

Graphical abstract

Abbreviations: AAS, atomic absorption spectroscopy; AD, Alzheimeŕs disease; LBs, Lewy bodies; LBD, Lewy body disease; PD, Parkinsońs disease; SN, substantia nigra
Keywords: Brain, Iron, AAS, Parkinsońs disease, Lewy body disease
Highlights
-
•
Sensitive detection of total iron content from cells, rodent and human brain extracts.
-
•
There are regional differences in iron concentrations in rodent and human brain.
-
•
Iron dyshomeostasis in the substantia nigra correlates with Lewy body disease.
-
•
Iron imaging studies may aid in clinical diagnosis and/or serve as biomarker for disease progression.
Abstract
Iron levels in the healthy human brain are known to be high in certain areas such as the substantia nigra (SN), and increase further with age. In addition, there is some evidence for a further increase in iron load in the SN of Parkinsońs disease (PD) patients as compared to controls, which correlates with motor disability. Here, we have analyzed total iron levels in cells as well as mouse and human brain samples by atomic absorption spectroscopy (AAS). Our data indicate that iron load is more pronounced in cells with dopaminergic features. Moreover, region-specific differences in iron load reflecting those in the human brain were detected in rodent brains as well. Whilst altered iron load was not observed in other regions also affected in PD patients, we report a significant increase in iron load in the SN of Lewy body disease patients as compared to Alzheimeŕs disease (AD) patients or controls, which correlates with neurodegeneration in this brain area.
1. Introduction
Iron is an important transition metal which plays a crucial role in many chemical reactions such as oxidative phosphorylation, myelin production or the synthesis and metabolism of neurotransmitters [1], [2]. However, excess iron can cause hydroxyl radical production via the Fenton reaction, leading to oxidative stress accompanied by the oxidation and modification of proteins, lipids, carbohydrates and DNA [3], [2]. Therefore, cellular iron levels tend to be tightly regulated.
During healthy ageing, iron is known to accumulate in several brain regions and cell types [2]. Generally the highest concentrations of iron are found in the substantia nigra (SN) as well as in the dopaminergic projection areas of the striatum including caudate nucleus and putamen [3], [4]. Interestingly, these are the sites most vulnerable to neurodegeneration underlying parkinsonian phenotypes, suggesting that excessive iron accumulation in those areas may contribute to neurodegeneration through generation of oxidative stress and resultant neuronal vulnerability [2], [5].
Neuropathologically, Parkinsońs disease (PD) is characterized by the relatively selective loss of dopaminergic neurons in the SN, with Lewy bodies (LBs) rich in α-synuclein present in surviving neurons. Additional brain areas, such as the caudate nucleus, are also known to be particularly affected during relatively early stages of disease development [6], [7]. Other neurodegenerative disorders such as Lewy Body Disease (LBD) are characterized by the presence of LBs in the SN, but in contrast to PD present with widespread LB pathology [8], [9]. Finally, Alzheimer disease (AD) pathology is characterized by senile plaques rich in Aβ and abnormal tau depositions in distinct areas throughout the brain [10]. The link between increased iron content and region-specific neuropathological changes and cell death in the distinct neurodegenerative diseases has remained largely unclear.
Given the particularly high levels of iron in the SN, many studies have aimed to determine alterations in total iron content in areas vulnerable to neurodegeneration in PD patients as compared to healthy controls. Distinct in vivo magnetic resonance imaging (MRI) approaches have reported increases in iron load in the SN of PD patients as compared to healthy controls, but contradictory results have been obtained in other brain areas such as the putamen or caudate nucleus (reviewed in [11]. However, there are limitations with respect to iron imaging using MRI approaches, as signals can be confounded by other factors such as calcium, lipid or myelin content [12], [13], [11]. Therefore, careful analysis of total iron content in postmortem samples is crucial towards establishing a clear correlation between increased iron load and neurodegeneration in PD. Analysis of iron content from postmortem samples have yielded conflicting outcomes, with either no change or an increase in total iron content in the SN and putamen of PD patients as compared to controls, and absolute values differing by a factor of almost 10 (reviewed in [14], [11]. Amongst those studies, iron determinations have been performed using distinct techniques such as X-ray fluorescent spectroscopy, inductively coupled plasma spectroscopy, atomic absorption spectroscopy (AAS), spectrophotometry, colorimetry or Mossbauer spectroscopy [15], [16], [14], [17], [18], [19], [20], [21], [22], [23], [11], [24], [25] reviewed in [14], [11]. At least in some cases, heterogeneity of the results may have been due to the differential sensitivities and specificities of the methods employed. However, other factors such as differences in age or disease state of patients, or methods of sample collection, sample storage or sample size may have contributed to the observed discrepant findings as well.
The aim of the present study was to develop a sensitive and reproducible method for detecting total iron levels in cellular, animal-derived and human-derived brain samples using AAS, and to gauge for region-specific differences in iron overload in various neurodegenerative diseases.
