

Abstract
Patient: Female, 53
Final Diagnosis: Apical hypertrophic cardiomyopathy
Symptoms: Chest pain • dizziness • palpitations
Medication: —
Clinical Procedure: —
Specialty: Cardiology
Objective:
Rare disease
Background:
Apical hypertrophic cardiomyopathy (ApHCM) is a relatively rare form of hypertrophic cardiomyopathy that predominantly affects the apex of the left ventricle and typically has a nonobstructive physiology. Its variable presentation and clinical course render ApHCM a commonly delayed or missed diagnosis.
Case Report:
A 53-year-old Caucasian woman presented with chronic progressive chest pain. She was initially started on treatment for acute coronary syndrome. Diagnosis of ApHCM was initially missed on echocardiography, but made on subsequent cardiac catheterization and cardiac MRI. She improved clinically with metoprolol, had a work-up for implantable cardioverter-defibrillator placement, and was referred for genetic testing.
Conclusions:
Despite earlier studies suggesting a more benign clinical course of ApHCM, recent studies report increased morbidity and mortality, which is comparable to the prognosis of other variants of hypertrophic cardiomyopathy such as hypertrophic obstructive cardiomyopathy. Thus, when formulating a differential diagnosis for chest pain, it is important to include structural heart disease including apical and other variants of hypertrophic cardiomyopathy as part of that differential, as appropriate management can prevent these devastating sequelae. Furthermore, when screening tests such as echocardiography cannot adequately establish the diagnosis of ApHCM, then cardiac MRI or invasive hemodynamic testing is necessary to establish or refute the diagnosis.
MeSH Keywords: Cardiomyopathies; Cardiomyopathy, Hypertrophic; Magnetic Resonance Imaging
Background
Apical hypertrophic cardiomyopathy (ApHCM) is a relatively rare form of hypertrophic cardiomyopathy that predominantly affects the apex of the left ventricle and typically has a nonobstructive physiology [1]. Its variable presentation and clinical course renders ApHCM a commonly delayed or missed diagnosis. Furthermore, since it was first described in Japan 40 years ago, there have been differing views on its clinical course, with initial studies supporting a more benign prognosis but more recent reports suggesting a cardiovascular morbidity of 25% to 30% and mortality in 4% to 29% of cases [1]. This case illustrates the clinical and diagnostic challenge of ApHCM, and reviews the current literature regarding implications for treatment and prognostic.
Case Report
A fifty-three-year old Caucasian woman presented to the emergency department with chest pain that woke her up at night. Over the past few months, she experienced substernal chest pressure which was initially brought on with exertion, but recently occurred more frequently and occurred with both with activity and rest. Over the past several weeks, these episodes were accompanied by palpitations and lightheadedness. She denied syncope, dyspnea, nausea, vomiting, and diaphoresis. Her past medical history was notable for hypertension, recent stroke, and obesity. Of note, she said she had occasional exertional chest discomfort as a child and was told she had asthma. Her family history was notable for sudden cardiac death in her paternal grandfather. She denied alcohol consumption, tobacco smoking, and illicit drug use.
On admission, the patient was afebrile, her heart rate was 81 beats per minute, respiratory rate was 16 breaths per minute, blood pressure was 111/73 mm Hg, and oxygen saturation was 98% on room air. She was alert and oriented and in no acute distress. She had no notable jugular venous distension. Cardiac examination revealed regular rate and rhythm and normal S1 and S2 with no murmur or extra heart sounds. Her lungs were clear to auscultation bilaterally. No peripheral edema was noted.
The first cardiac troponin-I drawn within 15 minutes of arrival was indeterminate at 0.3 ng/mL (normal reference range <0.04 ng/mL). Electrocardiogram exhibited deep T wave inversions in precordial leads and met criteria for left ventricular hypertrophy (Figure 1). Chest radiograph showed a normal cardiac silhouette and no pulmonary congestion. Given her progressive chest pain, indeterminate troponin, and T wave inversions, she was started on heparin drip, metoprolol 12.5 mg twice daily, and sublingual nitroglycerin 0.4 mg for presumed acute coronary syndrome, and continued on her home aspirin, atorvastatin, and losartan. Cardiology was consulted and recommended echocardiography. Transthoracic echocardiography demonstrated preserved ejection fraction (55% to 60%), grade 2 diastolic dysfunction (mild restrictive pattern), moderately dilated left atrium (63 mL volume), mildly elevated pulmonary artery systolic pressure (35 mm Hg), and possible left ventricular aneurysm. Right atrial size and pressure, and right ventricular systolic function were within normal limits. There was no evidence of wall motion abnormalities, dynamic left ventricular outflow tract obstruction, or valvular disease. Given the patient’s large body habitus and technically difficult images, the left ventricular apex was not well visualized and definitive diagnosis could not be established.
