

Abstract
A complex link between cell-wall recycling/repair and the manifestation of resistance to β-lactam antibiotics in many Enterobacteriaceae and Pseudomonas aeruginosa exists. This process is mediated by specific cell-wall-derived muropeptide products. These muropeptides are internalized into the cytoplasm and bind to the transcriptional regulator AmpR, which controls the cytoplasmic events that lead to expression of β-lactamase, an antibiotic-resistance determinant. The effector-binding domain (EBD) of AmpR was purified to homogeneity. We document that the EBD exists exclusively as a dimer even at a concentration as low as 1 μM. The EBD binds to the suppressor ligand UDP-N-acetyl-β-D-muramyl-L-Ala-γ-D-Glu-meso-DAP-D-Ala-D-Ala (4) and binds to two activator muropeptides, N-acetyl-β-D-glucosamine-(1–4)-1,6-anhydro-N-acetyl-β-D-muramyl-L-Ala-γ-D-Glu-meso-DAP-D-Ala-D-Ala (1c) and 1,6-anhydro-N-acetyl-β-D-muramyl-L-Ala-γ-D-Glu-meso-DAP-D-Ala-D-Ala (2c) as assessed by non-denaturing mass spectrometry. The EBD does not bind to 1,6-anhydro-N-acetyl-β-D-muramyl-L-Ala-γ-D-Glu-meso-DAP (2a). This binding selectivity revises the dogma in the field. The crystal structure of the EBD dimer was solved to 2.2 Å resolution. The EBD crystallizes in a “closed” conformation, in contrast to the “open” structure required to bind the muropeptides. Structural issues of this ligand recognition are addressed by molecular dynamics simulations, which reveal significant differences among the complexes with the effector molecules.
Graphical Abstract
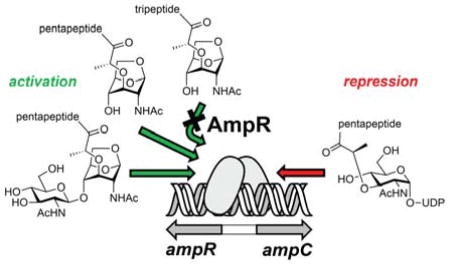
Nature has devised a complex inducible resistance mechanism to β-lactam antibiotics in many Enterobacteriaceae and Pseudomonas aeruginosa.1 Exposure of the bacterium to a β-lactam antibiotic damages its cell wall, the target of action of the β-lactam. The damaged cell wall is fragmented by a series of enzymes in a restoration effort. The products of these reactions are internalized to the cytoplasm by the AmpG permease for the purpose of recycling.2,3 In P. aeruginosa two of these recycling intermediates (compounds 1c and 2c, Figure 1), known collectively as muropeptides, induce β-lactam-antibiotic resistance (production of β-lactamase, the product of the gene ampC; Figure 1).4 The initiation of this response is regulated by AmpR, a member of the LysR-type transcriptional regulators (LTTRs) of Gram-negative bacteria.5
Figure 1.

Gram-negative peptidoglycan recycling and its role in AmpR-regulated AmpC β-lactamase induction is depicted. Cell wall is degraded by lytic transglycosylases (LTs) in the periplasm in a process that involves an oxocarbenium (the structure in the brackets), which entraps the C6 hydroxyl to give the 1,6-anhydromuramyl species.11 Compounds 1 are key muropeptides that are involved in signaling events that lead to induction of the AmpC β-lactamase.
Understanding of transcription factor-DNA regulation in general, and that of AmpR in particular, remains a significant challenge. AmpR is understood to be allosterically regulated by the internalized muropeptides and by the muropeptide recycling intermediates.6 These compounds bind differentially to the regulatory (effector-binding) domain of AmpR, whereby they either block or promote DNA transcription.7 Notwithstanding that the discovery of this pathway took place two decades ago,8 our current understanding of this process, including the nature of the effectors and the conformational states of AmpR, remains rudimentary. Here, we report the crystal structure of the regulatory domain of the AmpR transcription factor of P. aeruginosa, and the application of mass spectrometry for the identification of its effector muropeptides.
