

Abstract
Aims
The endothelium has emerged recently as a therapeutic target in the treatment of hypertension because endothelial dysfunction and subsequent vascular rarefaction cause target organ damage and further elevate blood pressure (BP). It led us to hypothesize that one of the endothelial survival factors, a potent derivative of angiopoietin-1 (cartilage oligomeric matrix protein, COMP-Ang-1), could be a novel class of antihypertensive agents that maintain endothelial integrity and function, thereby preventing the development of hypertension and target organ damage.
Methods and results
To study the role of COMP-Ang-1 in preventing hypertension and target organ damage, a COMP-Ang-1 plasmid was electroporated into adductor muscles of 6 weeks old, pre-hypertensive, spontaneously hypertensive rats (SHRs), and the secretion of its expressed protein into the bloodstream was confirmed by western blotting. In comparison with sham and reporter gene transfer, COMP-Ang-1 gene transfer significantly prevented increases in systolic BP and reduced microvascular rarefaction and tissue damage in the heart and kidney. However, overexpression of soluble Tie2 receptor completely abolished these beneficial effects of COMP-Ang-1 gene transfer on SHRs, indicating that expressed COMP-Ang-1 protein has antihypertensive effects in SHRs by binding Tie2 receptors on the vascular endothelium. In particular, COMP-Ang-1 gene-transferred SHRs had significantly higher plasma levels of nitrite than other controls, which was found to be due to that expressed COMP-Ang-1 protein promoted nitrite synthesis by activating endothelial nitric oxide synthase, one of the Tie2 downstream-signalling molecules.
Conclusion
The present study suggests a new potential of endothelial survival factor, COMP-Ang-1, as an antihypertensive agent that effectively reduces the hypertension-associated cardiovascular and renal damage, as well as prevents the further elevation of BP.
Keywords: Angiopoietin-1, Endothelium, Hypertension, Nitric oxide, Rarefaction, Target organ damage
1. Introduction
Hypertension is a significant medical problem because sustained high blood pressure (BP) has detrimental effects on target organs such as the heart and kidney, bringing about severe cardiovascular disease and end-stage renal disorders.1 Therefore, much of the focus in hypertension management has been on lowering BP, in the hope that it will result in a decrease in the incidence of hypertension-associated target organ damage. However, recent studies have revealed that controlling BP does not fully prevent cardiovascular and renal diseases, which indicates that hypertension is one of the risk factors in the development of target organ damage, but normalizing BP by itself may not be sufficient to prevent or cure hypertension-associated cardiovascular and renal diseases.2–4
Underlying target organ damage resulting from hypertension is the functional and structural change in the vasculature, particularly of microvessels (small arteries and arterioles) and capillaries, which become constricted and unperfused, and eventually disappear.5 This phenomenon, termed microvascular/capillary rarefaction, contributes to target organ damage by impairing blood flow. In addition, it further elevates BP by increasing peripheral vascular resistance in the microcirculation. According to recent reports, this disappearance of microvessels in hypertensive subjects as well as in hypertensive animal models, results from the increase in endothelial cell apoptosis upon exposure to a variety of vascular stresses.6,7 It implicates that inhibition of endothelial cell apoptosis could contribute to the prevention of microvascular rarefaction. In this regard, endothelial survival factors have potential as a novel class of antihypertensive agents by protecting the endothelium against a variety of vascular stresses, thereby preventing target organ damage and BP elevation in individuals with hypertension.1
A stable and potent variant of angiopoietin-1 (Ang-1), cartilage oligomeric matrix protein (COMP)-Ang-1, has strong endothelial protection activity.8 In particular, COMP-Ang-1 was found to protect microvascular endothelial cells in the intestinal villi from radiation-induced apoptosis and also was reported to attenuate renal fibrosis by preserving renal microvasculature in a unilateral ureteral obstruction model.9,10 These results may be explained by COMP-Ang-1 activating the anti-apoptotic Akt-signalling pathway through Tie2 receptors in the endothelium. These strong effects on endothelial protection against vascular injury led us to hypothesize that COMP-Ang-1-induced vascular protection could prevent both target organ damage and further elevation of systemic BP, compared with conventional antihypertensive agents. In this regard, the present study investigates the endothelium-targeted antihypertensive effects of COMP-Ang-1 in spontaneously hypertensive rats (SHRs), a model of essential hypertension that involves vascular fibrosis, target organ damage, and rarefaction.