Figure 1.
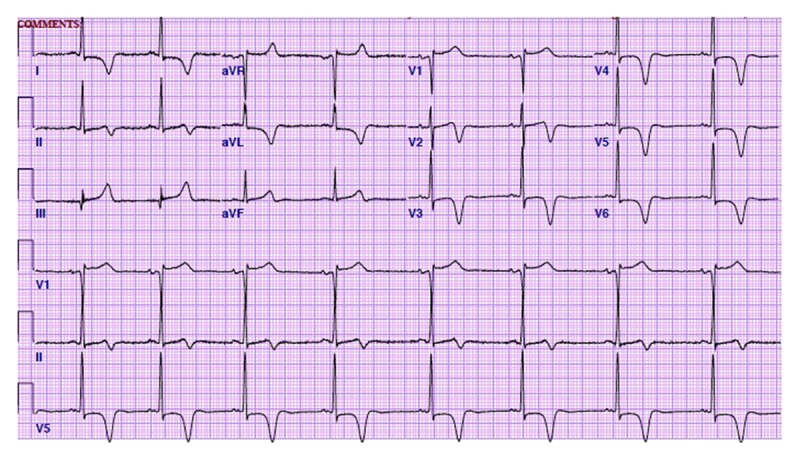
Electrocardiogram on admission exhibited deep T wave inversions in precordial leads (V2–V6) and met criteria for left ventricular hypertrophy.
To further evaluate for ischemic etiology of the patient’s chest pain, cardiac angiography was performed. Left heart catheterization demonstrated no significant epicardial coronary artery disease. Left ventriculogram was suggestive of a spade-like appearance (Figure 2); however, the interpreting cardiologist did not comment on this until later. During admission, our patient was noted to have several pre-syncopal episodes with non-sustained ventricular tachycardia (NSVT), the longest run lasting 25 beats. The second and third troponins were negative at 0.02 ng/mL.
Figure 2.
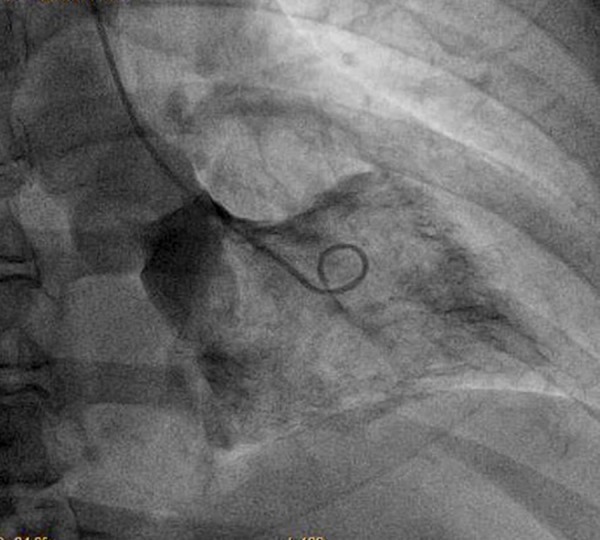
Left ventriculogram depicts a spade-shape silhouette of the left ventricle.
Given the patient’s symptoms, deep T wave inversions on electrocardiogram, symptomatic ventricular tachycardia episodes, family history of sudden cardiac death, and faint spade-shape silhouette on LV gram, cardiology recommended further cardiac imaging to evaluate for structural etiology of the patient’s chest pain and palpitations. Cardiac MRI with gadolinium enhancement demonstrated a spade-shape configuration of the left ventricle with apical hypertrophy (the thickest segment measuring 3 cm) and ventricular ectopy (Figure 3). LV aneurysm was not seen on cardiac MRI. These findings are consistent with apical hypertrophic cardiomyopathy.
Figure 3.