AmpR is a 32.6 kDa protein comprised of two domains. One domain is predicted to be a helix-turn-helix DNA-binding domain (DBD, amino acids 1–67; Pfam Family: HTH_1 (PF00126)) and the other is the effector-binding domain (EBD, amino acids 83–296; Pfam Family: LysR_substrate (PF03466)). The DBD binds to the intercistronic region of DNA between the ampR and ampC genes, encoding respectively the AmpR protein itself and the β-lactamase, which have overlapping promoters that transcribe DNA divergently.9,10
We cloned the full-length ampR gene (PA4109) from P. aeruginosa PAO1 in two constructs to give a histidine-tagged protein. One construct would express the protein in the cytoplasm and the other in the periplasm of Escherichia coli. Unfortunately, the protein expressed as insoluble inclusion bodies in both expression systems. The inclusion bodies were solubilized in a denaturant and the protein was purified to homogeneity. However, attempts to remove the denaturant and to refold the protein were unsuccessful. We segued our efforts toward the cloning and purification of the individual AmpR domains. The AmpR DBD failed to express. Its gene was annealed to the gene of three fusion proteins, but only one of the three constructs produced a soluble fusion protein (Supporting Information). Although this AmpR DBD-fusion protein was purified to homogeneity, the DBD precipitated during proteolytic separation of the fusion protein. In contrast, the clone for a His-tagged EBD of AmpR expressed well. The EBD was subsequently purified to homogeneity (Figure S1). Sedimentation velocity analysis showed that the EBD was exclusively dimeric in solution over the concentration range of 1–50 μM (Figure S2). The absence of monomeric protein in this analysis indicates the monomer-dimer KD as <1 μM.
The current hypothesis with respect to effector identity and cell-wall recycling posits that ampC transcription is regulated by occupancy of muropeptides at the effector-binding site of the EBD of AmpR.12 Previous studies demonstrated that the peptidoglycan biosynthetic precursor UDP-MurNAc pentapeptide (compound 4) serves as a transcriptional repressor.8,13 The repression by 4 is interpreted in terms of its elevated concentration indicating the completion of the repair to the cell wall. In contrast, the literature shows debate as to the structure(s) of the activator(s).8,14–16 We identified previously the muropeptides that increase in concentration upon exposure of P. aeruginosa to sub-lethal concentrations of a β-lactam antibiotic.4 Two muropeptides, 1c and 2c, induced resistance at the whole-cell level. The question became, whether or not these two muropeptides would bind to the EBD as their cytoplasmic target. We performed non-denaturing mass spectrometry17 of the EBD with four muropeptides (1c, 2a, and 2c, and 4). An earlier study had proposed compound 2a as an activator muropeptide.8 Compounds 1c, 2a, and 2c were prepared for this study by known multistep syntheses from our laboratory.18,19 Compound 4 is commercial.
Mass spectrometry analysis of the EBD in 1 M ammonium acetate (pH 6.6) in the absence of ligand showed only the dimer mass of 49281.5 ± 0.7 Da (calcd 49280.2 Da; Figure 2A), consistent with the sedimentation velocity analysis. The EBD was incubated individually with each of the four muropeptide samples (30 min at 25 °C) in the same ammonium acetate solution. Analysis of the EBD bound to the repressor (4) showed the free dimer of the EBD, the dimer bound to a single molecule of compound 4, and the dimer bound to two molecules of compound 4 (Figure 2B). Consistent with our observation that 1c and 2c activate the system for the resistance phenotype in whole-cell P. aeruginosa, the EBD binds to both (Figure 2C and 2D). Again, we see the free EBD, the EBD bound to one activator, and the EBD bound to two activators in each case. A determination of dissociation constants from this method was not possible as the supply of the ligands was extremely limited. Nonetheless, as a first approximation we can state that the repressor muropeptide 4 binds with greater affinity than do muropeptides 1c or 2c (Figure S3). This observation is intuitively reasonable, as the system is repressed until the activator muropeptides (1c and 2c) build up in concentration to displace the repressor muropeptide (4) from the AmpR EBD (discussed below). Muropeptide 2a was previously claimed to be the activator, and this identification has been the dogma in the literature for two decades.8 By this same mass spectrometry method no evidence is seen for 2a binding to the EBD (Figure 2E).