2. Methods
2.1. Animal experiments
Six-week-old male SHRs (Charles River Laboratories, Yokohama, Japan) were randomized for two electrophoretic transfers (eight electric pulses of 200 V/cm for 50 ms at 1 Hz using an ECM 830 electroporator on the first day of the experiment and once again a week later) of LacZ plasmid (pLacZ: pcDNA 3.1 backbone vector) or pCOMP-Ang-1 plasmid (pCOMP-Ang-1: pcDNA 3.1 backbone vector) into the adductor muscle (100 µg in half-saline). As a control group, 6-week-old male SHRs underwent a sham operation; their normotensive control, age-matched male Wistar Kyoto rats (Charles River Laboratories) were included. In the experiment with adenoviral soluble Tie2 receptor (Ad-sTie2), 1 × 109 pfu of Ad-sTie2 or Ad-LacZ (in 0.9% NaCl) was intravenously injected into SHRs 1 week before the gene transfer.11 This animal experiment was reviewed and approved by the Institutional Animal Care and Use Committee of Samsung Medical Center. All procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals published in the US National Institutes of Health (NIH publication no. 85–23, revised 1996). For surgical procedures, rats were anesthetized with an intraperitoneal injection of ketamine and xylazine (50 and 2 mg/kg, respectively). BP was measured every week using the tail-cuff method (IITC Life Science Instruments, Woundland Hills, CA, USA) on conscious rats on three different occasions.
2.2. Histological analysis of the heart and kidney
The hearts and kidneys were fixed in 4% paraformaldehyde and embedded in paraffin wax, or frozen in cryofreezing media for histological examination and immunohistochemical staining. Microscopic examination of Masson’s trichrome- and haematoxylin and eosin (H&E)-stained sections was performed to calculate the interventricular septal thickness and to evaluate histological changes in these organs in terms of the presence or absence of several specific pathological lesions (for detailed information, see Supplementary material online).
2.3. Proteinuria
Urine was collected weekly from each rat housed in a metabolic cage for 24 h. Urinary protein was measured using a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
2.4. Immunohistochemical staining
To quantify capillary density, the heart and kidney sections were stained with anti-rat CD31 IgG (BD Pharmingen, San Diego, CA, USA) or anti-rat RECA-1 IgG (Abcam, Cambridge, UK). After quenching the endogenous peroxidase activity and blocking with normal goat serum, sections were incubated with primary antibodies, and then with biotinylated secondary IgG (Jackson ImmunoResearch, West Grove, PA, USA). Positive immunoreactivity was visualized using ABC-peroxidase kits (ChemMate™ DAKO Envision™ Detection kit, DAKO, Carpinteria, CA, USA). Controls of immunostaining preparations were prepared by incubating with irrelevant class- and species-matched IgGs.
To assess hypoxic areas in the heart and kidney, 100 mg/kg of pimonidazole (Hydroxyprove-1™, Chemicon International, Temecula, CA, USA) was intravenously injected into SHRs 90 min prior to sacrifice. Paraffin sections of the heart and kidney were stained with hydroxyprobe-1 monoclonal IgG (for detailed information, see Supplementary material online).
2.5. Measurement of peripheral blood flow
Rats were anaesthetized, shaved, and depilated as described previously.12 Dermal blood flow above the tibialis muscle was measured using a non-invasive laser Doppler imager system (Moor Instruments, Axminster, Devon, UK). The laser beam (780 nm), reflected from moving erythrocytes in blood vessels, is detected and processed to provide a computerized image, in which the perfusion signal is displayed in colour codes ranging from dark blue (0) through red to white (1000). Using the image analysis program provided with LDI, mean flux values representing limb perfusion were calculated and normalized to the mean flux value of the normotensive control, WKY rats.
