

Abstract
Although large amounts of vitamin A and its metabolite all-trans retinoic acid (RA) are stored in the liver, how RA regulates liver immune responses during viral infection remains unclear. In this study, we demonstrated that IL-22, mainly produced by hepatic γδ T cells, attenuated liver injury in adenovirus-infected mice. RA can promote γδ T cells to produce mTORC1-dependent IL-22 in the liver, but inhibits IFN-γ and IL-17. RA also affected the aptitude of T cell responses by modulating dendritic cell (DC) migration and co-stimulatory molecule expression. These results suggested that RA plays an immunomodulatory role in viral infection. Proteomics data revealed that RA down-regulated S100 family protein expression in DCs, as well as NF-κB/ERK pathway activation in these cells. Furthermore, adoptive transfer of S100A4-repressed, virus-pulsed DCs into the hind foot of naïve mice failed to prime T cell responses in draining lymph nodes. Our study has demonstrated a crucial role for RA in promoting IL-22 production and tempering DC function through down-regulating S100 family proteins during viral hepatitis.
Keywords: retinoic acid, viral hepatitis, dendritic cells, IL-22
INTRODUCTION
Hepatitis caused by infection with one of the hepatitis A-E viruses can cause acute and chronic liver diseases (1, 2). Because the host immune response is important for control of viral replication, as well as liver injury, it is critical in an intricate balance that determines various disease outcomes among infected individuals. In the liver, many cell types can respond to viral infection by secreting a milieu of unique cytokines and chemokines, which either favor or hinder dendritic cell (DC) antigen sampling, egression and T cell functions. The liver stores the body’s majority of vitamin A (retinol) and is capable of producing large amounts of all-trans retinoic acid (RA). RA, a principal metabolite of retinol, can be secreted by activated hepatic stellate cells (HSCs) and preferentially induce Foxp3+ T regulatory (Tregs) cells, resulting in immune tolerance (3–5). RA also plays an important role in liver regeneration, fibrosis and tumors (6, 7); however, little is known about mechanistic actions of RA in regulating immune responses in physiological conditions and during viral hepatitis.
IL-22 belongs to the IL-10 family (8) and can be produced by various types of cells, including Th17, Th22, γδ T cells, NK cells, neutrophils, and group 3 innate lymphoid cells (ILC3) (9–14). RA can induce γδ T and ILC3 cells to produce IL-22, resulting in attenuated intestinal inflammation (15). IL-22 has also been shown to protect the liver by directly activating anti-apoptotic and proliferative programs in hepatocytes in several hepatitis models (16–19). Since IL-22 can promote recruitment of inflammatory cells by initiating the expression of acute phase proteins via the STAT3 pathway, it may also contribute to liver injury in certain contexts (20, 21). To date, the source and regulation of the liver-derived IL-22 are not well understood (22); the role of IL-22 in viral hepatitis remains debatable.
The enrichment of myeloid DCs is observed in the liver of patients with viral hepatitis (23). Under the appropriate liver microenvironment, these DCs have the unique capability of egress from the infective sites to draining lymphoid organs (24, 25). Since DC migration is a prerequisite for effective T cell priming during viral hepatitis, this process is subject to tight immunoregulatory mechanisms involving multiple intrahepatic players and molecular pathways (2, 26, 27). Recently, RA was reported to enhance both arginase (Arg)-1 and inducible nitric oxide synthase (iNOS) expression in IFN-γ-treated DCs, resulting in a tolerogenic phenotype (28). The latter study implies that RA can modulate antiviral T cell responses by regulating DC functions.
We hypothesized that RA plays a hepatoprotective role through promoting IL-22 production and modulating DC functions during viral hepatitis. In this study, we found that RA treatment inhibited multifunctional T cell responses and attenuated liver injury following adenovirus (Ad)-induced hepatitis. RA treatment increased IL-22 production from γδ T cells and double-negative (DN) T cells via a phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin complex 1 (mTORC1)-dependent fashion. Moreover, RA hindered DC functions by modulating novel S100 family proteins. Knockdown of S100A4 significantly impaired DC migratory capability, resulting in inefficient T cell priming. Together, these results demonstrated that RA protects the liver by promoting IL-22 production and modulating DC function in viral hepatitis.
MATERIALS AND METHODS
Animals
Female C57BL/6 (B6) mice were purchased from the Jackson Laboratory. IL-22-deficient mice on the B6 background were kindly provided by Dr. Wenjun Ouyang of Genentech. All mice were maintained and bred under specific pathogen-free conditions in the animal facility at The University of Texas Medical Branch; all procedures were reviewed and approved by the Institutional Animal Care and Use Committee. To induce hepatitis, we injected mice i.v. with 1× 109 pfu (low dose) or 3 × 109 pfu (high dose) replication-deficient recombinant Ad carrying the LacZ gene (purchased from Vector Development Laboratory, Baylor College of Medicine), as described previously (2, 29).
In vivo administration of RA or rIL-22
For RA treatment, mice were treated i.p. with 250 μg RA or DMSO daily after infection. For the analysis of DC function in vivo, mice were treated daily with RA 3 days prior to infection and euthanized at 2 days post-infection (dpi). For rIL-22 administration, mice were treated i.p. with 5 μg rIL-22 or PBS on 1, 3 and 5 dpi. Mice were euthanized at 6 dpi when the liver injury was at the peak.
Bone marrow-derived DC generation
Bone marrow-derived DCs were generated from B6 mice by cultivation with rGM-CSF (20 ng/ml), as described previously (30). Fresh GM-CSF-containing medium was added at days 3 and 6. RA (1 μM) was added at day 3, as described (31, 32). DCs were harvested at day 8 for function assay. The purity of CD11c+ cell at day 8 of culture was determined by flow cytometry analysis. The ERK inhibitor U0126 (10 nM, Cell Signaling) or control DMSO was added to the culture at day 7, and cells were harvested at day 8 for gene expression analysis.
DC transfection and inhibitor treatment
DCs were seeded in 24-well plates (5 × 105/well) and incubated with SignalSilence® S100A4 siRNA I and control siRNA (Cell Signaling) in the presence of Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer’s instructions. Cells were collected at 24 h for the analysis of S100A4 gene expression. At 72 h, cells were harvested and used for migration assay.
DC migration assay
DC (2 × 105) were re-suspended in complete medium with 2% FBS and placed in a Transwell upper chamber (Costar 3422, Corning). The complete medium with mouse rCCL21 (100 ng/ml) was placed in the lower chamber to induce the CCR7-dependent chemotaxis at 37°C. After 8 h, cells that migrated to the lower chamber were counted and photographed.
For in vivo analysis of DC migration, a modified protocol was used (33). DCs were infected with AdLacZ (MOI: 300) in vitro. Control DCs and RA-treated DCs were labeled with CFSE and cell proliferation dye eflour 670 respectively, and mixed by 1:1 ratio. DCs were injected s.c. into the hind foot. Popliteal draining lymph nodes (LNs) were harvested 1 day later and analyzed for DC migration in vivo.
Priming T cells in vivo by DC transfer
For in vivo T cell priming by DCs, a modified protocol was used (33). Briefly, control or RA-treated DCs were infected with Ad (MOI: 300) for 24 h. Virus-exposed DCs (5 × 105, in 20 μl of PBS) were injected s.c. into the hind foot. Popliteal draining LNs were harvested 7 days later and analyzed for T cell responses (2).
Abs and reagents
The following fluorochrome-labeled antibodies (Abs) specific to mouse markers and their corresponding isotype controls were purchased from eBioscience (San Diego, CA): PE-conjugated anti-IL-22 (1H8PWSR), APC-conjugated anti-IL-17A (eBio17B7), FITC- or APC-conjugated anti-IFN-γ (XMG1.2), APC-conjugated anti-TCRγδ (eBioGL3), PE-Cy7-conjugated anti-CD3 (17A2), Pacific Blue-conjugated anti-CD4 (GK1.5), PerCp-Cy5.5-conjugated anti-CD8 (53–6.7), FITC- or APC-conjugated anti-NK1.1 (PK136), FITC- or PerCp-Cy5.5-conjugated anti-CD11b (M1/70), FITC-conjugated anti-CD11c (N418), APC-conjugated anti-Gr-1 (clone: RB6-8C5), APC-conjugated anti-RORγt (B2D), Cell proliferation dye eFluor 670 and eFluor 506-conjugated fixable viability dye. Purified anti-CD16/32 (2.4G2) was purchased from BD Pharmingen (San Diego, CA). APC-Cy7-conjugated anti-CD3 (17A2) and Pacific Blue-conjugated anti-CD8 (53–6.7) were purchased from Biolegend (San Diego, CA). Recombinant murine IL-23, IL-22 and GM-CSF were from Peprotech (Rocky Hill, NJ). The all-trans RA was from Enzo Life Sciences (Farmingdale, NY). The Rapamycin, Ly294003, Wortmannin and rabbit mAb β-actin (D6A8), Phospho-p44/42 MAPK (Erk1/2), p44/42 MAPK (Erk1/2), Phospho-IκBα (Ser32) and IκBα (44D4)) were purchased from Cell Signaling Technology (Beverly, MA). Anti-S100A4 Ab (ab27957) was from Abcam (Cambridge, MA).
H&E and histological scores
Liver specimens were fixed in 10% buffered formalin. Paraffin-embedded sections were stained with H&E for histological evaluation by using a modified Knodell scoring system (34), as we previously performed (35).
Isolation of splenocytes and intrahepatic lymphocytes (IHLs)
Splenocytes were isolated by using red blood cell lysis buffer. Intrahepatic lymphocytes were isolated according to our previous method (26). Briefly, liver tissue was digested with collagenase IV (0.05%, Roche Applied Science, Indianapolis, IN) at 37°C for 30 min. Intrahepatic mononuclear cells were enriched by centrifugation (400 × g) at room temperature for 30 min over a 30/70% discontinuous Percoll gradient (Sigma). The total numbers of IHLs per liver were counted.
Isolation of hepatic stellate cells (HSCs)
HSCs were isolated from mice by the modified methods as described (36, 37). The liver was perfused with PBS first to remove the blood, followed by in situ digestion (0.04% collagenase type IV and 0.2% pronase (Gibco Life Technologies)) for 10 min. Then, the liver was further digested with 0.08% collagenase type IV, 0.08% pronase and 10 U/ml DNase I (Sigma-Aldrich) at 37°C bath shaking for 30 min. Ten percent Optiprep (Axis-Shield PoC AS) was used for density gradient centrifugation to isolate HSCs. The gating of HSCs on flow cytometry was estimated based on the autofluorescence of the cells by ultraviolet-excited fluorescence.