Figure 2.

Representative extracted-ion chromatogram (+14 charge state) of non-denaturing mass spectrometry displaying the AmpR EBD as an intact dimer and in complexes with muropeptides. The spikes in the signals represent multiple adducts of ammonium acetate from the matrix to the various species. (A) AmpR EBD (20 μM) alone. (B) AmpR EBD (20 μM) + 4 (500 μM). (C) AmpR EBD (20 μM) + 1c (500 μM). (D) AmpR EBD (20 μM) + 2c (1 mM). (E) AmpR EBD (20 μM) + 2a (1 mM). Refer to Figure S3 for additional substrate concentrations.
We turned our attention to the EBD structure. The AmpR EBD (12 mg/mL) was crystallized by the sitting-drop vapor-diffusion method at 17 °C. Its structure at 2.2 Å resolution (PDB code: 5MMH; Table S1 and Figure S4) showed four monomers in the asymmetric unit. The four dimers are symmetry-related in the crystal, with all binding sites oriented on the same side of the quaternary structure. In congruence with the other available structures of the LTTR family, the EBD of AmpR is a homodimer. Each monomer is comprised of two subdomains that assume α/β Rossmann-like folds, linked by a hinge. Two effector-binding sites are found in the dimer. Our structure for the EBD of P. aeruginosa is shown in Figure 3A superimposed with the structure of the Citrobacter freundii EBD.13 The structure for the C. freundii EBD was in complex with muropeptide 4 (PDB code: 4WKM). In this structure the N-acetylmuramic acid and pentapeptide stem segments are seen, indicating that the rest of 4 was mobile. Our structure of the P. aeruginosa EBD (PDB code: 5MMH) revealed a closed protein conformation, as assessed by the positions of Y192 and R133 “gatekeeper” residues bridging the binding site. Their appearance contrasts with the “open” crystal structure of the C. freundii AmpR EBD (Figure 3A). Hence, these two crystal structures demarcate two distinct conformational states for the EBD. Importantly, residue 133 of K. pneumonia and A. baumannii is a histidine and the residues flanking H133 are strictly conserved, suggesting that H133 may serve as the gatekeeper residue in these organisms. The opening motion of Y192/R133 is concurrent with motion of a neighboring loop to enables access of the effector molecules to the effector-binding site (shown by an arrow in Figure 3A). The residues within the peptide-binding cavity and dimer interface are highly conserved in all of the AmpR proteins of the Gram-negative ESKAPE (Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and the Enterobacter species) pathogens (Figure S5).20,21 This conservation implies an universality among these organisms for the structural identity of their effectors. The deep pocket for the peptide in the effector-binding site accommodates the pentapeptide, terminating with a polar anchor—the side chains of Tyr264, Thr103 and Ser221—which hydrogen bonds to the terminal carboxylate. This anchor was first noted by Vadlamani et al. for the C. freundii EBD.12 These three residues are strictly conserved. They are likely the reason that muropeptides 4, 1c, and 2c bind, whereas 2a does not. Repeated attempts at both soaking and co-crystallization of compounds 2c, 4, and N-acetylglucosamine failed to produce crystalline complexes.
Figure 3.

(A) Structure overlay of the P. aeruginosa AmpR EBD monomer in gold (PDB code: 5MMH) with that of C. freundii bound to 4 in green (PDB code: 4WKM; no electron density for UDP was seen). The diaminopimelate (third amino acid in the stem peptide) ion-pairs with the gatekeeper R133 once R133 repositions to the open conformation. Models of P. aeruginosa AmpR EBD dimer bound to (B) 4, (C) 1c, and (D) 2c.
