, Golosov A, Grosche P, Yasoshima K, Zhao H, Hu QY, Imase H, Parker DT. US20130196899. A1. Novartis AG; 2013. Synthetic apelin mimetics for the treatment of heart failure; pp. 1–50. [Google Scholar]
- 75.Macaluso NJ, Pitkin SL, Maguire JJ, Davenport AP, Glen RC. Discovery of a competitive apelin receptor (APJ) antagonist. ChemMedChem. 2011;6:1017–1023. doi: 10.1002/cmdc.201100069. [DOI] [PubMed] [Google Scholar]
- 76.Margathe JF, Iturrioz X, Alvear-Perez R, Marsol C, Riche S, Chabane H, Tounsi N, Kuhry M, Heissler D, Hibert M, Llorens-Cortes C, Bonnet D. Structure activity relationship studies toward the discovery of selective apelin receptor agonists. J Med Chem. 2014;57:2908–2919. doi: 10.1021/jm401789v. [DOI] [PubMed] [Google Scholar]
- 77.Khan P, Maloney PR, Hedrick M, Gosalia P, Milewski M, Li L, Roth GP, Sergienko E, Suyama E, Sugarman E, Nguyen K, Mehta A, Vasile S, Su Y, Shi S, Stonich D, Nguyen H, Zeng FY, Novo AM, Vicchiarelli M, Diwan J, Chung TDY, Pinkerton AB, Smith LH. Probe Reports from the NIH Molecular Libraries Program. National Center for Biotechnology Information; Bethesda, MD: 2010. Functional agonists of the apelin (APJ) receptor; pp. 1–9. [PubMed] [Google Scholar]
- 78.Hachtel S, Wohlfart P, Weston J, Mueller M, Defossa E, Mertsch K, Weng JH, Binnie RA, Abdul-Latif F, Bock WJ, Walser A. Benzimidazole carboxylic acid amide derivatives as APJ receptor modulators. WO2014044738. 2014;A1:1–443. Sanofi [Fr] [Google Scholar]
- 79.Maloney PR, Khan P, Hedrick M, Gosalia P, Milewski M, Li LD, Roth GP, Sergienko E, Suyama E, Sugarman E, Nguyen K, Mehta A, Vasile S, Su Y, Stonich D, Nguyen H, Zeng FY, Novo AM, Vicchiarelli M, Diwan J, Chung TDY, Smith LH, Pinkerton AB. Discovery of 4-oxo-6-((pyrimidin-2-ylthio)methyl)-4H-pyran-3-yl 4-nitrobenzoate (ML221) as a functional antagonist of the apelin (APJ) receptor. Bioorg Med Chem Lett. 2012;22:6656–6660. doi: 10.1016/j.bmcl.2012.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]