RALDH1 activity assay
Cell RALDH1 activity was determined by using the ALDEFLUOR staining kit (STEMCELL Technologies Inc.) (5). Briefly, cells were resuspended at 106 cells/ml in ALDEFLUOR assay buffer, containing activated ALDEFLUOR substrate with or without the inhibitor DEAB, and incubated at 37°C for 30 min. ALDEFLUOR-reactive cells were detected in the FITC channel.
Viral copy number
The copy number of adenovirus in the liver was detected by qPCR, as we previously reported (29).
Intracellular staining
Intracellular staining was performed according to our previous methods (35). Briefly, for detection of IFN-γ, TNF-α and IL-2, cells were incubated for 4 h with PMA (50 ng/ml) and ionomycin (750 ng/ml) in the presence of GolgiStop (BD Bioscience). For IL-17 and IL-22 detection, cells were cultured with rIL-23 (20 ng/ml) for 16 h. GolgiStop was added at the last 4 h of culture. The inhibitors of the mTORC1/PI3K pathway were added in selected experiments. After incubation, cells were collected, stained with fixable viability dye, blocked with FcγR blocker (CD16/32), and stained for specific surface molecules. After surface staining, cells were fixed, permeabilized and stained for intracellular cytokines by using a fixation/permeabilization kit (eBioscience). Samples were processed on an LSRII FACSFortessa (Becton Dickinson, San Jose, CA) and analyzed by using FlowJo software (TreeStar, Ashland, OR).
Mass Spectrometric Analysis
DCs were lysed on ice for 2 h in a radio-immunoprecipitation assay buffer (Santa Cruz Biotechnology, CA) containing 1% Nonidet P40, PMSF (0.2 mM), and protease inhibitor cocktail (Roche™). After centrifugation, the protein concentration in the supernatant was determined by BCA Protein Assay (Thermo Scientific Pierce, Rockford, IL). Approximately 100 μg of each protein sample was re-suspended in 25 mM triethylammonium bicarbonate buffer, pH 7.8. The protein was reduced by addition of 10 mM DTT and incubated at 50°C for 30 min, followed by carbaminomethylation achieved by adding 25 mM iodoacetamide and incubating the mixture in the dark for 1 h. The proteins were precipitated by adding 4 volumes of pre-cooled (−20°C) acetone and stored at −20°C overnight. The protein was pelleted at 14,000 rpm for 10 min at 4°C and the supernatant discarded. The protein pellet was dissolved in 25 mM triethylammonium bicarbonate buffer and partially digested by trypsin (Sigma-Aldrich) at a protein/trypsin enzyme ratio of 25:1 (by mass) for 10 h at 37°C.
A Tandem Mass Tagging isobaric (TMT) kit (Thermo-Fisher Scientific) was used to label the peptides following the manufacturer’s recommended conditions. The peptides were separated by reversed phase liquid chromatography (RPLC) using an Easy-UPLC equipped with an auto-sampler (Thermo Scientific). A PicoFrit 150-mm × 75-μM, 5-μm particle size analytical column (New Objective) was used for the RPLC with a 275-min gradient (solvent A, 0.1% FA in water; solvent B, 0.1% FA in ACN). 5–30% solvent B was used for separating the peptides. The QExactive mass analyzer was set to acquire data at resolution 35,000 for the parent full-scan mass spectrum followed by data-dependent, high collision-energy dissociation (HCD) MS/MS spectra for the top 15 most abundant ions acquired at R=17500 resolution.
Proteins were identified and quantified through the Proteome Discoverer 1.4 platform (Thermo-Fisher Scientific) by using the Sequest HT searching engine that employed the UniProt mouse.fasta database with 51532 peptide sequence entries (released in July 2014). Sequest searching parameters were used as follows. Carbamidomethylation of cysteine and TMT-6 modification of peptide N-terminus and lysine were set as fixed modifications and oxidation of methionine and deamination of asparagine and glutamine as variable modifications; trypsin was the protease selected and up to two missed cleavages were used. Mass tolerance for the precursor ions was 10 ppm and for the MS/MS 0.2 Da. Peptides were filtered for a maximum false discovery rate of 1%. At least one unique peptide with a posterior error probability of less than 0.05 was accepted for quantification by using the TMT-reporter ions, and proteins were grouped.
Ingenuity Pathway Analysis (IPA) of DC proteome
Predicted protein-protein interaction networks and canonical pathways were generated from mass spectrometry data analysis by Ingenuity Pathway Analysis Software (Ingenuity Systems, Qiagen, USA) by using log2 fold-changes and p-values between two-group comparisons. The ratios of significant protein expression levels were determined at r = 1.50.
Real-time RT-PCR
Total RNA of frozen tissues were extracted with an RNeasy Mini kit (Qiagen), digested with DNase I (Ambion), used for cDNA systhesis by using an iScript™ Reverse Transcription Kit (Bio-Rad). The iQ SYBR Green Supermix (Bio-Rad) and gene-specific primers were used for PCR in CFX96 Touch real-time PCR detection system (Bio-Rad). Relative quantitation of mRNA expression was calculated by using the 2−ΔΔCt method. The sequences of the primers used are listed here: GAPDH Forward 5′-TGGAAAGCTGTGGCGTGAT-3′, Reverse 5′-TGCTTCACCACCTTCTTGAT-3′; RALDH1 Forward 5′-ATACTTGTCGGATTTAGGAGGCT-3′, Reverse 5′- GGGCCTATCTTCCAAATGAACA-3′; IFN-γ Forward 5′-ATGAACGCTACACACTGCATC-3′, Reverse 5′-CCATCCTTTTGCCAGTTCCTC-3′; TNF-α Forward 5′-CCCTCACACTCAGATCATCTTCT-3′, Reverse 5′-CTTTGAGATCCATGCCGTTG-3′; Arg-1 Forward 5′-CTCCAAGCCAAAGTCCTTAGAG-3′, Reverse 5′-AGGAGCTGTCATTAGGGACATC-3′; IL-22 Forward 5′-GTGGGATCCCTGATGGCTGTCCTGCAG-3′, Reverse 5′-AGCGAATTCTCGCTCAGACTGCAAGCAT-3′; S100A4 Forward 5′-TCCACAAATACTCAGGCAAAGAG-3′; Reverse 5′-GCAGCTCCCTGGTCAGTAG-3′; S100A10 Forward 5′-TGGAAACCATGATGCTTACGTT-3′, Reverse 5′-GAAGCCCACTTTGCCATCTC-3′; Adeonvirus Hexon Forward 5′-GAGCCAGCATTAAGTTTGATAGCA-3′, Reverse 5′-AGATAGTCGTTAAAGGACTGGTCGTT-3′.
ELISA assays
Liver proteins were extracted from frozen tissues by homogenization on ice in the RIPA buffer (Cell Signaling) with a protease inhibitor cocktail (Sigma). After centrifugation at 20,000 × g for 15 min, the supernatant was collected and protein concentration measured with a protein assay kit (Bio-Rad). Equal amounts of the liver proteins (100 μg) were loaded for ELISA assays. For detecting IL-17 and IL-22 in culture supernatants, IHLs were isolated from mice at 3 dpi and cultured with or without rIL-23 (20 ng/ml) for 3 days. RA (100 nM) and inhibitors (Rapamycin, 25 nM; Ly294002, 5 μM; Wortmannin, 100 nM) were selectively added at the beginning of the culture. The supernatants collected at the end of the culture were measured for IL-17 and IL-22 levels by using ELISA kits (eBioscience).
Western blotting
Whole cell protein was extracted with a Radio-Immunoprecipitation Assay (RIPA, Cell signaling Technology). Protein samples were separated by 12.5 % SDS-PAGE and transferred to PVDF membranes (0.45 lm, Millipore) and blocked for 1 h with 5% bovine serum albumin (BSA), 0.1 % Tween-20 in TBS. The membranes were then washed and incubated with primary antibodies overnight. The goat anti-rabbit or goat anti-mouse IgG secondary antibodies (1:5000 dilution) were used to incubate for 1 h. After washing, immunoreactive bands were visualized by chemiluminescent detection (ECL, Roche Diagnostics, Penzberg, Germany) and exposure to X-ray film (Thermo Fisher Scientific, MA, US).
Statistical analysis
The difference between the two different groups was determined by using Student’s t test. One-way ANOVA was used for multiple group comparisons (GraphPad Software v4.0). P values < 0.05 were considered significant*, < 0.01 as significant**, and <0.001 as highly significant***.
RESULTS
RA alleviates liver injury and modulates T cell responses
We first analyzed the hepatic expression of retinaldehyde dehydrogenase 1 (RALDH1), which is a rate-limiting enzyme for vitamin A metabolism. Viral infection increased the level of RALDH1 in both liver and hepatic stellate cells (HSCs) (Fig. 1A and B). To investigate the role of RA in viral hepatitis, we infected mice with Ad and treated them i.p. with 250 μg RA or solvent control. We found that at 6 dpi (at the peak of liver damage), RA-treated mice displayed attenuated liver inflammation and damage, as judged by their significant reduction in serum alanine transaminase (ALT), aspartate aminotransferase (AST) levels and inflammatory infiltration (Figs. 1C and S1A). RA treatment significantly decreased the numbers of IHLs and splenocytes (Fig. 1D), the percentage and number of intrahepatic CD4+ and CD8+ T cells (Fig. 1E), the numbers of multifunctional CD4+ and CD8+ T cells (Fig. 1F), as well as IFN-γ and TNF-α transcripts in liver tissues (Fig. S1B). However, the viral load was not changed in the liver by RA treatment (Fig. S1B). Although RA treatment did not change the percentages of intrahepatic CD3+ Foxp3+ cells, their number was decreased due to the reduced inflammatory cells in the liver (Fig. S1C). RA treatment also remarkably decreased the percentages of both CXCR3+ and CD44+CD62L− T effector cells in the spleen (Fig. S1D). Lower dose of RA treatment (25 μg per day) also reduced the serum ALT and AST levels, as well as decreased the numbers of IFN-γ-producing cells in both the spleen and liver (Fig. S1E–G). In vitro treatment of splenocytes and IHLs with RA for 24 h inhibited IFN-γ expression on CD4+ and CD8+ T cells in a dose-dependent manner (Fig. S2). Overall, RA treatment decreased cytokine-producing T cells and alleviated liver injury in Ad-induced hepatitis.
Figure 1. Retinoic acid (RA) alleviated liver injury in Ad-induced viral hepatitis.