As mentioned previously the C. freundii EBD complex with 4 gave no electron density for the uridine diphosphate (UDP) segment.13 Our molecular dynamics (MD) simulations support the conclusion that this absence is due to conformational flexibility of this segment. We generated computational models for the complexes of compounds 4 (Figure 3B), 1c (Figure 3C), and 2c (Figure 3D) using our structure of the P. aeruginosa EBD. Two 30 ns MD simulations were performed for each complex. In the case of the complex with 4, the segment of the ligand absent in the structure of the C. freundii protein could assume two starting positions. One allowed for interactions of the substituent saccharides across the dimer interface with one another (see Figure 3B), and the other promoted interactions of the saccharides with two potential saccharide-binding pockets at 11 o’clock and at 5 o’clock (shown by arrows in Figure 3B). Neither of the two binding orientations for 4 were maintained throughout the course of the simulations. Indeed, the two interconverted. The disaccharide component of 1c in complex with the EBD (Figure 3C) was also mobile. In the course of the dynamics simulations, the two disaccharides in Figure 3C interacted with one another, but also individually with the surface of the EBD. In contrast, the EBD complex with 2c (Figure 3D)—which is truncated in the saccharide component—could not allow for interactions between the saccharides of each muropeptide. Importantly, in all of the simulations, binding of the pentapeptide stem to the effector-binding site was stable regardless of the saccharide component. This observation implies that the free pentapeptide (compound 3c)—the product of the reaction of cytoplasmic AmpD22 with either compound 1c or 2c and of periplasmic AmpDh223 or AmpDh324 with the cell wall (both of P. aeruginosa)—might also serve as an AmpR effector molecule. In a seminal work, Cho et al. documented that crosslinking of nascent peptidoglycan is inhibited by exposure to β-lactam antibiotics and the peptidoglycan then experiences degradation by an LT to generate the muropeptide products.25 Insofar as nascent peptidoglycan is comprised of the pentapeptide stems, the muropeptide pool will be enriched by the pentapeptide species. This report is consistent with our identification of 1c and 2c as activator muropeptides,4 as well as the results of mass spectrometry presented in the present report.
Jacobs et al. had proposed a model in which the repressor is bound to AmpR in homeostasis, but the activator (of which two are now known) would displace the repressor in promotion of transcription.8 Although they proposed that 2a is the activator of AmpR, we see no evidence of its recognition; either in our quantitative analyses of the muropeptides after exposure of P. aeruginosa to a sub-lethal concentration of a β-lactam antibiotic4 or by our binding assays to the EBD (the present study). Nonetheless, the overall model appears reasonable, based on the collective data, barring the nature of the activator molecule(s). The AmpR transcription factor regulates not only β-lactam resistance, but also quorum sensing, metabolism, stress-response, second-messenger-mediated signaling, and cellular phosphorylation events. The role of AmpR in antibiotic resistance has been studied the most, especially in reference to efflux pumps.26 The present study identifies the muropeptide activators that bind to the P. aeruginosa AmpR EBD and addresses the structural issues for the binding. The full understanding of the conformational issues that would also involve the DBD and its interactions with the DNA must await the future elucidation of the structure of the full-length AmpR.
Supplementary Material
Acknowledgments
This work in the USA was supported by grants from the NIH (GM61629 and T32GM075762) and in Spain by grants from the Spanish Ministry of Economy and Competitiveness (BFU2014-59389-P). The Mass Spectrometry & Proteomics Facility of the University of Notre Dame is supported by grant CHE0741793. We thank the staff from ALBA synchrotron for help in data collection.
Footnotes
No competing financial interests have been declared.