2. Materials and methods
2.1. Cells and sample preparation
HEK293T and rat dopaminergic neuroendocrine PC12 cells were cultured as described before [28]. Cells were treated with ferric ammonium citrate (FAC; Sigma Aldrich, F5879) using the indicated concentrations and times, and cell extracts prepared from a 100 mm dish by scraping cells off the dish in the presence of 1 ml ice-cold phosphate-buffered saline (PBS). Resuspended cells were centrifuged at 16,100g for 10 min at 4 °C, and wet weight of the cell pellet was determined before sample digestion using 500 μl of pure HNO3 at 65 °C during 2 h. The resulting lysates were subjected to AAS determination without further dilution. Total iron content was normalized to either wet weight or to total protein content as determined by Bradford assay (Thermo Scientific, 23227) upon appropriate dilution of samples [29], [30].
2.2. Animals and sample preparation
Both inbred C57BL/6 and outbred CD-1 mice were obtained from Charles River, and housed under standard conditions of light, temperature and humidity with unlimited access to water and food. All experimental animal protocols and animal procedures complied with National guidelines for the care and use of laboratory animals, and were according to principles and directives of the European Communities Council Directives. Animals were either two or nine months old at the time of sample preparation. Animals were sacrificed by cervical dislocation, and brain tissue was quickly dissected on ice. Cortex, cerebellum and substantia nigra were obtained, wet weight determined, cut into smaller pieces if necessary and samples immediately frozen and stored under liquid nitrogen. Frozen samples from each region were resuspended in 500 μl of pure HNO3 for 2 h at 65 °C. Four additional protocols were employed, which included: 1) resuspension of samples in 500 μl of a mix of pure NHO3 and H2O2 (1:1) and incubation at 90 °C for 2 h; 2) resuspension of slices in 500 μl of pure HNO3 overnight at room temperature, followed by heating samples for 20 min at 90 °C the following day, addition of an equal volume of H2O2 and incubation for 15 min at 70 °C; 3) resuspension of samples in 500 μl cell lysis buffer (1% SDS in PBS, pH 7.4 containing 1 mM PMSF, 1 mM Na3VO4 and 5 mM NaF) Dounce homogenized and sonicated; 4) resuspension of samples in 250 μl cell lysis buffer, Dounce homogenization and sonication, followed by addition of 250 μl pure HNO3. In all cases, lysates were mixed and sonicated twice during incubations, and centrifuged at 3500 × g for 30 min at 4 °C. An aliquot was taken for quantification of iron content and the remainder was frozen down. Samples were subjected to AAS upon a 1:10 dilution in MilliQ H2O. Total iron content was normalized to either wet weight or to total protein content as determined by Bradford assay (Thermo Scientific, 23227) upon appropriate dilution of samples [31].
2.3. Human postmortem tissue and sample preparation
Samples from three patients with PD and three age-matched controls, or from five patients with LBD, five patients with AD and five age-matched controls were obtained at postmortem following informed consent from next of kin, and under a protocol approved by the Local Ethics Committee of the Bellvitge University Hospital (June 2014, reference number PR059/14). The postmortem delay between death and tissue processing was betweeen 4 and 9 h, and the pH of the brain was between 6.6 and 6.9 in all cases. One-half of the brain was immediately cut on coronal 1 cm-thick sections, frozen in dry ice and stored at −80 °C until use.
All sporadic PD cases had suffered from classical parkinsonism and none of them had apparent cognitive impairment or dementia. Cases analyzed here were tested for the G2019S LRRK2 mutation and were found negative for this mutation.
A complete neuropathological examination was performed in all cases using formalin-fixed tissue which was embedded in paraffin no less than three weeks later basically as previously described [32], [33].
Neuropathological characterization of PD was according to established criteria [7], and PD cases corresponded to stage 4 of Braak and Braak, implying involvement of selected nuclei of the medulla oblongata, pons and midbrain, amygdala and nucleus basalis of Meynert. All PD cases showed marked loss of neurons in the substantia nigra pars compacta exceeding 60%, whereas moderate pathology occurred in the caudate and putamen. In no case did Lewy pathology involve the frontal cortex. Diagnostic determination of LBD was carried out following consensus guidelines [9], and staging of AD changes was performed following Braak and Braak criteria [10]. According to those criteria, LBD cases were stages 4/5, and AD cases were stage 5. Control subjects showed absence of neurological symptoms, metabolic and vascular diseases, and the neuropathological study disclosed no abnormalities including lack of PD- and AD-related pathology.
Postmortem slices (around 0.07 g) were resuspended in pure HNO3 (500 μl) and incubated at 65 °C for 2 h, and lysates were mixed and sonicated twice during this incubations, followed by centrifugation at 3500 x g for 30 min at 4 °C. An aliquot was taken for quantification of iron content and the remainder was frozen down. Samples were subjected to AAS upon a 1:10 dilution in MilliQ H2O. Total iron content was normalized to tissue wet weight. Three independent sections were analyzed per brain region and patient to obtain an averaged value of total iron content.