2.6. Western blot analysis
To determine the amount of expressed COMP-Ang-1 protein in the blood, 500 µL of retro-orbital blood was collected and centrifuged to obtain the plasma. Briefly, diluted plasma samples were run in a 10% sodium dodecyl sulphate (SDS)–polyacrylamide gel (PAGE) and electrophoretically transferred to nitrocellulose membranes. After incubating the membrane in blocking buffer, the membrane was blotted with monoclonal anti-FLAG IgG (diluted at 1:1000; Sigma, St Louis, MO, USA). Immunoreactive bands were visualized by chemiluminescence using a horseradish peroxidase-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and ECL kit (Amersham, Piscataway, NJ, USA). Quantitative analysis of the blots was performed using Image-Lab (MCM Design, Birkerød, Denmark). To evaluate the phosphorylated degree of Tie2 receptor in SHRs, the lung of gene-transferred SHRs was lysed in the buffer containing protease and phosphatase inhibitors. Then lung lysates were incubated with anti-Tie2 or anti-endothelial nitric oxide synthase (eNOS) IgG and subsequently immunoprecipitated using protein G-sepharose. Immunoprecipitates were resolved by SDS–PAGE and immunoblotted with antiphosphotyrosine IgG. To examine the amount of total protein loaded into gel, blots were stripped and re-immunoblotted with anti-Tie2 (R&D Systems, Minneapolis, MN, USA) or anti-eNOS (Zymed Laboratories, San Francisco, CA, USA) IgG. To investigate the phosphorylation of eNOS upon COMP-Ang-1 treatment, serum-starved human umbilical vein endothelial cells (HUVECs; Cambrex, East Rutherford, NJ, USA) were treated with 100 ng/mL of COMP-Ang-1 protein for 1 h and then lysed with lysis buffer. Total proteins were separated by SDS–PAGE and transferred to nitrocellulose membrane as described earlier. Briefly, the membrane was blotted with anti-eNOS IgG or phospho-eNOS IgG (Upstate Cell Signaling Solutions, Lake Placid, NY, USA). They were then incubated with horseradish peroxidase-conjugated IgG and developed using a chemiluminescence kit.
2.7. Nitrite measurement
The concentration of nitrite, a metabolite of nitric oxide, in the blood and in the conditioned media from HUVECs was measured with an NO assay kit (R&D Systems) according to the manufacturer’s instruction. To deplete COMP-Ang-1 protein in serum from SHRs, 200 ng of soluble Tie2 (R&D Systems) was added into serum obtained in the third week of the experiment. After overnight incubation at 4°C, protein A/G agarose beads (Pierce Chemical Co., Rockford, IL, USA) were added and incubated for another 2 h at 4°C. Then, the supernatant obtained after the centrifugation was used for the experiment.
2.8. Statistical analysis
All data are presented as mean ± SEMs. One-way analysis of variance was used to determine the significance of differences between groups; where appropriate, data were analysed using post hoc Student’s t-tests for unpaired observations and the Bonferroni correction for multiple comparisons. P < 0.05 was accepted as significant. The number of samples examined in each experiment is indicated by n.