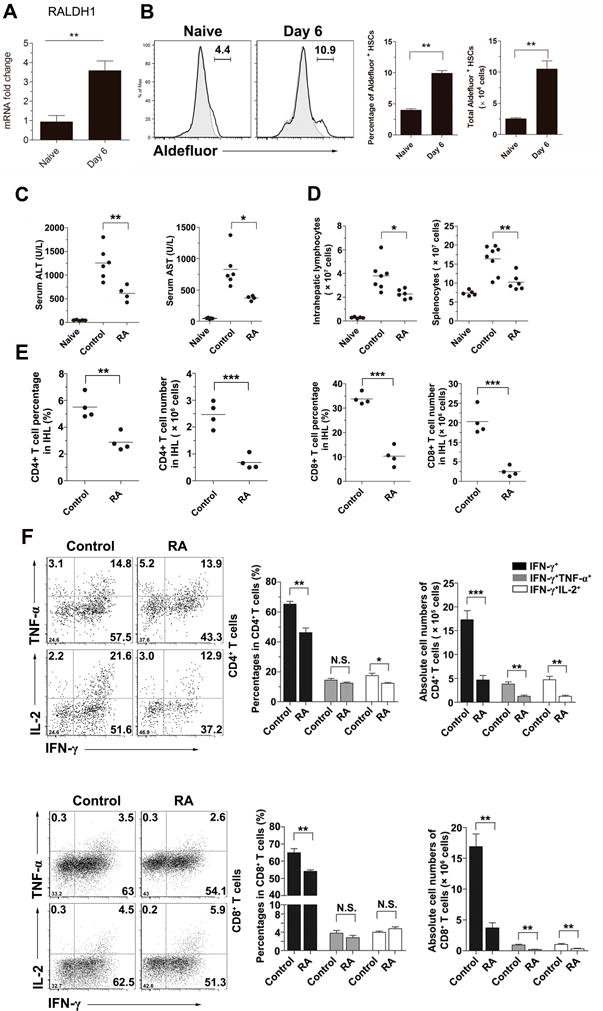
C57BL/6 mice (B6) were i.v. injected with 3 × 109 pfu of AdLacZ. (A) The mRNA levels of RALDH1 in the livers were analyzed at 6 days post infection (dpi). (B) Hepatic stellate cells (HSCs) were isolated and analyzed for the RALDH1 expression. (C-F) The AdLacZ-infected mice were injected i.p. with DMSO (control) or 250 μg RA daily and sacrificed at 6 days post infection (dpi). (C) Serum ALT and AST levels. (D) Total numbers of IHLs and splenocytes. (E) Percentages and absolute numbers of hepatic CD4+ and CD8+ T cells. (F) Percentages and absolute numbers of cytokine-producing T cells in the liver. The experiment was repeated three times independently, and representative graphs are shown (4–6 mice/group). *p < 0.05; **p < 0.01.
RA promotes IL-22-producing γδ T cells and DN T cells in the liver
Hepatic IL-22 protein levels were increased following Ad infection (Fig. 2A). A higher frequency of intrahepatic IL-22+ cells were found at 1 dpi compared to those in naïve mice, returning to baseline at 6 dpi (Fig. 2A). Most of the hepatic IL-22-producing cells were CD3+, but CD4− and CD8− T cells (Fig. 2B). CD3− cells, presumably ILCs, expressed few IL-22 (0.1% of total IHLs, Fig. 2B). Other cell types, including CD11b+, CD11c+, F4/80+, Ly6G+, and NK+ cells, did not express IL-22 (Fig. S3A). The major producers of hepatic IL-22 were γδ T cells (Fig. 2B) and IL-22+ γδ T cells can also produce IL-17 (Fig. 2C). We found that around 48% of hepatic γδ T cells belonged to the Vγ4 subset, among which around 15.5% cells were IL-22+IL-17+ and 25.9% cells were IL-17+ (Fig. S3B) (2, 38). In addition to γδ T cells, CD3+TCRγδ− T cells also produced IL-22, which expressed neither CD4 nor CD8 (Fig. 2B). Among these DN T cells, there were 6% IL-17+IL-22+ and 1.3% IL-22+ cells (Fig. 2D). Thus, γδ T cells and DN T cells were the main sources of IL-22 in the liver.
Figure 2. RA promoted IL-22 expression by γδ T cells and double negative (DN) T cells in the liver of viral hepatitis.
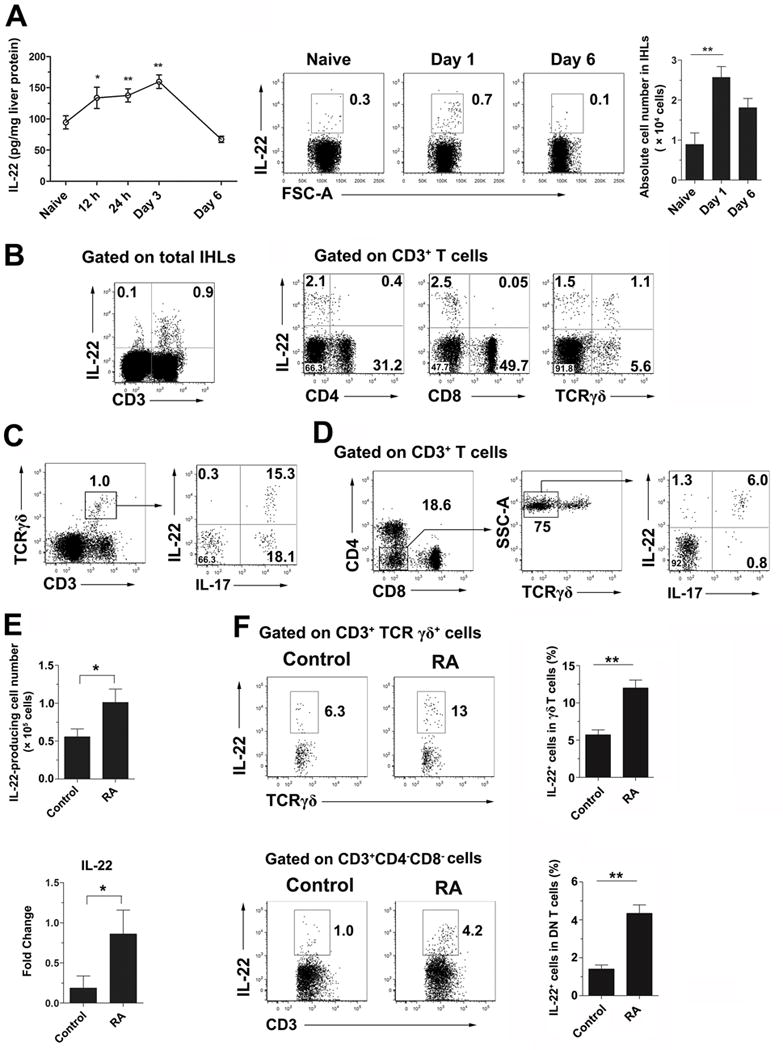
B6 mice were i.v. injected with 3 × 109 pfu of AdLacZ and sacrificed at the indicated time points. (A) Kinetic analysis of hepatic IL-22 production by an ELISA assay. IHLs were stimulated with recombinant IL-23 (rIL-23, 20 ng/ml) for 16 h plus GolgiStop for the last 4 h. The cells were examined for intracellular IL-22. Right panel: Cumulative statistical results of the absolute number of IL-22+ cells in the liver. (B) IHLs from day 1 infected mice were gated on the CD3+ population. IL-22-producing cells were further analyzed on CD4+, CD8+ and TCRγδ+ T cells. (C) The IHLs were gated on γδ T cells for IL-17 and IL-22 expression. (D) The IHLs were gated on DN T cells (CD3+ CD4− CD8− TCRγδ− cells) for IL-17 and IL-22 expression. (E) Mice were infected and treated with RA as shown in Fig. 1. Hepatic IL-22-producing cells were analyzed at 6 dpi. Left panel: absolute number of IL-22+ cells. Right panel: IL-22 mRNA level of IHLs. (F) Intracellular IL-22 expression in hepatic γδ T and DN T cells. The experiment was repeated two to three times independently, and a representative graph is shown (6–8 mice/group). *p < 0.05, **p < 0.01.
To investigate whether RA can enhance hepatic IL-22 production during viral hepatitis (15), we treated mice with RA daily starting at 1 dpi and sacrificed mice at 6 dpi. RA-treated mice displayed a 2-fold increase in IL-22+ cells in the liver compared to those in controls (Fig. 2E). Liver IL-22 mRNA levels were significantly elevated after RA treatment (Fig. 2E). Further study showed that RA can promote γδ T (> 2-folds) and DN T cells (> 4-folds) to produce IL-22 in the liver (Fig. 2F). Thus, RA can promote IL-22 production from γδ T and DN T cells in the liver in viral hepatitis.
PI3K/mTORC1 signal pathway regulates IL-22 production
The PI3K/mTORC1 pathway regulates cytokine production in monocytes, macrophages and DCs to bacterial infection (39); however, it is unclear whether this pathway regulates IL-17 and IL-22 expression in viral hepatitis. To address this issue, we isolated IHLs at 2 dpi for culture in the presence of Rapamycin (a mTORC1 inhibitor) and Ly294002 or Wortmannin (PI3K inhibitors) in vitro (40), followed by analysis of IL-17 and IL-22 production. Inhibition of the PI3K/mTORC1 markedly reduced the percentages of IL-17+ and IL-22+ γδ T and DN T cells (Fig. 3A). Additionally, RA alone markedly reduced IL-17, without significantly changing IL-22 production (Fig. 3B); RA also synergized with PI3K/mTORC1 inhibitors to further inhibit IL-17, but not IL-22, production (Fig. 3B). These results suggest that the PI3K/mTORC1 signaling pathway is essential for IL-17 and IL-22 expression in innate immune cells such as γδ T and DN T cells in viral hepatitis.
Figure 3. IL-17 and IL-22 production in γδ T and DN T cells was dependent on the PI3K/mTORC1 signaling pathway.