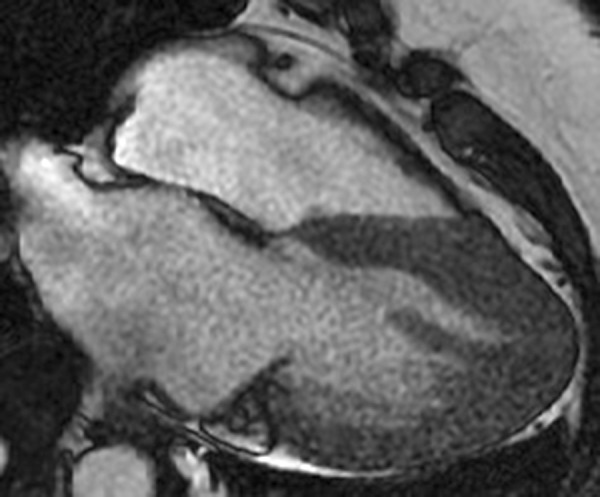
Cardiac MRI with gadolinium enhancement demonstrates a spade-like silhouette of the left ventricle with focal apical hypertrophy, confirming the diagnosis of apical hypertrophic cardiomyopathy.
Hospital course was complicated by uptrending creatinine (peaked at 1.8 mg/dL) following cardiac catheterization, which was thought to be secondary to contrast-induced nephropathy. Nephrology was consulted and recommended supportive management. Creatinine was 1.3 mg/dL on discharge. The patient was chest-pain free and ambulating without symptoms prior to discharge.
Our patient was discharged on metoprolol to prevent ventricular arrhythmias and chest pain, had a work-up for implantable cardioverter defibrillator (ICD) placement for primary prevention of sudden cardiac death, and was referred for genetic testing. Given the genetic predisposition of ApHCM, the patient’s family members were also encouraged to undergo genetic testing and echocardiography screening. At 6-month follow-up, the patient remained symptom free. Though she had not yet undergone confirmatory genetic testing, the presence of apex-predominant LV hypertrophy with a spade-like silhouette of the LV chamber, symptomatic ventricular arrhythmias, family history of sudden cardiac death, and her overall clinical presentation, strongly suggested ApHCM as the etiology of her clinical presentation.
Discussion
ApHCM typically has an autosomal dominant inheritance pattern but can also be sporadic. Recent studies suggest the association of ApHCM with genetic mutations in ACTC1, TPM1, MYBPC3 and MYH7, and current studies are underway to identify other contributory genes [1]. It is characterized by hypertrophy of the left ventricular apex, deep T wave inversions on electrocardiogram, and an ace-of-spades silhouette of the left ventricular chamber [1]. Phenotypic variants of ApHCM include apex-predominant hypertrophy, mid-ventricular obstruction with cavity obliteration, and left ventricular aneurysm.
The heterogeneity of its presentation and variability of its clinical course render apical hypertrophic cardiomyopathy a commonly missed or delayed diagnosis [1], as in our patient’s case. About 54% of patients are symptomatic. Symptoms include chest pain, palpitations, dyspnea, lightheadedness, and syncope [1,2]. Our patient’s chronic intermittent chest discomfort may have been an early manifestation of ApHCM. In the absence of epicardial coronary artery disease, the chest pain of ApHCM is caused by myocardial ischemia due to narrowing of intramural small vessel coronary arteries from the hypertrophied myocardium as well as increased myocardial oxygen demand from myocyte hypertrophy [3]. Ventricular arrhythmias, as seen in our patient, can be precipitated by asymmetric hypertrophy of the left ventricle, which can result in light-headedness, syncope, or sudden cardiac death. Impaired filling of the left ventricle from stiffness of the wall and subsequent decreased cardiac output can lead to diastolic heart failure. Increased atrial pressures and dilatation from diastolic dysfunction can lead to atrial fibrillation, which predisposes to stroke.
Management of ApHCM is geared towards symptom control, genetic testing, and prevention of adverse cardiovascular sequelae [1,2]. Beta-blockers are the recommended initial therapy in ApHCM patients to treat angina by decreasing myocardial oxygen, prevent non-sustained ventricular arrhythmias, and to slow heart rate to improve diastolic filling. Long-term outcomes of beta-blocker therapy in ApHCM patients are unknown and further studies are needed. Non-dihydropyridine calcium channel blockers are second-line therapy if patients cannot tolerate beta-blockers. Recent studies have explored the role of ACE-inhibitors and angiotensin II receptor blockers in reducing adverse cardiac remodeling and fibrosis in ApHCM and other variants of hypertrophic cardiomyopathy. It is important to manage co-morbidities such as hypertension and valvular disease, which function as mechanical stressors, to prevent further pathological ventricular hypertrophy [4]. Further management of ApHCM includes work-up for ICD placement for primary prevention of sudden cardiac death in high-risk patients, genetic testing for both the patient and first-degree family members, and electrocardiographic and echocardiographic screening of both patients and first-degree relatives [1,2]. According to the 2011 American College of Cardiology/American Heart Association (ACC/AHA) guidelines for hypertrophic cardiomyopathy, the recommendations for genetic screening and follow-up are similar for both ApHCM and its counterpart hypertrophic obstructive cardiomyopathy (HOCM) [2]. First-degree family members who test positive for the genotype but do not display the phenotype on initial screening should undergo serial electrocardiography and echocardiography every three to four years for adults and every 12 to 18 months for pediatric patients [1]. Family members who are genotype-negative do not need further follow-up.