2.4. Atomic absorption spectroscopy
An AAnalyst 800 atomic absorption spectrometer (Perkin Elmer, Norwalk, USA) equipped with a flame was used for iron determination. Mono-elementary hollow cathode lamps (Perkin Elmer) for iron, operating at the recommended currents, were employed as light source. Certified reference solutions of iron for AAS (titrisol grade, Merck) were used to generate standard curves, and dilutions prepared using reverse osmosis type quality water produced by Milli-RO 12 plus Milli-Q purification system (Millipore, Bedford, MA, USA). Standard curves ranged from zero to 5 mg iron/l (linear equation through zero, correlation coefficient 0.997), and the total iron concentrations determined from the standard curve were normalized to either total protein concentration or wet tissue weight as indicated. Analytical method for the determination of iron was previously validated. The validation parameters for the analytical procedure included calibration curve range, LOD/LOQ, precision (minimal, intermediate and reproducibility), and recovery percentage.
2.5. Sample preparation and analysis by western blotting
Postmortem brain lysates were prepared from slices of individual tissues, extracts were resolved by sodium dodecylsulfate-polyacrylamide gel electrophoresis followed by Western blotting as previously described [33]. The following antibodies were employed: a mouse monoclonal anti-LAMP2 (H4B4) (1:2000, Santa Cruz Biotechnology, sc18822), a mouse monoclonal anti-LAMP1 (H4A3) (1:2000, Santa Cruz Biotechnology, sc20011), a rabbit monoclonal anti-ACTIN (1:400, Sigma-Aldrich, A2066/030M4844), a monoclonal anti-α-Tubulin (clone DM1A, 1:10,000, Sigma-Aldrich T6199), a rabbit polyclonal Anti-Phospho-AKT473 antibody (1:500, Cell Signaling, 9271), a rabbit polyclonal Anti-AKT antibody (1:500, Cell Signaling, 9272), a rabbit polyclonal Anti-Phospho-ERK antibody (1:500, Cell Signaling, 4370) or a rabbit polyclonal anti-ERK antibody (1:500, Cell Signaling, 4695). The final detection was performed as previously described [33].
2.6. Statistical analysis
Data are represented as mean ± SEM. Statistical comparisons were made using analysis of variance (ANOVA), and a p value < 0.05 was considered statistically significant.
3. Results
3.1. Effects of ferric ammonium citrate (FAC) on iron concentration in cultured cells
Our previous studies have shown that treatment of cultured cells with ferric ammonium citrate (FAC) causes an increase in oxidative stress associated with endolysosomal alterations and impaired autophagy which may be associated with disease pathomechanism(s) [28]. To corroborate that these effects were due to underlying increases in total cellular iron content, we subjected HEK293T cells to increasing concentrations of FAC for either 24 h or 48 h, and determined total iron concentration by AAS (Fig. 1A,B) as previously validated (calibration curve range: 0–5 mg L−1; LOD/LOQ: 0.16/0.54 mg L−1; precision (minimal-intermediate-reproducibility) 1.03-2.57-4.46; % recovery 101). There was a dose- and time-dependent increase in iron content which matched the previously determined dose- and time-dependent increase in apoptosis, indicating that an increase in total iron was the underlying cause for the observed changes in cell viability [28]. Since cells with dopaminergic features such as PC12 cells have been found to display increased apoptosis upon FAC treatment as compared to HEK293T cells [28], we compared total iron concentrations in the presence or absence of FAC treatment amongst the two distinct cell types. PC12 cells were found to display increased cellular iron load as compared to HEK293T cells (Fig. 1C), suggesting that the underlying differences in cell viability are due to differences in intracellular iron load in those two distinct cell lines, rather than due to differential iron sensitivities.
Fig. 1.

FAC causes a dose- and time-dependent increase in total iron levels in cultured cells, and a further increase in cultured cells possessing dopaminergic features. (A) HEK293T cells were exposed to the indicated concentrations of FAC for 24 h, and iron content quantified by AAS. (B) HEK293T cells were exposed to the indicated concentrations of FAC for 48 h, and iron content quantified by AAS. (C) HEK293T cells or dopaminergic PC12 cells were exposed to the indicated concentrations of FAC for 48 h, and iron content analyzed by AAS. Bars represent mean ± SEM (n = 3; * p < 0.05; ** p < 0.01; *** p < 0.005).
3.2. Region-specific differences in total iron in mouse brain
Various protocols for tissue solubilization have been described, including pure nitric acid either in the presence or absence of H2O2, or lysis buffers containing detergents [34], [29], [31]. In addition, different times and temperatures have been employed, ranging from 2 − 12 h and 65 − 90 °C. A careful repetitive and side-by side analysis of the different protocols was performed using cortical tissue from mice. Most accurate and reproducible AAS results were obtained using pure nitric acid for 2 h at 65 °C, which completely dissolved tissue sections, and normalization of total iron content determined by AAS to tissue wet weight rather than to estimations of total protein concentrations, and this methodology was employed in all further studies.