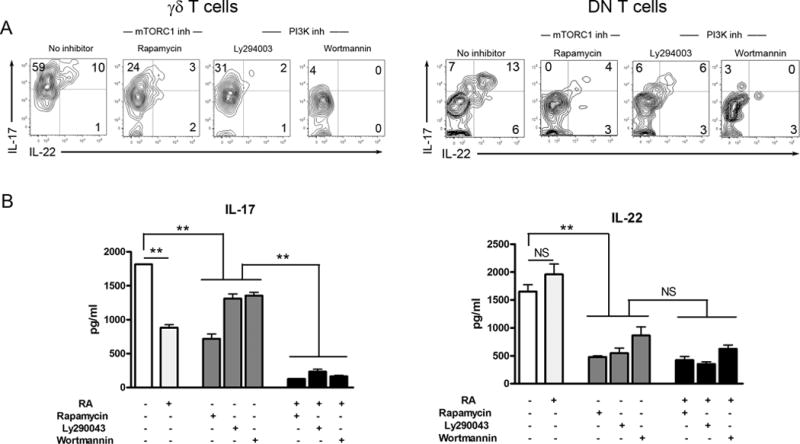
B6 mice were i.v. injected with 3 × 109 pfu of AdLacZ and sacrificed at 2 dpi. IHLs were isolated and stimulated with rIL-23 (20 ng/ml) for flow cytometry. The culture system was initially added with or without RA (100 nM), Rapamycin (25 nM), Ly294002 (5 μM) or Wortmannin (100 nM). DMSO was used as solvent control. For intracellular staining, the cells were cultured for 16 h plus GolgiStop for the last 4 h. (A) γδ T and DN T cells were gated first, followed by the analysis of intracellular IL-17 and IL-22 expression. (B) For the detection of cytokines in the supernatant, the cells were cultured for 3 days. The supernatants were collected for an ELISA assay. The experiment was repeated three times independently. **p < 0.01, ***p < 0.001.
IL-22 ameliorates liver injury in viral infection
To investigate whether IL-22 is pro-inflammatory or hepatoprotective in Ad-induced hepatitis, we treated infected mice with rIL-22 daily starting 1 dpi and analyzed the outcomes at 7 dpi. While IL-22-treated mice displayed reduced serum ALT and AST levels (Fig. 4A), the hepatic infiltration of lymphocytes as well as the IFN-γ and TNF-α cytokine expression were comparable between treated- and control groups (data not shown). To further validate these observations, we performed Ad infection (1 × 109 pfu) in IL-22 knockout (KO) mice. Compared to wild-type controls, the IL-22 KO mice had 2- to 3-fold higher serum ALT and AST levels (Fig. 4B), higher numbers of IHLs (Fig. 4C), accompanied by higher percentages and numbers of intrahepatic IFN-γ+ CD4+ T cells (Fig. 4D). Collectively, these data demonstrate a hepatoprotective role for IL-22 in viral hepatitis.
Figure 4. IL-22 ameliorated liver injury during viral hepatitis.
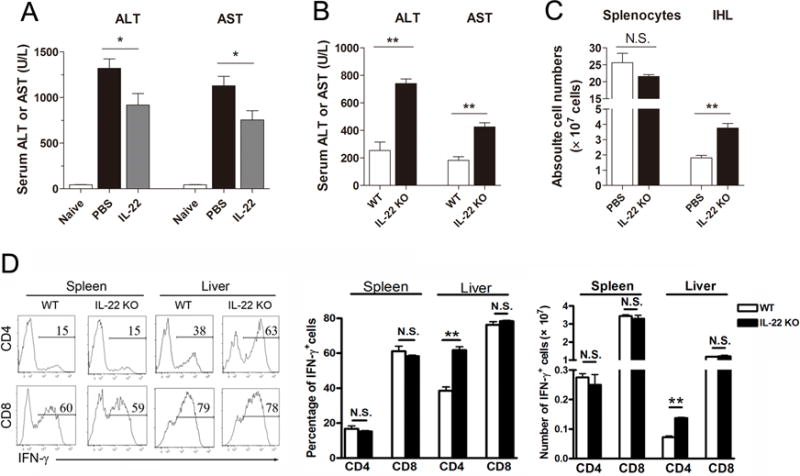
(A) B6 mice were i.v. injected with 3 × 109 pfu of AdLacZ, followed by i.p. injection with 5 μg recombinant IL-22 (rIL-22) or PBS (control) on 1, 3 and 5 dpi. Mice were harvested at 6 dpi. Serum ALT and AST levels were measured. (B) WT and IL-22 KO mice were i.v. injected with 1 × 109 pfu of AdLacZ and sacrificed at 6 dpi. Serum ALT and AST were detected. (C) Numbers of splenocytes and IHLs, as well as the (D) percentages and total numbers of IFN-γ-producing CD4+ and CD8+ T cells. The experiment was repeated three times independently, and a representative graph is shown (3–8 mice/group). *p < 0.05, **p <0.01, N.S. no statistical difference.
RA inhibits DC activation and T cell priming in vitro and in vivo
Inhibition of DC function has been shown to contribute to T cell exhaustion during chronic hepatitis C virus infection (41, 42). To test whether RA can modulate T cell responses by regulating DC functions, we treated mice with RA daily starting at −3 dpi and sacrificed the animals at 2 dpi. RA-treated mice displayed lower levels of co-stimulatory molecules (CD80, CD86 and CD40 expression) on DCs compared with those in control animals (Fig. 5A). To confirm these observations in vitro, we treated DCs with RA (1 μM) at days 3 and 6 of culture and examined DC phenotypes and functions at day 8. RA treatment resulted in significant lower level of CD11c, CD86 and CD40 expression (Fig. 5B and C) and increased PD-L2 expression levels compared to findings in control DCs (Fig. 5C). RA treatment did not alter the viral infection of DCs (Fig. S4A), but reduced DC ability to migrate in a CCL21-dependent chemotactic assay carried in a two-chamber cell culture system (Fig. 5D). We confirmed the chemotactic assay in vivo by using a DC-transferring system (33, 43). Briefly, RA-treated and control DCs were infected with Ad in vitro for 24 h and labeled with different dyes respectively, followed by mixing of 1:1 ratio. DCs were injected s.c. into the hind foot of naïve mice and draining LNs were harvested 24 h and analyzed for DC migration in vivo. We found that control DCs, but not the RA-treated DCs were found in draining LNs after 24 h (Fig. S4B). Finally, to validate the immunobiological effects of RA, we analyzed T cell responses in this in vivo T cell priming system. The unlabeled control DCs and RA-treated DCs were injected into the hind foot of naive mice. Draining LNs were harvested at 7 dpi, and T cell functions were subsequently analyzed. Mice receiving RA-treated DCs in footpads displayed a marked reduction in the percentages of IFN-γ+, IL-2+ TNF-α+ CD4+ T cells, as well as those of IFN-γ+ CD8+ T cells in draining LNs (Fig. 5E). These results suggested to us that locally produced RA can directly repress DC functions in migration and priming CD4+ and CD8+ T cell responses in distant secondary lymphoid organs.
Figure 5. RA inhibited dendritic cell (DC) activation and T cell priming in vivo.
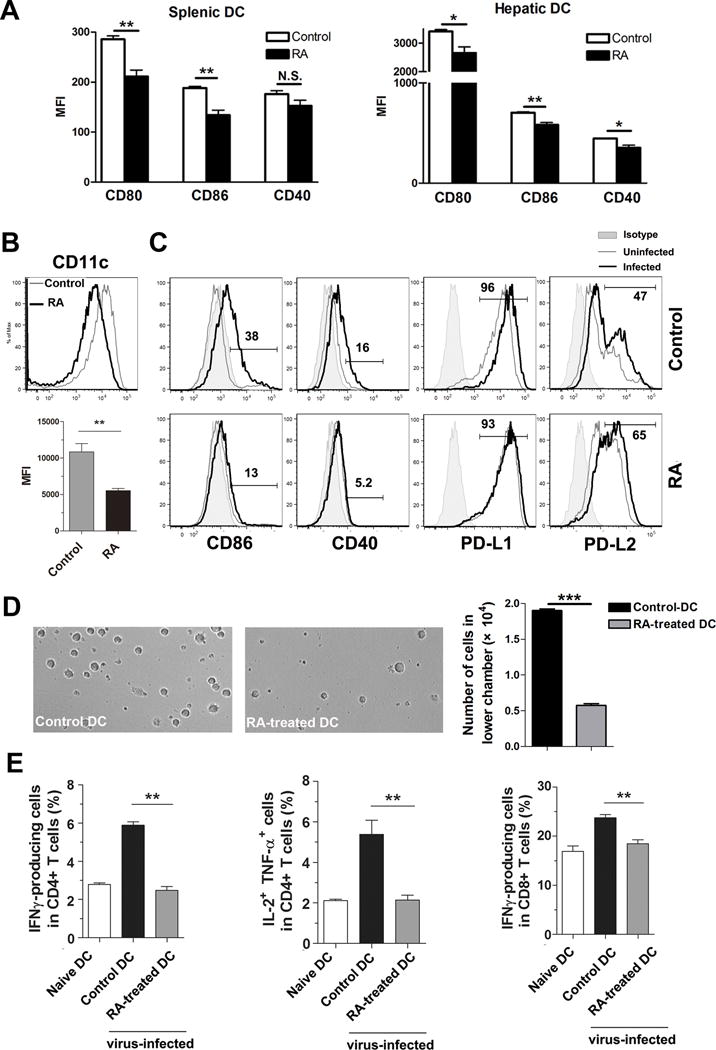
Mice were i.p. injected with RA (250 μg) or DMSO (control) daily from -3 to 1 dpi. These animals were i.v. injected with 3 × 109 pfu of AdLacZ and sacrificed at 2 dpi. (A) DCs in the spleen and liver were first gated on CD11b+CD11c+ cells and the mean fluorescence intensity (MFI) of co-stimulatory molecules was examined. (B) Bone marrow-derived DCs were generated from B6 mice by using complete RPMI 1640 with 20 ng/ml rGM-CSF. RA (1 μM) was added at day 3 of an 8-day DC culture. Upper panel: Surface CD11c expression level of control and RA-treated DCs. Grey line: control DCs; black line: RA-treated DCs. Lower panel: MFI of CD11c on DCs. (C) RA-treated and control DCs were infected with Ad-LacZ (MOI: 300) for 12 h. Surface molecules were analyzed by flow cytometry. Shaded: Isotype control; grey line: uninfected DCs; black line: Ad-infected DCs. (D) 2 × 105 control or RA-treated DCs were cultured, respectively, in the upper chambers of 24-well transwell plates. The lower chambers contained complete medium with rCCL21 (100 ng/ml). After 8 h of culture, the cells in the lower chambers were collected for photographing and counting. (E) Virus-exposed DCs or naïve DCs (5 × 105 in 20 μl of PBS) were injected s.c. into the hind footpad of naïve mice. Three types of DC were transferred: naïve DCs, virus-exposed control DCs, and virus-exposed RA-treated DCs (prepared as in Fig 5B and C). Popliteal draining lymph nodes (LNs) were harvested at 7 dpi. Lymphocytes were isolated and stimulated by PMA and Ionomycin plus Golgistop for 4 h. Percentages of cytokine-producing CD4+ and CD8+ T cells in draining LNs were examined by flow cytometry. Each experiment was repeated at least three times independently. Shown are representative flow cytometric results. *p < 0.05, **p < 0.01, ***p<0.001.