Supporting Information. Experimental procedures, supplementary figures, and tables are given. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lindberg F, Westman L, Normark S. Proc Natl Acad Sci USA. 1985;82:4620–4624. doi: 10.1073/pnas.82.14.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lindquist S, Weston-Hafer K, Schmidt H, Pul C, Korfmann G, Erickson J, Sanders C, Martin HH, Normark S. Mol Microbiol. 1993;9:703–715. doi: 10.1111/j.1365-2958.1993.tb01731.x. [DOI] [PubMed] [Google Scholar]
- 3.Cheng Q, Park JT. J Bacteriol. 2002;184:6434–6436. doi: 10.1128/JB.184.23.6434-6436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee M, Dhar S, De Benedetti S, Hesek D, Boggess B, Blazquez B, Mathee K, Mobashery S. Angew Chem Int Ed. 2016;55:6882–6886. doi: 10.1002/anie.201601693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maddocks SE, Oyston PCF. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 6.Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. EMBO J. 1994;13:4684–4694. doi: 10.1002/j.1460-2075.1994.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher JF, Mobashery S. Bioorg Chem. 2014;56:41–48. doi: 10.1016/j.bioorg.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs C, Frère JM, Normark S. Cell. 1997;88:823–832. doi: 10.1016/s0092-8674(00)81928-5. [DOI] [PubMed] [Google Scholar]
- 9.Lodge J, Busby S, Piddock L. FEMS Microbiol Lett. 1993;111:315–320. doi: 10.1111/j.1574-6968.1993.tb06404.x. [DOI] [PubMed] [Google Scholar]
- 10.Caille O, Zincke D, Merighi M, Balasubramanian D, Kumari H, Kong KF, Silva-Herzog E, Narasimhan G, Schneper L, Lory S, Mathee K. J Bacteriol. 2014;196:3890–3902. doi: 10.1128/JB.01997-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson JW, Fisher JF, Mobashery S. Ann N Y Acad Sci. 2013;1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanson ND, Sanders CC. Curr Pharm Des. 1999;5:881–894. [PubMed] [Google Scholar]
- 13.Vadlamani G, Thomas MD, Patel TR, Donald LJ, Reeve TM, Stetefeld J, Standing KG, Vocadlo DJ, Mark BL. J Biol Chem. 2015;290:2630–2643. doi: 10.1074/jbc.M114.618199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dietz H, Pfeifle D, Wiedemann B. Antimicrob Agents Chemother. 1997;41:2113–2120. doi: 10.1128/aac.41.10.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiedemann B, Dietz H, Pfeifle D. Clin Infect Dis. 1998;27(Suppl 1):S42–S47. doi: 10.1086/514921. [DOI] [PubMed] [Google Scholar]
- 16.Park JT, Uehara T. Microbiol Mol Biol Rev. 2008;72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hernández H, Robinson CV. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 18.Suvorov M, Lee M, Hesek D, Boggess B, Mobashery S. J Am Chem Soc. 2008;130:11878–11879. doi: 10.1021/ja805482b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hesek D, Lee M, Zhang W, Noll BC, Mobashery S. J Am Chem Soc. 2009;131:5187–5193. doi: 10.1021/ja808498m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 21.Rice LB. J Infect Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 22.Lee M, Zhang W, Hesek D, Noll BC, Boggess B, Mobashery S. J Am Chem Soc. 2009;131:8742–8743. doi: 10.1021/ja9025566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Caballero S, Lee M, Artola-Recolons C, Carrasco-López C, Hesek D, Spink E, Lastochkin E, Zhang W, Hellman LM, Boggess B, Mobashery S, Hermoso JA. J Am Chem Soc. 2013;135:10318–10321. doi: 10.1021/ja405464b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M, Artola-Recolons C, Carrasco-López C, Martínez-Caballero S, Hesek D, Spink EE, Lastochkin E, Zhang W, Hellman LM, Boggess B, Hermoso JA, Mobashery S. J Am Chem Soc. 2013;135:12604–12607. doi: 10.1021/ja407445x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho H, Uehara T, Bernhardt TG. Cell. 2014;159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balasubramanian D, Kumari H, Mathee K. Pathog Dis. 2015;73:1–14. doi: 10.1111/2049-632X.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.