We probed for region-specific and age-dependent alterations in total iron content in mouse brain tissue. AAS analysis was performed on distinct brain regions obtained from either an inbred (Fig. 2A) or an outbred (Fig. 2B) mouse strain. Total iron content was found to be higher in SN as compared to cortex or cerebellum in both strains, and no differences were observed between the two genotypes (Fig. 2A,B). Furthermore, no differences in iron content in the distinct brain regions were observed between female and male samples, or between samples from 2 month-old versus 9 month-old mice, respectively (Fig. 2). Altogether, these data show increased iron content in rodent SN as compared to other brain areas without genotype-specific, sex-specific or age-specific alterations.
Fig. 2.

Increase in iron content in the SN of mice as determined by AAS. (A) Cortex, cerebellum and SN iron content was determined by AAS from female or male, and 2-month or 9-month old outbred CD1 animals as indicated. Bars represent mean ± SEM (n = 3; * p < 0.05). (B) Cortex, cerebellum and SN iron content was determined by AAS from female or male 9-month old inbred C57BL/6 mice as indicated. Bars represent mean ± SEM (n = 4; * p < 0.05).
3.3. Region-specific differences in total iron content in human brain samples from control and PD patients
To minimize variability, samples were carefully chosen according to the following criteria: a) control and disease patients were age- and sex-matched; b) samples were collected within a short postmortem interval, and the pH of the cerebral spinal fluid was within a normal range; c) patient samples were collected from one hemisphere, directly frozen and kept at − 80 °C, whilst the other hemisphere was subjected to a detailed neuropathological analysis; d) samples had comparable disease stage; e) frozen sections were dissolved with pure nitric acid for 2 h at 65 °C in all cases; f) independent iron determinations were performed on 3 individual sections per region per patient; and g) iron concentration was normalized to wet weight of the frozen sections rather than to total protein content (for further details see Materials and methods).
We first analyzed iron content in the cortex, cerebellum and caudate from age- and sex-matched control versus PD patients. Iron content was significantly higher in the caudate as compared to cortex and cerebellum, but there were no differences observed between PD and control samples (Fig. 3). Thus, at least in this sample set, there are no discernible differences in iron content in the caudate nucleus, even though LBs in that area are present during the PD disease stage analyzed here.
Fig. 3.
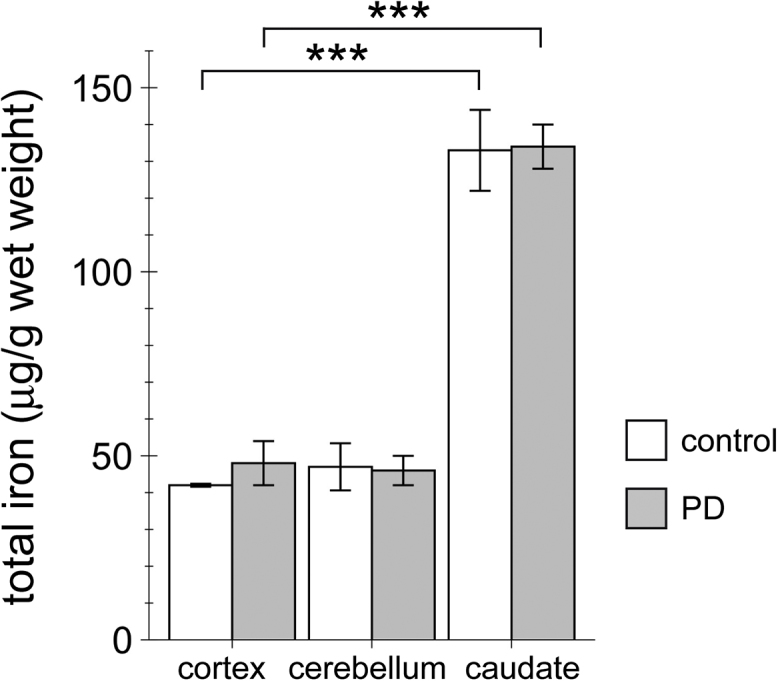
Iron content in the cortex, cerebellum and caudate nucleus from healthy control and age- and sex-matched PD patients. Iron content from the distinct brain areas was determined by AAS. Bars represent mean ± SEM (n = 3; *** p < 0.005).
3.4. Analysis of signalling cascades and endolysosomal alterations in human brain samples from control and PD patients
As another means to determine a link between iron-related events and cellular demise, we performed a biochemical analysis on patient samples. Animal studies using distinct oxidative stress-induced PD models have shown activation of select signalling cascades including kinases of the MAPK superfamily such as Erk1/2 [35]. Furthermore, in SN from PD patients a link was observed between aberrant kinase activation and LB pathology in non-apoptotic cells, suggesting an early role for some of those kinases in LB formation [32], [36]. Similarly, alterations in the phosphorylation status of Akt/PKB have been reported in animal models as well as in the SN of PD patients [37], [38], [39]. Akt/PKB is known to regulate mTORC1 activity which in turn modulates autophagy, a process whereby cytoplasmic constituents such as protein aggregates are engulfed within specialized double-membrane vesicles (autophagosomes) and delivered to the lysosome for degradation, and SN samples from PD patients display altered levels of endolysosomal markers [40], [41]. Therefore, we determined levels of total and/or phosphorylated (activated) forms of Erk1/2 and Akt/PKB, as well as total levels of endolysosomal markers (LAMP1, LAMP2) from postmortem extracts of control and PD subjects (Fig. 4). Even though affected by disease pathology, no significant changes in the levels of the respective proteins were detectable in the caudate nucleus of PD versus control patients (Fig. 4), in agreement with a detectable lack of total iron content as measured by AAS.