RA regulates expression of S100 family proteins in DCs
To investigate the molecular mechanisms responsible for RA-mediated DC functions, we performed a mass spectrometric study on in vitro RA- or PBS-treated DCs. Among 2,329 proteins identified, 38 proteins were down-regulated and 26 proteins were up-regulated in RA-treated DCs by at least 1.5 fold (Table 1). Among the top 15 down-regulated proteins, three (3) proteins belonged to the S100 family of calcium-binding proteins (S100A4, S100A6, and S100A10). These proteins are present in myeloid cells such as macrophages, which regulate their cellular chemotaxis (44). DCs and malignant tumors with a strong metastasis potential are also reported to express S100 proteins (45–48). Ingenuity Pathway Analysis (IPA) based on our data demonstrated that S100A4/A6/A10 proteins are modulated by the NF-κB and ERK signaling pathways (Fig. 6A). These proteins are likely to be involved in DC maturation and IL-12 signaling. RT-PCR analyses confirmed that RA-treated DCs expressed significantly lower mRNA levels of S100A4 and S100A10, but higher levels of Arg-1 (top 1 of the upregulated proteins) compared with those of control DCs (Fig. 6A and B). Western blotting further confirmed that RA treatment markedly inhibited the protein expression of S100A4, as well as p-IκBα and p-ERK, in DCs (Fig. 6C). These results established that RA suppresses S100 proteins and down-regulates signaling pathways that control DC maturation.
Table 1.
Differentially expressed proteins in RA-treated and control dendritic cells
| Accession | Description | RA/Ctrl-1 | RA/Ctrl-2 | RA/Ctrl-3 | RA/Ctrl-4 |
|---|---|---|---|---|---|
| D3Z2R5 | Protein Sepn1 OS=Mus musculus GN=Sepn1 PE=4 SV=1 - [D3Z2R5_MOUSE] | 0.180 | 0.396 | 0.368 | 0.808 |
| E9PWW9 | Protein Rsf1 OS=Mus musculus GN=Rsf1 PE=1 SV=1 - [E9PWW9_MOUSE] | 0.251 | 0.283 | 0.560 | 0.630 |
| Q9EQC5 | N-terminal kinase-like protein OS=Mus musculus GN=Scyl1 PE=1 SV=1 - [NTKL_MOUSE] | 0.268 | 0.797 | 0.663 | 1.968 |
| Q0VGP9 | Calpain-3 OS=Mus musculus GN=Capn3 PE=2 SV=1 - [Q0VGP9_MOUSE] | 0.354 | 0.460 | 1.025 | 1.332 |
| P07091 | Protein S100-A4 OS=Mus musculus GN=S100a4 PE=1 SV=1 - [S10A4_MOUSE] | 0.356 | 0.326 | 0.388 | 0.340 |
| E9QNG1 | Intersectin-2 OS=Mus musculus GN=Itsn2 PE=1 SV=1 - [E9QNG1_MOUSE] | 0.387 | 0.403 | 1.164 | 1.212 |
| Q3TL72 | NEDD8-activating enzyme E1 catalytic subunit OS=Mus musculus GN=Uba3 PE=2 SV=1 - [Q3TL72_MOUSE] | 0.406 | 0.640 | 0.751 | 1.182 |
| D3YWB3 | Transmembrane channel-like protein OS=Mus musculus GN=Tmc4 PE=3 SV=1 - [D3YWB3_MOUSE] | 0.436 | 0.503 | 0.670 | 0.773 |
| Q68EF0 | Rab-3A-interacting protein OS=Mus musculus GN=Rab3ip PE=1 SV=1 - [RAB3I_MOUSE] | 0.450 | 0.380 | 1.513 | 1.276 |
| Q61878 | Bone marrow proteoglycan OS=Mus musculus GN=Prg2 PE=1 SV=1 - [PRG2_MOUSE] | 0.454 | 0.567 | 0.468 | 0.585 |
| Q3KPB0 | Olfactory receptor OS=Mus musculus GN=Olfr616 PE=2 SV=1 - [Q3KPB0_MOUSE] | 0.455 | 0.552 | 0.558 | 0.676 |
| Q67BT3 | Solute carrier family 13 member 5 OS=Mus musculus GN=Slc13a5 PE=2 SV=1 - [S13A5_MOUSE] | 0.485 | 0.626 | 0.563 | 0.671 |
| P14069 | Protein S100-A6 OS=Mus musculus GN=S100a6 PE=1 SV=3 - [S10A6_MOUSE] | 0.509 | 0.457 | 0.557 | 0.502 |
| Q8C7V8 | Coiled-coil domain-containing protein 134 OS=Mus musculus GN=Ccdc134 PE=2 SV=1 - [CC134_MOUSE] | 0.516 | 0.747 | 0.708 | 1.025 |
| Q9R0A0 | Peroxisomal membrane protein PEX14 OS=Mus musculus GN=Pex14 PE=1 SV=1 - [PEX14_MOUSE] | 0.518 | 1.022 | 0.636 | 0.623 |
| Q64444 | Carbonic anhydrase 4 OS=Mus musculus GN=Ca4 PE=1 SV=1 - [CAH4_MOUSE] | 0.547 | 0.429 | 0.554 | 0.481 |
| P08207 | Protein S100-A10 OS=Mus musculus GN=S100a10 PE=1 SV=2 - [S10AA_MOUSE] | 0.547 | 0.571 | 0.554 | 0.586 |
| A2AQ89 | SH2 domain-containing adapter protein F (Fragment) OS=Mus musculus GN=Shf PE=1 SV=1 - [A2AQ89_MOUSE] | 0.560 | 0.618 | 0.550 | 0.606 |
| Q8BNU0 | Armadillo repeat-containing protein 6 OS=Mus musculus GN=Armc6 PE=2 SV=1 - [ARMC6_MOUSE] | 0.574 | 0.627 | 0.875 | 0.956 |
| Q9QZW0-2 | Isoform 2 of Phospholipid-transporting ATPase 11C OS=Mus musculus GN=Atp11c - [AT11C_MOUSE] | 0.577 | 0.525 | 0.758 | 0.690 |
| E9PYI6 | Protein Simc1 OS=Mus musculus GN=Simc1 PE=4 SV=1 - [E9PYI6_MOUSE] | 0.579 | 0.614 | 0.747 | 0.747 |
| P08101-2 | Isoform IIB2 of Low affinity immunoglobulin gamma Fc region receptor II OS=Mus musculus GN=Fcgr2 - [FCGR2_MOUSE] | 0.580 | 0.587 | 0.621 | 0.628 |
| P20152 | Vimentin OS=Mus musculus GN=Vim PE=1 SV=3 - [VIME_MOUSE] | 0.581 | 0.609 | 0.663 | 0.681 |
| Q5PPR2 | Exocyst complex component 1 OS=Mus musculus GN=Exoc1 PE=1 SV=1 - [Q5PPR2_MOUSE] | 0.583 | 0.531 | 0.929 | 0.846 |
| Q8BG67-2 | Isoform 2 of Protein EFR3 homolog A OS=Mus musculus GN=Efr3a - [EFR3A_MOUSE] | 0.588 | 0.651 | 0.241 | 0.267 |
| Q8K4S1 | 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase epsilon-1 OS=Mus musculus GN=Plce1 PE=1 SV=3 - [PLCE1_MOUSE] | 0.593 | 0.606 | 0.555 | 0.566 |
| A2AP58 | Protein Sptbn5 (Fragment) OS=Mus musculus GN=Sptbn5 PE=4 SV=1 - [A2AP58_MOUSE] | 0.599 | 0.769 | 0.572 | 0.733 |
| P04117 | Fatty acid-binding protein, adipocyte OS=Mus musculus GN=Fabp4 PE=1 SV=3 - [FABP4_MOUSE] | 0.605 | 0.598 | 0.597 | 0.567 |
| E9QKK1 | Centromere-associated protein E OS=Mus musculus GN=Cenpe PE=3 SV=1 - [E9QKK1_MOUSE] | 0.614 | 0.691 | 0.686 | 0.762 |
| Q99MV1 | Tudor domain-containing protein 1 OS=Mus musculus GN=Tdrd1 PE=1 SV=2 - [TDRD1_MOUSE] | 0.615 | 0.885 | 0.715 | 0.926 |
| E9Q264 | Protein Myh15 OS=Mus musculus GN=Myh15 PE=4 SV=1 - [E9Q264_MOUSE] | 0.644 | 0.572 | 0.757 | 0.671 |
| Q8R1H8-2 | Isoform 2 of Basic leucine zipper transcriptional factor ATF-like 2 OS=Mus musculus GN=Batf2 - [BATF2_MOUSE] | 0.646 | 0.680 | 0.654 | 0.752 |
| Q8R1F1 | Niban-like protein 1 OS=Mus musculus GN=Fam129b PE=2 SV=2 - [NIBL1_MOUSE] | 0.657 | 0.715 | 0.710 | 0.773 |
| E9Q0F0 | Protein Krt78 OS=Mus musculus GN=Krt78 PE=4 SV=1 - [E9Q0F0_MOUSE] | 0.658 | 0.535 | 1.125 | 0.914 |
| Q497J0 | MCG130175, isoform CRA_b OS=Mus musculus GN=BC100530 PE=2 SV=1 - [Q497J0_MOUSE] | 0.659 | 0.687 | 0.617 | 0.678 |
| S4R2S8 | Ankyrin-3 OS=Mus musculus GN=Ank3 PE=4 SV=1 - [S4R2S8_MOUSE] | 0.662 | 0.702 | 0.701 | 0.634 |
| P07356 | Annexin A2 OS=Mus musculus GN=Anxa2 PE=1 SV=2 - [ANXA2_MOUSE] | 0.663 | 0.670 | 0.690 | 0.697 |
| B8XCJ6 | Protein unc-80 homolog OS=Mus musculus GN=Unc80 PE=2 SV=1 - [B8XCJ6_MOUSE] | 0.664 | 0.756 | 0.721 | 0.820 |
| P10605 | Cathepsin B OS=Mus musculus GN=Ctsb PE=1 SV=2 - [CATB_MOUSE] | 1.505 | 1.558 | 1.391 | 1.460 |
| O89017 | Legumain OS=Mus musculus GN=Lgmn PE=1 SV=1 - [LGMN_MOUSE] | 1.535 | 1.426 | 1.560 | 1.308 |
| D3Z7I2 | Ataxin-7-like protein 1 OS=Mus musculus GN=Atxn7l1 PE=1 SV=1 - [D3Z7I2_MOUSE] | 1.538 | 1.124 | 1.441 | 1.052 |