Since ApHCM was first described in Japan 40 years ago, there have been differing views regarding its clinical and prognostic course. ApHCM constitutes roughly 13% to 25% of all cases of hypertrophic cardiomyopathy in Japanese patients, however it is much less often described in non-Japanese populations [5]; our patient was Caucasian. The data in Western populations is limited, but recent studies suggest that the risk of sudden cardiac death, myocardial ischemia, advanced heart failure, atrial fibrillation, stroke, and ventricular arrhythmias is comparable to patients with other variants of hypertrophic cardiomyopathy such as HOCM [1]. Predictors of cardiovascular morbidity include female gender, age under 41 years at presentation, left atrial enlargement, hypertension, diabetes, apical aneurysm, and New York Heart Association (NYHA) class II or above at presentation [6]. Studies done by the Mayo Clinic suggest that mortality in advanced ApHCM is comparable to ischemic cardiomyopathy, and propose the role of left ventricular assist device therapy as a bridge to transplant in ApHCM patients with advanced heart failure [7]. Future randomized control trials are needed evaluating outcomes and the impact of appropriate medical therapy on patients with ApHCM.
Conclusions
Despite earlier studies suggesting a more benign clinical course of ApHCM, recent studies report increased morbidity and mortality, which is comparable to the prognosis of other variants of hypertrophic cardiomyopathy such as HOCM. Thus, when formulating a differential diagnosis for chest pain, it is important to include structural heart disease including apical and other variants of hypertrophic cardiomyopathy as part of that differential, as appropriate management can prevent these devastating sequelae. Furthermore, when screening tests such as echocardiography cannot adequately establish the diagnosis of ApHCM, then cardiac MRI or invasive hemodynamic testing is necessary to establish or refute the diagnosis [1,2]. The present case illustrates these points.
Acknowledgments
The authors are grateful to Dr. Jie Ren and Dr. Eric Chou of Cardiology for their general support in the writing of this paper.
Footnotes
Conflicts of interest
The authors have nothing to disclose.
References:
- 1.Jan MF, Todaro MC, Oreto L, Tajik AJ. Apical hypertrophic cardiomyopathy: Present status. Int J Cardiol. 2016;222:745–59. doi: 10.1016/j.ijcard.2016.07.154. [DOI] [PubMed] [Google Scholar]
- 2.Gersh B, Maron BJ, Bonow RO, Dearani JA, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58(25):e212–60. doi: 10.1016/j.jacc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Pasternac A, Noble J, Streulens Y, et al. Pathophysiology of chest pain in patients with cardiomyopathies and normal coronary arteries. Circulation. 1982;65(4):778–89. doi: 10.1161/01.cir.65.4.778. [DOI] [PubMed] [Google Scholar]
- 4.Samak M, Fatullayev J, Sabashnikov A, et al. Cardiac hypertrophy: An introduction to molecular and cellular basis. Med Sci Monit Basic Res. 2016;22:75–79. doi: 10.12659/MSMBR.900437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitaoka H, Doi Y, Casey SA, et al. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. 2003;92:1183–86. doi: 10.1016/j.amjcard.2003.07.027. [DOI] [PubMed] [Google Scholar]
- 6.Ericksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39(4):638–45. doi: 10.1016/s0735-1097(01)01778-8. [DOI] [PubMed] [Google Scholar]
- 7.Muthiah K, Phan J, Robson D, et al. Centrifugal continuous-flow left ventricular device in patients with hypertrophic cardiomyopathy: A case series. ASAIO J. 2013;59:183–87. doi: 10.1097/MAT.0b013e318286018d. [DOI] [PubMed] [Google Scholar]