Fig. 4.

Analysis of activation status and total levels of several protein kinase pathways and endolysosomal proteins implicated in PD. Protein extracts from caudate nucleus were analyzed by Western blotting for differences in the phosphorylation status and/or total protein levels between control and PD patient samples as indicated.
3.5. Increase in total iron content in SN from LBD as compared to AD and healthy control patients
LBD is clinically associated with memory/cognitive deficits as well as a movement disorder phenotype. As sharing clinical features with AD, it can be misdiagnosed in the early stages of the disease. Neuropathologically, it is characterized by LB in the SN as well as many other brain areas [8], [9], and the common form of LBD also presents with AD pathology. Thus, comparing iron content in the SN from LBD versus AD samples allows for correlating altered iron concentration with LB pathology and parkinsonian features in LBD patients. SN samples from moderate (stage 4/5) LBD or moderate/severe (stage 5) AD were compared to age- and sex-matched healthy controls. AAS determination of total iron content revealed a significant increase in SN from LBD, but not AD patients, as compared to controls (Fig. 5). Thus, the presence of LB pathology and neurodegeneration in the SN of LBD samples, absent from AD samples, correlates with an increase in iron content in this brain area.
Fig. 5.
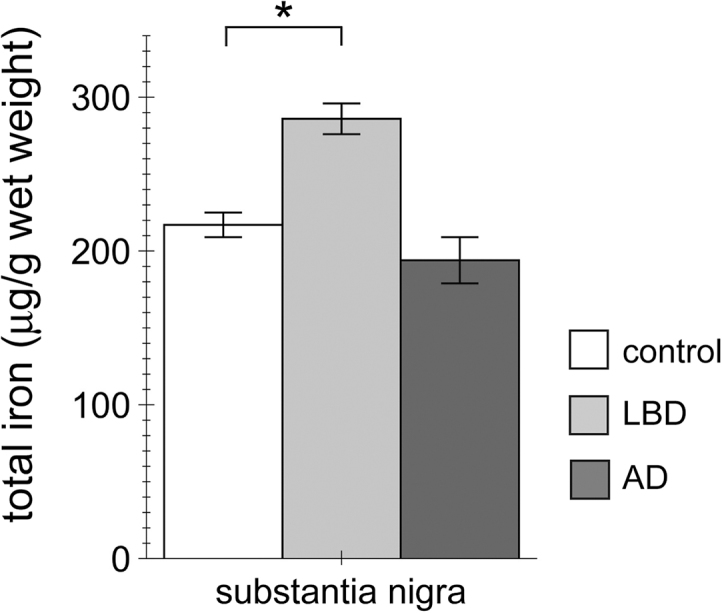
Increase in total iron content in the SN from LBD patients as compared to AD patients or healthy controls. Total iron content in the SN was determined by AAS from control, LBD and AD patients as indicated. Bars represent mean ± SEM (n = 5; * p < 0.05).
4. Discussion
Here, we have analyzed total iron content in cellular systems as well as in mouse and human brain samples to determine region-specific differences in total iron content in the context of several neurodegenerative disorders. Our results show regional differences in iron concentration in distinct rodent and human brain areas. Importantly, our refined detection approach allowed us to determine significant increases in total iron in the SN of LBD patients as compared to AD patients or healthy controls. Our data indicate a positive correlation between iron dyshomeostasis in the SN and neurodegeneration associated with LBD, which may aid in establishing iron-based imaging methods for clinical diagnoses.
Our data indicate that dopaminergic cells are more prone to iron-induced cytotoxicity as compared to non-dopaminergic cells because they display an increase in iron load, rather than an increase in inherent susceptibility towards iron. Several factors may contribute to this observation, including differences in iron uptake and/or export or altered iron storage. Iron is stored in non-toxic form in ferritin, and is also present in mitochondria and the endolysosomal system [5]. In dopaminergic neurons, large amounts of iron are additionally sequestered in neuromelanin granules [5], and oxidative stress has been shown to increase tyrosine hydroxylase activity and thus neuromelanin production in PC12 cells [42]. Thus, whilst further studies will be required to elucidate the mechanism(s) underlying the differential iron load capacities, the increase in iron concentration in cell lines with dopaminergic as compared to non-dopaminergic features may be, at least in part, due to the presence of neuromelanin.