| B1AR51 | Dynein, axonemal, heavy chain 9 OS=Mus musculus GN=Dnah9 PE=4 SV=1 - [B1AR51_MOUSE] | 1.597 | 1.326 | 1.537 | 1.275 |
| D6RJ84 | Uridine-cytidine kinase OS=Mus musculus GN=Uckl1 PE=3 SV=1 - [D6RJ84_MOUSE] | 1.612 | 1.687 | 1.448 | 1.484 |
| Q8C419 | Probable G-protein coupled receptor 158 OS=Mus musculus GN=Gpr158 PE=1 SV=2 - [GP158_MOUSE] | 1.624 | 1.573 | 1.581 | 1.531 |
| Q8BP18 | Protein Zfp101 OS=Mus musculus GN=Zfp101 PE=2 SV=1 - [Q8BP18_MOUSE] | 1.629 | 1.531 | 1.620 | 1.521 |
| P27784 | C-C motif chemokine 6 OS=Mus musculus GN=Ccl6 PE=1 SV=1 - [CCL6_MOUSE] | 1.648 | 1.129 | 1.524 | 1.043 |
| P63254 | Cysteine-rich protein 1 OS=Mus musculus GN=Crip1 PE=1 SV=2 - [CRIP1_MOUSE] | 1.669 | 1.643 | 1.744 | 1.765 |
| Q91Z53 | Glyoxylate reductase/hydroxypyruvate reductase OS=Mus musculus GN=Grhpr PE=1 SV=1 - [GRHPR_MOUSE] | 1.707 | 0.774 | 1.815 | 0.822 |
| A2BGH0 | BPI fold-containing family B member 4 OS=Mus musculus GN=Bpifb4 PE=2 SV=1 - [BPIB4_MOUSE] | 1.709 | 1.927 | 1.884 | 2.123 |
| P20065-2 | Isoform Short of Thymosin beta-4 OS=Mus musculus GN=Tmsb4x - [TYB4_MOUSE] | 1.730 | 1.679 | 1.529 | 1.537 |
| D3Z7R7 | Minor histocompatibility protein HA-1 OS=Mus musculus GN=Hmha1 PE=1 SV=1 - [D3Z7R7_MOUSE] | 1.756 | 0.829 | 1.687 | 0.796 |
| Q059P4 | Filamin A interacting protein 1 OS=Mus musculus GN=Filip1 PE=2 SV=1 - [Q059P4_MOUSE] | 1.772 | 1.485 | 1.839 | 1.540 |
| H7BWZ9 | Protein dopey-1 OS=Mus musculus GN=Dopey1 PE=4 SV=1 - [H7BWZ9_MOUSE] | 1.816 | 2.073 | 1.625 | 1.854 |
| B1AY85 | Plexin domain-containing protein 2 OS=Mus musculus GN=Plxdc2 PE=4 SV=1 - [B1AY85_MOUSE] | 1.822 | 1.870 | 1.991 | 2.042 |
| E9QKG2 | Katanin p60 ATPase-containing subunit A1 OS=Mus musculus GN=Katna1 PE=1 SV=1 - [E9QKG2_MOUSE] | 1.872 | 1.514 | 2.014 | 1.628 |
| O35639 | Annexin A3 OS=Mus musculus GN=Anxa3 PE=1 SV=4 - [ANXA3_MOUSE] | 1.927 | 1.916 | 1.822 | 1.826 |
| Q91XV3 | Brain acid soluble protein 1 OS=Mus musculus GN=Basp1 PE=1 SV=3 - [BASP1_MOUSE] | 1.972 | 1.737 | 1.870 | 1.656 |
| P08249 | Malate dehydrogenase, mitochondrial OS=Mus musculus GN=Mdh2 PE=1 SV=3 - [MDHM_MOUSE] | 2.087 | 2.076 | 1.950 | 1.932 |
| Q8BIG7 | Catechol O-methyltransferase domain-containing protein 1 OS=Mus musculus GN=Comtd1 PE=2 SV=1 - [CMTD1_MOUSE] | 2.211 | 1.021 | 1.104 | 0.764 |
| P70695 | Fructose-1,6-bisphosphatase isozyme 2 OS=Mus musculus GN=Fbp2 PE=2 SV=2 - [F16P2_MOUSE] | 2.560 | 2.264 | 2.149 | 1.899 |
| Q61176 | Arginase-1 OS=Mus musculus GN=Arg1 PE=1 SV=1 - [ARGI1_MOUSE] | 2.938 | 3.026 | 2.877 | 2.891 |
| Q8K3H0 | DCC-interacting protein 13-alpha OS=Mus musculus GN=Appl1 PE=1 SV=1 - [DP13A_MOUSE] | 4.298 | 1.034 | 1.669 | 0.401 |
| Q8R0W0 | Epiplakin OS=Mus musculus GN=Eppk1 PE=1 SV=2 - [EPIPL_MOUSE] | 4.557 | 3.937 | 3.681 | 3.179 |
| S4R1P5 | Dystonin OS=Mus musculus GN=Dst PE=1 SV=1 - [S4R1P5_MOUSE] | 5.939 | 5.118 | 4.314 | 3.616 |
Figure 6. RA regulated S100A protein expression and downstream signaling pathway of DCs.
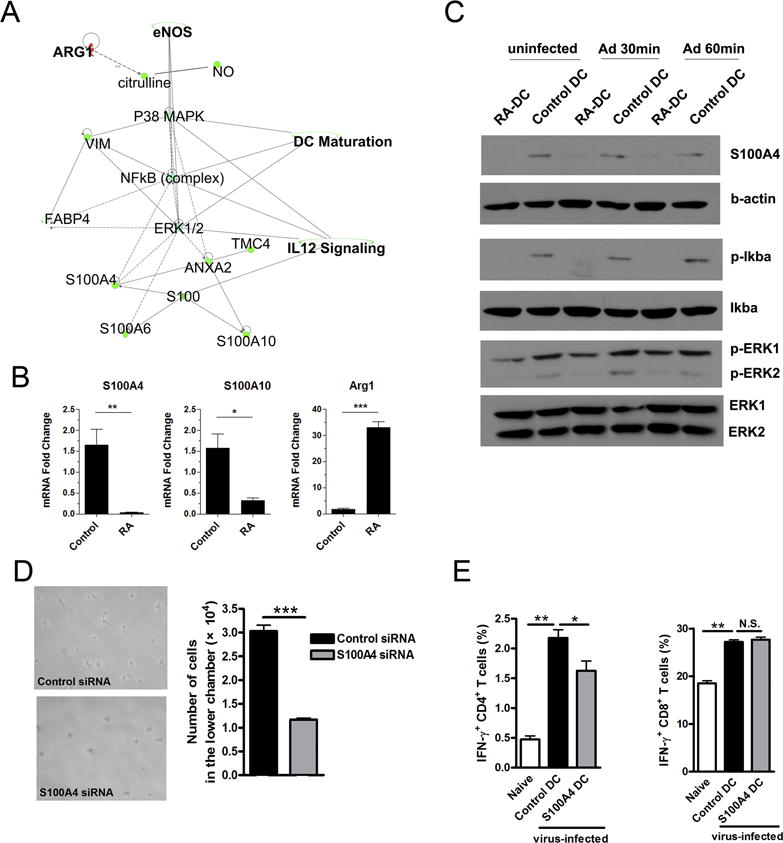
The proteins from control and RA-treated DC were extracted, digested by trypsin, and labeled (see Materials and Methods section). Labeled peptides were analyzed by using mass spectrometry. (A) Predicted canonical pathways were generated from mass spectrometry data analysis by Ingenuity Pathway Analysis (IPA) Software. (B) Transcript levels of arginase 1, S100A4 and S100A10 in DCs. (C) Control and RA-treated DCs were infected by Ad-LacZ (MOI: 300) for 30 and 60 min, respectively. Shown are western blot analysis of S100A4, p-Iκbα and p-ERK. (D) 2 × 105 control or S100A4 koncodown-DCs were cultured, respectively, in the upper chambers of 24-well transwell plates. The lower chambers contained complete medium with rCCL21 (100 ng/ml). After 8 h of culture, the cells in the lower chambers were collected for photographing and counting. (E) DCs were incubated with S100A4 siRNA or control siRNA for 6 h and cultured for another 3 days, followed by transwell assay (as shown in Fig. 5D) and in vivo T cell priming analysis (as shown in Fig. 5E). Each experiment was repeated at least three times independently. *p < 0.05, **p < 0.01, ***p < 0.001.
S100A4 is critical for DC functions in vitro and in vivo
S100A4 has been shown to bind to intracellular myosin IIA in macrophages and regulate their cytoskeleton constituents (49). We speculated that RA can control cellular polarization and mobility of DCs by suppressing S100A4 expression. To test this hypothesis, we inhibited S100A4 expression in DCs using siRNA to S100A4 or the ERK inhibitor U0126 (Fig. S4C and D). Interestingly, inhibition of S100A4 did not alter the expression level of CCR7 on DCs, but slightly decreased the CD80 and CD86 expression levels (Fig. S4E). By using a CCL21-dependent transwell assay, as described above (Fig. 5D), we found that S100A4-siRNA treatment resulted in significant fewer DCs in the lower chamber compared to those found in control siRNA-treated DCs (Fig. 6D). By using the in vivo T cell priming assay, as shown in Fig. 5E, we found that S100A4 down-regulation by siRNA reduced the capability of DCs to egress and prime CD4+ T cells in distant LNs (Fig. 6E). S100A4-repressed DCs led to decreased frequencies of IFN-γ+ CD4+ T cells in draining LNs; no differences in CD8+ T cell functionality were observed (Fig. 6E). These data argue that RA can down-regulate NF-κB/ERK/S100 proteins and suppress DC migration and functions in vivo.
DISCUSSION
The liver stores the body’s majority of vitamin A. Its principal metabolite, all-trans RA, has been linked with T cell exhaustion in hepatitis (4, 5, 50, 51). However, it is unknown how RA in the liver can compromise T cell priming in distant lymphoid organs. In this study, we used a murine model of adenovirus infection to mimic viral infection in human. Although recombinant Ad5 strain can’t present the full life cycle of a coiled-type virus, it can induce virus-specific T cells and cause the liver injury, which are similar with the responses in viral hepatitis in humans (52). We observed that viral infection can increase the RALDH1 activity and found that exogenous RA greatly attenuated liver injury by inhibiting inflammatory infiltration and decreasing T cell responses (Fig. 1). RA also promoted hepatoprotective IL-22 production, but inhibited IL-17 production, from γδ T and DN T cells through a PI3K/mTORC1-dependent mechanism. In addition, RA strongly modulated DC maturation and migration and compromised T cell priming in distant lymphoid organs. RA achieved this, in part, by repressing the expression of novel S100 family proteins and activation of the NF-kB/ERK pathways. Our study has revealed a previously unappreciated role and multipronged mechanism of RA in hepatoprotection and immune regulation in viral hepatitis.