3. Results
3.1. COMP-Ang-1 gene transfer prevents elevation of blood pressure in spontaneously hypertensive rats
To study the role of COMP-Ang-1 in preventing hypertension, COMP-Ang-1 plasmid was electroporated into adductor muscles of 6-week-old, pre-hypertensive SHRs whose BP, however, continued to rise to the fully hypertensive stage with the appearance of hypertension-related histopathological changes. The electroporation-mediated gene transfer allowed us to avoid the repeated administration of COMP-Ang-1 protein to maintain its significant plasma level until the end of the experiment. As shown in Figure 1A, the secretion of expressed COMP-Ang-1 protein into the bloodstream was confirmed by western blotting, where COMP-Ang-1 protein reached its peak concentration 2 weeks after the first gene transfer and remained at significant levels until the fifth week of the experiment. When the systolic BP of SHRs was monitored using the tail-cuff method (Figure 1B), untreated or pLacZ-transferred SHRs developed hypertension to the fully hypertensive levels (164.5 ± 7.4 mmHg in untreated and 164.7 ± 7.1 mmHg in pLacZ SHRs), whereas pCOMP-Ang-1-transferred SHRs exhibited significantly lower systolic BP until 5 weeks after the first gene transfer (144.1 ± 4.4 mmHg, P < 0.05). However, heart rate and body weight were similar in the pCOMP-Ang-1-treated group and other controls (data not shown).
Figure 1.
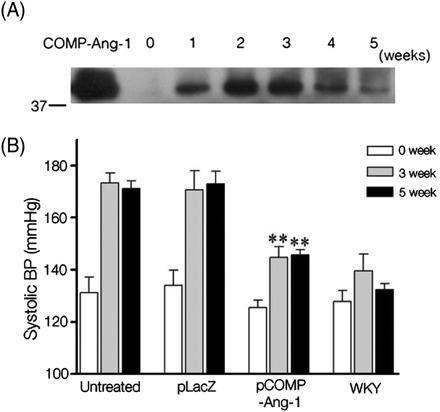
COMP-Ang-1 gene transfer prevents the blood pressure (BP) elevation in spontaneously hypertensive rats (SHRs). (A) COMP-Ang-1 protein secreted into the bloodstream was quantified by western blotting using IgG against the FLAG sequence (DYKDDDDK) tagged at the N-terminus of COMP-Ang-1. As a positive control, 100 ng of COMP-Ang-1 protein was loaded with 7 μL of blood serum from spontaneously hypertensive rats. (B) Systolic BP (in mmHg) of each group was measured using the tail-cuff method in the third and fifth weeks after the first gene transfer. **P < 0.01 vs. pLacZ-transferred or untreated spontaneously hypertensive rats (n = 6).
3.2. COMP-Ang-1 gene transfer attenuates target organ damage in spontaneously hypertensive rats
Because tissue damage in target organs is frequently accompanied by elevated BP in both SHRs and human subjects, histopathological changes in the heart and kidney were carefully investigated and compared in pCOMP-Ang-1-transferred and other control SHRs. As shown in Figure 2, untreated and pLacZ-treated SHRs exhibited severe accumulation of inflammatory cells and multifocal myocyte necrosis, similar to other reports on SHRs at hypertensive stage. In particular, the interventricular septum in the heart was significantly thickened, which indicates that left ventricular hypertrophy, a typical characteristic of the hypertension-induced cardiac remodelling had occurred. In contrast to the significant tissue damage in control SHRs, pCOMP-Ang-1-transferred SHRs exhibited much less infiltration of inflammatory cells and no significant increase in the interventricular septal thickness. In the kidney, similar results were observed. As shown in Figure 3, the pCOMP-Ang-1-transferred group did not exhibit the severe tubulointerstitial inflammation and tubular fibrosis/necrosis that were, however, frequently observed in pLacZ-transferred and untreated SHRs (Figure 3A and B). These histopathological changes in the kidney account for the difference in the renal function of SHRs, as shown by the amount of protein excreted into urine (Figure 3C).
Figure 2.
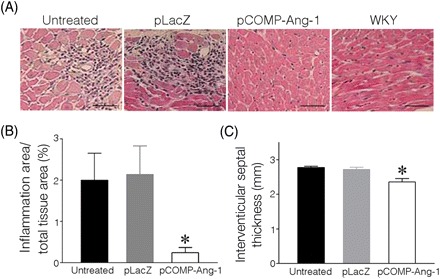
COMP-Ang-1 gene transfer reduces myocardial damage in SHRs. (A) Representative images of H&E-stained heart. (B) Inflammatory infiltrates shown in H&E sections were quantified as a percentage of the total tissue area. (C) Interventricular septal thickness was measured on H&E-stained heart images using ImagePro. *P < 0.05 vs. untreated or pLacZ (n = 6). Scale bar is 20 µm.