Our study describes refined conditions to determine total iron content by AAS from distinct rodent brain areas. This allowed us to detect increased iron in the rodent SN as compared to other brain areas, which has not been previously detected [31]. The region-specific differences in iron levels in the mouse brain were not dependent on genetic background (inbred versus outbred strains), and no sex-specific differences were observed, indicating that total iron content in rodent model systems is comparable amongst different genetic backgrounds and sexes. Furthermore, no differences in iron content were observed between two-month and nine-month old mice. In humans, total iron concentrations in the SN are known to linearly increase with healthy ageing [2], and an age-dependent increase in total iron in the SN of mice has been previously reported [43]. However, this study compared brain iron in animals aged 5-months versus 14-months old using another methodology, and also reported age-dependent increases in iron load in the cerebellum. Therefore, it currently remains unclear whether animal models can faithfully recapitulate the region-specific and age-specific increase in total iron content observed in human brain during healthy ageing.
The total levels of iron determined in the cortex, cerebellum and caudate nucleus from age- and sex-matched human control patients were similar to a subset of previous reports [11], [15], [18], [20], with caudate nucleus displaying an almost three-fold increase in total iron content as compared to cortex or cerebellum. Several postmortem and especially MRI-based in vivo studies have reported an increase in iron content in the caudate nucleus of PD patients, and this brain area is affected during relatively early disease stage [6], [7]. However, we did not observe significant changes in total iron content between control and PD patients in the caudate, with all PD patient samples from the same disease stage. Similarly, even though we have previously been able to detect alterations in the phosphorylation state of select proteins implicated in PD pathogenesis in this brain area [33], no alterations in the activation state and/or levels of a set of kinases known to be activated upon oxidative stress, or in the levels of proteins implicated in endolysosomal events were detected. Thus, the reported discrepancies in total iron content in the caudate nucleus may have been due to the different methodologies employed, and/or due to differences in disease stage [2], with changes possibly only becoming evident in severe disease cases.
In AD patients, iron has been shown to accumulate in the cortex and hippocampus, but not the cerebellum [44], [1]. In contrast, there are virtually no reports on brain iron levels from LBD patients [3]. Apart from sharing many clinical features with AD patients as related to cognitive decline, LBD patients also display parkinsonian features, which correlates with LBs in the SN as well as other brain areas upon postmortem analysis. We determined iron levels in the SN from control, AD and LBD cases. A significant increase in total iron content was observed in age- and disease stage-matched SN samples from LBD cases as compared to AD or control.
The lack of iron accumulation in the SN of AD patients is consistent with the pathological profile of neurodegeneration in AD. Conversely, an increase in total iron content in the SN of LBD patients is consistent with the correlation between increased iron and movement disorder phenotype in PD patients. Interestingly, an in vivo imaging study has shown that iron accumulation in the SN may be a predictor of parkinsonism during the course of AD [45]. In addition, quantitative susceptibility mapping (QSM), a novel sensitive method for determining iron concent in vivo, has recently been shown to detect regionally progressive iron accumulation in a manner dependent on disease severity [13], [46]. Whilst future studies will be required to corroborate the present observations, our data suggest that iron imaging studies in patients suffering from AD, AD with parkinsonism, PD or LBD may aid in clinical diagnosis and/or serve as biomarker for disease progression.
Acknowledgements
This work was supported by grants from FEDER, The Spanish Ministry of Economy and Competitiveness (MINECO; SAF2014-58653-R), the BBVA Foundation and the Michael J. Fox Foundation. B.F. was supported by CEI Biotic Granada (CAEP2-13) and by a Juan de la Cierva Fellowship (JCI-2010-07703).
References
- 1.Hare D., Ayton S., Bush A., Lei P. A delicate balance: iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013;18(July (5)):34. doi: 10.3389/fnagi.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ward R.J., Zucca F.A., Duyn J.H., Crichton R.R., Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–1060. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg D., Hochstrasser H. Iron metabolism in Parkinsonian syndromes. Mov. Disord. 2006;21:1299–1310. doi: 10.1002/mds.21020. [DOI] [PubMed] [Google Scholar]