RA is essential for T cell differentiation and effector responses (53–55); however, it can also suppress Th1 polarization and T effector cell proliferation (56–58). RA was found to preferentially induce TGF-β-dependent Treg cells, playing a role in the tolerogenic nature (3, 4, 59–61). Most Treg cell studies were performed in vitro by using cell cultures in the presence of RA. In a colitis mouse model, however, RA treatment failed to enhance intestinal Foxp3 gene expression (15). Here, we found that RA inhibited T cell responses (Fig. 1), but did not induce Treg cells in viral hepatitis (Fig. S1). Both high-dose (250 μg) and moderate-dose (25 μg) of RA can reduce the number of cytokine-producing cells in the spleen and liver. In addition to inducing IL-22, RA can suppress IL-17 and IFN-γ production, suggesting that RA may reduce the liver inflammation by inhibiting the pro-inflammatory cytokines. Our results are consistent with other reports (15, 58), therefore pointing to possible additional underlying mechanisms for RA-induced immune tolerance (62). RA treatment did not significantly change viral copy numbers in mouse liver at 6 dpi (Supp. Fig. 1B). These findings are consistent with previous reports that most of adenovirus is eliminated rapidly by the innate immune mechanisms 24 h post-infection; the remaining is cleared by T cells slowly in the subsequent 3 weeks (35, 63). Although RA can induce IL-22 to protect the liver from damage, it also inhibits T cell responses and may presumably delay viral clearance in patients. Therefore, antiviral therapy needs to combine with potential RA treatment to prevent fulminant viral hepatitis.
IL-22 expression is upregulated in the liver of HBV and HCV patients (64, 65), and single-nucleotide polymorphisms in the IL-22 gene influence treatment response and viral clearance (66), possibly meaning that IL-22 can modulate antiviral immune response. In this study, we demonstrated that γδ T cells and DN T cells, but not the Th17 cells (20, 67), act as the source of liver-derived IL-22. We report, for the first time, that IL-22 attenuates liver injury in viral infection mainly through its regulation of intrahepatic infiltration and CD4+ T cell activation (Fig. 4). The contradiction between our findings and those reported by others (20, 67) may be attributed to the differences of animal models and absence of Th17 cells in early viral infection. The mTOR signaling pathway is necessary for immune cell homeostatic and functional fates (39, 68). Here, we found that IL-17 and IL-22 production in γδ T cells and DN T cells were controlled by the PI3K/mTORC1 signaling pathway. These results have led us to suggest for the first time that PI3K/mTORC1 signals not only control adaptive T cell differentiation fate decisions (68), but also modulate γδ T cell and DN T cell functions. Interestingly, RA can synergize with PI3K/mTORC1 inhibitors to further suppress IL-17 production (Fig. 3B). This finding may imply that, in addition to the mTOR/STAT3 pathway of IL-17 regulation (68), RA may also regulate IL-17 production through an mTOR/STAT3-independent means (57).
Highly effective antigen presentation is accomplished in two distant organs during viral hepatitis (42, 69, 70). Upon encountering antigens in the inflamed tissues, DCs rapidly mature and egress to draining LNs to prime naïve T cells. Activated T cells subsequently migrate from the lymphoid organs to the inflamed organs (71). Here, we demonstrated that RA treatment down-regulated the co-stimulatory molecules CD86 and CD40, decreased the chemotactic ability of DCs in vitro (Fig. 5A–D) and impaired their capacity of T cell priming in vivo (Fig. 5E). Using a non-hypothesis-driven, proteomic approach, we found that RA treatment significantly down-regulated the expression of a family of calcium-binding proteins, including S100A4, S100A6 and S100A10 in DCs (Fig. 6A and B). RA treatment down-regulated NFκB and ERK pathways, which are involved in S100 protein regulation, DC maturation, and IL-12 signaling. Given that S100A4 can bind to intracellular myosin IIA in macrophages and regulate dynamics of cytoskeleton constituents (44, 49), we hypothesized that the presence of RA in the liver can directly modulate cellular polarization and motility of intrahepatic DCs. We knocked down S100A4 and found that downregulation of S100A4 led to impaired capacity of DC migration in vitro and of priming T cells in an in vivo transfer study. In addition, RA-treated DCs displayed high levels of Arg-1, which is similar to myeloid-derived suppressor cells (72). These results confirm that the DC inhibition caused by high levels of RA in the liver microenvironment may lead to the T cell dysfunction, including tolerance, anergy, exhaustion or senescence in chronic viral hepatitis (42, 73). Our data demonstrated, for the first time, that RA controls DC migration through modulating S100 family proteins. Interestingly, RA has also been shown to regulate the migration of Treg cells in T cell-mediated acute hepatitis (74), which suggested to us that, in addition to DCs, RA may regulate other immune cells migration. It is also possible that RA blockade resulted in the release of DCs from liver to secondary lymphoid organs, which promoted the activation or expansion of Tregs.
In summary, we found that exogenous RA can attenuate liver injury in viral hepatitis by promoting the PI3K/mTORC1-dependent IL-22 production in liver γδ and DN T cells. We further identified a novel pathway by which RA inhibits DC function through a S100A4-mediated mechanism, resulting in down-regulation of T cell responses. This study has demonstrated the importance of innate intrahepatic subpopulations of γδ and DN T cells and their cytokine profiles during the early stages of viral infection in the liver. Since the cross-talk among RA and DCs plays a key role in modulating adaptive immune responses, further modifying these communications may represent a potential therapeutic approach to treat viral hepatitis.
Supplementary Material
Acknowledgments
We thank Dr. Wenjun Ouyang for the IL-22 KO mice, Dr. Netanya Sandler Utay for critical reading of the manuscript, and Ms. Linsey Yeager for assistance with manuscript preparation. The authors wish to express gratitude to other members of the UTMB Joint Immunology Working Group (Drs. Robin Stephens, Ricardo Rajsbaum, Haitao Hu, and their trainees) for many helpful discussions.
This work was supported in part by grants from the NIH (AI109100 and AI126371 to JS). Panpan Yi was partially supported by the Department of Infectious Diseases, Xiangya Hospital, China and the Natural Science Foundation of Hunan Province (#14JJ6003).
Abbreviations
- Ad
adenovirus
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- DCs
dendritic cells
- DN T
double-negative T
- γδ
gamma delta
- IHL
intrahepatic lymphocyte
- LNs
lymph nodes
- mTOR
mammalian target of rapamycin
- PI3K
phosphoinositide 3-kinase
- RA
all-trans retinoic acid
Footnotes
Disclosures: The authors declare no financial or commercial conflict of interest.
References
- 1.Wu Z, Han M, Chen T, Yan W, Ning Q. Acute liver failure: mechanisms of immune-mediated liver injury. Liver Int. 2010;30:782–794. doi: 10.1111/j.1478-3231.2010.02262.x. [DOI] [PubMed] [Google Scholar]
- 2.Hou L, Jie Z, Desai M, Liang Y, Soong L, Wang T, Sun J. Early IL-17 Production by Intrahepatic T Cells Is Important for Adaptive Immune Responses in Viral Hepatitis. J Immunol. 2013;190:621–629. doi: 10.4049/jimmunol.1201970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC. Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal Immunol. 2015;8:265–278. doi: 10.1038/mi.2014.64. [DOI] [PubMed] [Google Scholar]
- 4.Dunham RM, Thapa M, Velazquez VM, Elrod EJ, Denning TL, Pulendran B, Grakoui A. Hepatic Stellate Cells Preferentially Induce Foxp3+ Regulatory T Cells by Production of Retinoic Acid. J Immunol. 2013;190:2009–2016. doi: 10.4049/jimmunol.1201937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng T, Cong Y, Qin H, Benveniste EN, Elson CO. Generation of mucosal dendritic cells from bone marrow reveals a critical role of retinoic acid. J Immunol. 2010;185:5915–5925. doi: 10.4049/jimmunol.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809–816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 7.Lee YS, Jeong WI. Retinoic acids and hepatic stellate cells in liver disease. J Gastroenterol Hepatol. 2012;27(Suppl 2):75–79. doi: 10.1111/j.1440-1746.2011.07007.x. [DOI] [PubMed] [Google Scholar]
- 8.Fickenscher H, Hor S, Kupers H, Knappe A, Wittmann S, Sticht H. The interleukin-10 family of cytokines. Trends Immunol. 2002;23:89–96. doi: 10.1016/s1471-4906(01)02149-4. [DOI] [PubMed] [Google Scholar]
- 9.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 10.Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, Cianfarani F, Odorisio T, Traidl-Hoffmann C, Behrendt H, Durham SR, Schmidt-Weber CB, Cavani A. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119:3573–3585. doi: 10.1172/JCI40202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP. gammadelta T cells protect against lung fibrosis via IL-22. J Exp Med. 2010;207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar P, Thakar MS, Ouyang W, Malarkannan S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013;6:69–82. doi: 10.1038/mi.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci U S A. 2013;110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mielke LA, Jones SA, Raverdeau M, Higgs R, Stefanska A, Groom JR, Misiak A, Dungan LS, Sutton CE, Streubel G, Bracken AP, Mills KH. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117–1124. doi: 10.1084/jem.20121588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radaeva S, Sun R, Pan H-n, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 17.Park O, Wang H, Weng H, Feigenbaum L, Li H, Yin S, Ki SH, Yoo SH, Dooley S, Wang FS, Young HA, Gao B. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology. 2011;54:252–261. doi: 10.1002/hep.24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: Role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Zhang Y, Wang L, Fan F, Zhu L, Li Z, Ruan X, Huang H, Wang Z, Huang Z, Huang Y, Yan X, Chen Y. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol. 2010;53:339–347. doi: 10.1016/j.jhep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Cobleigh MA, Lian JQ, Huang CX, Booth CJ, Bai XF, Robek MD. A Proinflammatory Role for Interleukin-22 in the Immune Response to Hepatitis B Virus. Gastroenterology. 2011;141:1897–1906. doi: 10.1053/j.gastro.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang SC, Nickerson-Nutter C, Pittman DD, Carrier Y, Goodwin DG, Shields KM, Lambert AJ, Schelling SH, Medley QG, Ma HL, Collins M, Dunussi-Joannopoulos K, Fouser LA. IL-22 induces an acute-phase response. J Immunol. 2010;185:5531–5538. doi: 10.4049/jimmunol.0904091. [DOI] [PubMed] [Google Scholar]