- 4.Hallgren B., Sourander P. The effect of age on the non-haemin iron in the human brain. J. Neurochem. 1958;3:41–51. doi: 10.1111/j.1471-4159.1958.tb12607.x. [DOI] [PubMed] [Google Scholar]
- 5.Zecca L., Youdim M.B.H., Riederer P., Connor J.R., Crichton R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 6.Braak H., Ghebremedhin E., Rub U., Bratzke H., Del Tredici K. Stages in the development of Parkinsońs disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 7.Jellinger K.A., Mizuno Y. Parkinsońs disease. In: Dickson D., editor. Neurodegeneration: the Molecular Pathology of Dementia and Movement Disorders. ISN Neuropath Press; Basel: 2003. pp. 159–187. [Google Scholar]
- 8.Mc Keith I.G., Galasko D., Kosaka K., Perry E.K., Dickson D.W., Hansen L.A., Salmon D.P., Lowe J., Mirra S.S., Byrne E.J., Lennox G., Quinn N.P., Edwardson J.A., Ince P.G., Bergeron C., Burns A., Miller B.L., Loveston S., Collerton D., Jansen E.N., Ballard C., de Vos R.A., Wilcock C.K., Jellinger K.A., Perry R.H. Consensus guidelines for the clinical and pathological diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB International Workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 9.Mc Keith I. Dementia with lewy bodies. Lancet Neurol. 2004;3:19–28. doi: 10.1016/s1474-4422(03)00619-7. [DOI] [PubMed] [Google Scholar]
- 10.Braak H., Braak E. Temporal sequence of Alzheimeŕs disease-related pathology. In: Peters A., Morrison J.H., editors. vol. 14. Kluver Aademic/Plenum Publishers; New York Boston Dordrecht London Moscow: 1999. pp. 475–512. (Cerebral cortex, Neurodegenerative and Age-related Changes in Structure and Function of the Cerebral Cortex). [Google Scholar]
- 11.Wang J.Y., Xuang Q.Q., Zhu L.B., Zu H., Li T., Li R., Chen S.F., Huang C.P., Zhang X., Zhu J.H. Meta-analysis of brain iron levels of Parkinsońs disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016;6:36669. doi: 10.1038/srep36669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dougherty A., Raz N. Age-related differences in iron content of subcortical nuclei observed in vivo: a meta-analysis. Neuroimage. 2013;70:113–121. doi: 10.1016/j.neuroimage.2012.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du G., Liu T., Lewis M.M., Kong L., Wang Y., Connor J., Mailman R.B., Huang X. Quantitative susceptibility mapping of the midbrain in Parkinsońs disease. Mov. Disord. 2016;31:317–324. doi: 10.1002/mds.26417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman A., Galazka-Friedman J. The history of the research of iron in parkinsonian substantia nigra. J. Neural Transm. 2012;119:1507–1510. doi: 10.1007/s00702-012-0894-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J.C., Hardy P.A., Kucharczyk W., Clauberg M., Joshi J.G., Vourlas A., Dhar M., Henkelman R.M. MR of human postmortem brain tissue: correlative study between T2 and assays of iron and ferritin in Parkinson and Huntington disease. Am. J. Neuroradiol. 1993;14:275–281. [PMC free article] [PubMed] [Google Scholar]
- 16.Earle K.M. Studies on Parkinsońs disease including X-ray fluorescent spectroscopy of formalin fixed brain tissue. J. Neuropathol. Exp. Neurol. 1968;27:1–13. doi: 10.1097/00005072-196801000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Galazka-Friedman J., Bauminger E.R., Friedman A., Barcikowska M., Hechel D., Nowik I. Iron in parkinsonian and control substantia nigra-Mossbauer spectroscopy study. Mov. Disord. 1996;11:8–16. doi: 10.1002/mds.870110104. [DOI] [PubMed] [Google Scholar]
- 18.Griffiths P.D., Crossman A.R. Distribution of iron in the basal ganglia and neocortex in postmortem tissue in Parkinsońs disease and Alzheimeŕs disease. Dementia. 1993;4:61–65. doi: 10.1159/000107298. [DOI] [PubMed] [Google Scholar]
- 19.Loeffler D.A., Connor J.R., Juneau P.L., Snyder B.S., Kanaley L., DeMaggio A.J., Nguyen H., Brickman C.M., LeWitt P.A. Transferrin and iron in normal, Alzheimeŕs disease, and Parkinsońs disease brain regions. J. Neurochem. 1995;65:710–716. doi: 10.1046/j.1471-4159.1995.65020710.x. [DOI] [PubMed] [Google Scholar]
- 20.Mann V.M., Cooper J.M., Daniel S.E., Srai K., Jenner O., Marsden C.D., Schapira A.H.V. Complex I, iron, and ferritin in Parkinsońs disease substantia nigra. Ann. Neurol. 1994;36:876–881. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- 21.Riederer P., Sofic E., Rausch W.D., Schmidt B., Reynolds G.P., Jellinger K., Youdim M.B.H. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 22.Sofic E., Riederer P., Heinsen H., Beckmann H., Reynolds G.P., Hebenstreit G., Youdim M.B.H. Increased iron (111) and total iron content in post mortem substantia nigra of parkinsonian brain. J. Neural Trans. 1988;74:199–205. doi: 10.1007/BF01244786. [DOI] [PubMed] [Google Scholar]
- 23.Visanji N.P., Collingwood J.F., Finnegan M.E., Tandon A., House E. Iron deficiency in Parkinsonism: region-specific iron dysregulation in Parkinsońs disease and multiple system atrophy. J. Parkinsons Dis. 2013;3:523–537. doi: 10.3233/JPD-130197. [DOI] [PubMed] [Google Scholar]
- 24.Wypijewska A., Galazka-Friedman J., Bauminger E.R., Wszolek Z.K., Schweitzer K.J., Dickson D.W., Jaklewicz A., Elbaum D., Friedman A. Iron and reactive oxygen species activity in parkinsonian substantia nigra. Park. Reat. Disord. 2010;16:329–333. doi: 10.1016/j.parkreldis.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Yu X., Du T., Song N., He Q., Shen Y., Jiang H., Xie J. Decreased iron levels in the temporal cortex in postmortem human brains with Parkinson disease. Neurology. 2013;80:492–495. doi: 10.1212/WNL.0b013e31827f0ebb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salvador G.A., Oteiza P.I. Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. Neurotoxicology. 2011;32:75–82. doi: 10.1016/j.neuro.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Lei P., Ayton S., Finkelstein D.I., Spoerri L., Ciccotosto G.D., Wright D.K., Wong B.X.W., Adlard P.A., Cherny R.A., Lam L.Q., Roberts B.R., Volitakis I., Egan G.F., McLean C.A., Cappai R., Duce J.A., Bush A.I. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012;18:291–296. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 30.Ferrer I., Blanco R., Carmona M., Puig B., Barrachina M., Gómez C., Ambrosio S. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinsońs disease and Dementia with Lewy bodies. J. Neural. Transm. 2001;108:1383–1396. doi: 10.1007/s007020100015. [DOI] [PubMed] [Google Scholar]
- 31.Rubio de la Torre E., Luzón-Toro B., Forte-Lago I., Minguez-Castellanos A., Ferrer I., Hilfiker S. Combined kinase inhibition modulates parkin inactivation. Hum. Mol. Genet. 2009;18:809–823. doi: 10.1093/hmg/ddn407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.House M.J., St. Pierre T.G., Kowdley K.V., Montine T., Connor J., Beard J., Berger J., Siddaiah N., Shankland E. Correlation of proton transverse relaxation rates (R2) with iron concentrations in postmortem brain tissue from Alzheimeŕs disease patients. Magn. Reson. Med. 2007;57:172–180. doi: 10.1002/mrm.21118. [DOI] [PubMed] [Google Scholar]
- 33.Dzamko N., Zhou J., Huang Y., Halliday G.M. Parkinsońs disease-implicated kinases in the brain; insights into disease pathogenesis. Front. Mol. Neurosci. Jun. 2014;24(7):57. doi: 10.3389/fnmol.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J.H., Kulich S.M., Oury T.D., Chu C.T. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am. J. Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greene L.A., Lewy O., Malagelada C. Akt as a victim, villain and potential hero in Parkinsońs diseasse pathophysiology and treatment. Cell Mol. Neurobiol. 2011;31:969–978. doi: 10.1007/s10571-011-9671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malagelada C., Jin Z.H., Greene L.A. RTP801 is induced in Parkinsońs disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 2008;28:14363–14371. doi: 10.1523/JNEUROSCI.3928-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timmons S., Coakley M.F., Moloney A.M., ÓNeill C. Akt signal transduction dysfunction in Parkinsońs disease. Neurosci. Lett. 2009;467:30–35. doi: 10.1016/j.neulet.2009.09.055. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez-Erviti L., Rodriguez-Oroz M.C., Cooper J.M., Caballero C., Ferrer I., Obeso J.A., Schapira A.J. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010;67:1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 39.Chu Y., Dodiya H., Aebischer P., Olanow C.W., Kordower J.H. Alterations in lysosomal and proteasomal markers in Parkinsońs disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 40.He A.Y., Qiu L.J., Gao Y., Zhu Y., Xu Z.W., Xu J.M., Zhang Z.H. The role of oxidative stress in neuromelanin synthesis in PC12 cells. Neuroscience. 2011;189:43–50. doi: 10.1016/j.neuroscience.2011.05.040. [DOI] [PubMed] [Google Scholar]
- 41.Ayton S., Lei P., Adlard P.A., Volitakis I., Cherny R.A., Bush A.I., Finkelstein D.I. Iron accumulation confers neurotoxicity to a vulnerable population of nigral neurons: implications for Parkinsońs disease. Mol. Neurodeg. 2014;9:27. doi: 10.1186/1750-1326-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dexter D.T., Wells F.R., Lees A.J., Agid F., Agid Y., Jenner P., Marsden C.D. Increased nigral iron content and alterations in other metal ions occuring in brain in Parkinsońs disease. J. Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- 43.Uitti R.J., Rajput A.H., Rozdilsky B., Bickis M., Wollin T., Yuen W.K. Regional metal concentrations in Parkinsońs disease, other chronic neurological diseases, and control brains. Can. J. Neurol. Sci. 1989;16:310–314. doi: 10.1017/s0317167100029140. [DOI] [PubMed] [Google Scholar]
- 44.Belaidi A.A., Bush A.I. Iron neurochemistry in Alzheimeŕs disease and Parkinsońs disease: targets for therapeutics. J. Neurochem. 2016;139:179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- 45.Brar S., Henderson D., Schenck J., Zimmerman E.A. Iron accumulation in the substantia nigra of patients with Alzheimer disease and Parkinsonism. Arch. Neurol. 2009;66:371–374. doi: 10.1001/archneurol.2008.586. [DOI] [PubMed] [Google Scholar]
- 46.Guan X., Xuan M., Gu Q., Huang P., Liu C., Wang N., Xu X., Luo W., Zhang M. Regionally progressive accumulation of iron in Parkinsońs disease as measured by quantitative susceptibility mapping. NMR Biomed. 2016 doi: 10.1002/nbm.3489. (10.002/nbm.3489) [DOI] [PMC free article] [PubMed] [Google Scholar]