- 22.Foster R, Golden-Mason L, Rutebemberwa A, Rosen H. Interleukin (IL)-17/IL-22-Producing T cells Enriched Within the Liver of Patients with Chronic Hepatitis C Viral (HCV) Infection. Digest Dis Sci. 2012;57:381–389. doi: 10.1007/s10620-011-1997-z. [DOI] [PubMed] [Google Scholar]
- 23.Velazquez VM, Hon H, Ibegbu C, Knechtle SJ, Kirk AD, Grakoui A. Hepatic enrichment and activation of myeloid dendritic cells during chronic hepatitis C virus infection. Hepatology. 2010;56:2071–2081. doi: 10.1002/hep.25904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 25.MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang Y, Jie Z, Hou L, Yi P, Wang W, Kwota Z, Salvato M, de Waal Malefyt R, Soong L, Sun J. IL-33 promotes innate IFN-gamma production and modulates dendritic cell response in LCMV-induced hepatitis in mice. Eur J Immunol. 2015;45(11):3052–63. doi: 10.1002/eji.201545696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jie Z, Liang Y, Hou L, Dong C, Iwakura Y, Soong L, Cong Y, Sun J. Intrahepatic Innate Lymphoid Cells Secrete IL-17A and IL-17F That Are Crucial for T Cell Priming in Viral Infection. J Immunol. 2014;192:3289–3300. doi: 10.4049/jimmunol.1303281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhatt S, Qin J, Bennett C, Qian S, Fung JJ, Hamilton TA, Lu L. All-trans retinoic acid induces arginase-1 and inducible nitric oxide synthase-producing dendritic cells with T cell inhibitory function. J Immunol. 2014;192:5098–5108. doi: 10.4049/jimmunol.1303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan J, Jie Z, Hou L, Wanderley JL, Soong L, Gupta S, Qiu S, Chan T, Sun J. Parenchymal expression of CD40 exacerbates adenovirus-induced hepatitis in mice. Hepatology. 2011;53:1455–1467. doi: 10.1002/hep.24270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xin L, Li K, Soong L. Down-regulation of dendritic cell signaling pathways by Leishmania amazonensis amastigotes. Mol Immunol. 2008;45:3371–3382. doi: 10.1016/j.molimm.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderson SE, Santangelo KS, Bertone AL. Chondrogenic effects of exogenous retinoic acid or a retinoic acid receptor antagonist (LE135) on equine chondrocytes and bone marrow-derived mesenchymal stem cells in monolayer culture. Am J Vet Res. 2011;72:884–892. doi: 10.2460/ajvr.72.7.884. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Wang H, Liang T, Cai X, Rao X, Huang Z, Sheng G. Retinoic acid promotes neural conversion of mouse embryonic stem cells in adherent monoculture. Mol Biol Rep. 2012;39:789–795. doi: 10.1007/s11033-011-0800-8. [DOI] [PubMed] [Google Scholar]
- 33.Qi H, Popov V, Soong L. Leishmania amazonensis-dendritic cell interactions in vitro and the priming of parasite-specific CD4(+) T cells in vivo. J Immunol. 2001;167:4534–4542. doi: 10.4049/jimmunol.167.8.4534. [DOI] [PubMed] [Google Scholar]
- 34.Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, Wollman J. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 35.Liang Y, Jie Z, Hou L, Aguilar-Valenzuela R, Vu D, Soong L, Sun J. IL-33 Induces Nuocytes and Modulates Liver Injury in Viral Hepatitis. J Immunol. 2013;190:5666–5675. doi: 10.4049/jimmunol.1300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang YJ, Luo J, Yuan Q, Zheng D, Liu YP, Shi L, Zhou Y, Chen AL, Ren YY, Sun KY, Sun Y, Wang Y, Zhang ZS. New insight into the antifibrotic effects of praziquantel on mice in infection with Schistosoma japonicum. PLoS One. 2011;6:e20247. doi: 10.1371/journal.pone.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305–315. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roark CL, Simonian PL, Fontenot AP, Born WK, O’Brien RL. gammadelta T cells: an important source of IL-17. Curr Opin Immunol. 2008;20:353–357. doi: 10.1016/j.coi.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Muller M, Saemann MD. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–577. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 40.Nauc V, De Lamirande E, Leclerc P, Gagnon C. Inhibitors of phosphoinositide 3-kinase, LY294002 and wortmannin, affect sperm capacitation and associated phosphorylation of proteins differently: Ca2+-dependent divergences. J Androl. 2004;25:573–585. doi: 10.1002/j.1939-4640.2004.tb02828.x. [DOI] [PubMed] [Google Scholar]
- 41.Belz G, Mount A, Masson F. Dendritic cells in viral infections. Handb Exp Pharmacol. 2009;188:51–77. doi: 10.1007/978-3-540-71029-5_3. [DOI] [PubMed] [Google Scholar]
- 42.Rodrigue-Gervais IG, Rigsby H, Jouan L, Sauve D, Sekaly RP, Willems B, Lamarre D. Dendritic cell inhibition is connected to exhaustion of CD8+ T cell polyfunctionality during chronic hepatitis C virus infection. J Immunol. 2010;184:3134–3144. doi: 10.4049/jimmunol.0902522. [DOI] [PubMed] [Google Scholar]
- 43.Inaba K, Metlay JP, Crowley MT, Steinman RM. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med. 1990;172:631–640. doi: 10.1084/jem.172.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li ZH, Dulyaninova NG, House RP, Almo SC, Bresnick AR. S100A4 regulates macrophage chemotaxis. Mol Biol Cell. 2010;21:2598–2610. doi: 10.1091/mbc.E09-07-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boomershine CS, Chamberlain A, Kendall P, Afshar-Sharif AR, Huang H, Washington MK, Lawson WE, Thomas JW, Blackwell TS, Bhowmick NA. Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut. 2009;58:1267–1274. doi: 10.1136/gut.2008.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hilly O, Koren R, Raz R, Rath-Wolfson L, Mizrachi A, Hamzany Y, Bachar G, Shpitzer T. The role of s100-positive dendritic cells in the prognosis of papillary thyroid carcinoma. Am J Clin Pathol. 2013;139:87–92. doi: 10.1309/AJCPAKYDO56NKMYZ. [DOI] [PubMed] [Google Scholar]
- 47.Zeid NA, Muller HK. S100 positive dendritic cells in human lung tumors associated with cell differentiation and enhanced survival. Pathology. 1993;25:338–343. doi: 10.3109/00313029309090853. [DOI] [PubMed] [Google Scholar]
- 48.Wilson AJ, Maddox PH, Jenkins D. CD1a and S100 antigen expression in skin Langerhans cells in patients with breast cancer. J Pathol. 1991;163:25–30. doi: 10.1002/path.1711630106. [DOI] [PubMed] [Google Scholar]
- 49.Li ZH, Bresnick AR. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res. 2006;66:5173–5180. doi: 10.1158/0008-5472.CAN-05-3087. [DOI] [PubMed] [Google Scholar]
- 50.Lu L, Lan Q, Li Z, Zhou X, Gu J, Li Q, Wang J, Chen M, Liu Y, Shen Y, Brand DD, Ryffel B, Horwitz DA, Quismorio FP, Liu Z, Li B, Olsen NJ, Zheng SG. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A. 2014;111:E3432–3440. doi: 10.1073/pnas.1408780111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ichikawa S, Mucida D, Tyznik AJ, Kronenberg M, Cheroutre H. Hepatic Stellate Cells Function as Regulatory Bystanders. J Immunol. 2011;186:5549–5555. doi: 10.4049/jimmunol.1003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang Y, Ertl HC, Wilson JM. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 53.Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, Grigg ME, Kastenmayer R, Schwartzberg PL, Belkaid Y. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity. 2011;34:435–447. doi: 10.1016/j.immuni.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allie SR, Zhang W, Tsai CY, Noelle RJ, Usherwood EJ. Critical role for all-trans retinoic acid for optimal effector and effector memory CD8 T cell differentiation. J Immunol. 2013;190:2178–2187. doi: 10.4049/jimmunol.1201945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan X, Sande JL, Pufnock JS, Blattman JN, Greenberg PD. Retinoic acid as a vaccine adjuvant enhances CD8+ T cell response and mucosal protection from viral challenge. J Virol. 2011;85:8316–8327. doi: 10.1128/JVI.00781-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwata M, Eshima Y, Kagechika H. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol. 2003;15:1017–1025. doi: 10.1093/intimm/dxg101. [DOI] [PubMed] [Google Scholar]
- 57.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O’Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–2284. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. 2007;179:3724–3733. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 62.Thatcher JE, Isoherranen N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin Drug Metab Toxicol. 2009;5:875–886. doi: 10.1517/17425250903032681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004;15:1157–1166. doi: 10.1089/hum.2004.15.1157. [DOI] [PubMed] [Google Scholar]
- 64.Xiang X, Gui H, King NJ, Cole L, Wang H, Xie Q, Bao S. IL-22 and non-ELR-CXC chemokine expression in chronic hepatitis B virus-infected liver. Immunol Cell Biol. 2012;90:611–619. doi: 10.1038/icb.2011.79. [DOI] [PubMed] [Google Scholar]
- 65.Dambacher J, Beigel F, Zitzmann K, Heeg MH, Goke B, Diepolder HM, Auernhammer CJ, Brand S. The role of interleukin-22 in hepatitis C virus infection. Cytokine. 2008;41:209–216. doi: 10.1016/j.cyto.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 66.Hennig BJ, Frodsham AJ, Hellier S, Knapp S, Yee LJ, Wright M, Zhang L, Thomas HC, Thursz M, Hill AV. Influence of IL-10RA and IL-22 polymorphisms on outcome of hepatitis C virus infection. Liver Int. 2007;27:1134–1143. doi: 10.1111/j.1478-3231.2007.01518.x. [DOI] [PubMed] [Google Scholar]
- 67.Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, Jin L, Zhou C, Fu J, Gao B, Fu Y, Wang FS. Pathological functions of interleukin-22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology. 2014;59:1331–1342. doi: 10.1002/hep.26916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lau AH, Thomson AW. Dendritic cells and immune regulation in the liver. Gut. 2003;52:307–314. doi: 10.1136/gut.52.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barbier L, Tay SS, McGuffog C, Triccas JA, McCaughan GW, Bowen DG, Bertolino P. Two lymph nodes draining the mouse liver are the preferential site of DC migration and T cell activation. J Hepatol. 2012;57:352–358. doi: 10.1016/j.jhep.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 71.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–628. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 72.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 74.Lee YS, Yi HS, Suh YG, Byun JS, Eun HS, Kim SY, Seo W, Jeong JM, Choi WM, Kim MH, Kim JH, Park KG, Jeong WI. Blockade of Retinol Metabolism Protects T Cell-Induced Hepatitis by Increasing Migration of Regulatory T Cells. Mol Cells. 2015;38:998–1006. doi: 10.14348/molcells.2015.0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.