Figure 3.
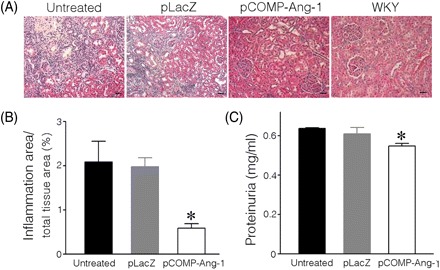
COMP-Ang-1 gene transfer reduces renal damage in SHRs. (A) Representative images of H&E-stained kidney. (B) Inflammatory infiltrates shown in H&E sections were quantified as a percentage of the total tissue area. (C) The amount of protein in urine collected at the end of the experiment. *P < 0.05 vs. pLacZ-transferred or untreated SHRs (n = 6). Scale bar is 20 µm.
3.3. COMP-Ang-1 gene transfer attenuates the capillary rarefaction in spontaneously hypertensive rats
Target organ damage in hypertensive SHRs as shown in Figures 2 and 3, and in human subjects as well, is caused by microvascular rarefaction, which decreases blood flow into surrounding tissues, resulting in ischaemia and inflammation. In addition, COMP-Ang-1 acts as a strong endothelial survival factor by binding and activating Tie2, an endothelial-specific receptor, so that pCOMP-Ang-1 transfer was expected to prevent microvascular rarefaction and to subsequently attenuate target organ damage. The vascular density in target organs was evaluated by immunohistochemistry using endothelial-specific CD31 or RECA IgG. As a result, pCOMP-Ang-1-transferred SHRs were found to exhibit greater capillary density in the heart and more intact endothelial lining in glomeruli of the kidney than in other controls (Figure 4A–C; Supplementary material online, Figure S1). As the disappearance of capillaries decreases blood flow and causes ischaemia in the surrounding tissues, the extent of tissue hypoxia was examined using hydroxyprobe-1. Consistent with a high density of intact capillaries, it was found that pCOMP-Ang-1- transferred SHRs had a much lesser and smaller hypoxic area in the heart and kidney, compared with the control SHRs. To confirm whether the capillary rarefaction evaluated by immunohistochemistry indeed represents the decrease in blood flow, dermal blood flow was assessed in the hindlimb of SHRs by LDI that detects blood cells moving within the depth of the skin layer. The dermal blood flow has been considered to represent the magnitude of microvascular rarefaction in hypertensive patients.13 In accordance with other results, Figure 4D shows that dermal blood flow in the pCOMP-Ang-1-transferred group was significantly higher than in controls, which demonstrates that pCOMP-Ang-1 transfer substantially prevented hypertension-induced microvascular rarefaction in SHRs.
Figure 4.
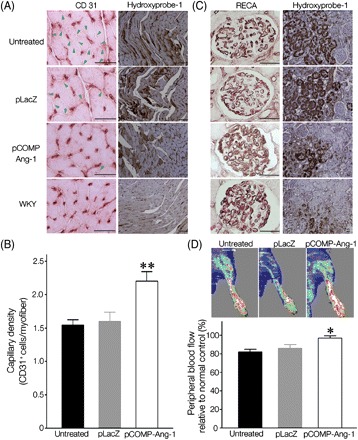
COMP-Ang-1 gene transfer attenuates the microvascular rarefaction and tissue ischaemia in the heart and kidney of SHRs. The microvasculature of the heart (A) [arrowheads in (A) indicate the absence of capillaries], (B) and kidney (C) was visualized and quantified by immunohistochemical staining with CD31 IgG on frozen sections or RECA IgG on paraffin sections, respectively. In addition, the extent of hypoxia in the heart and kidney was visualized by immunohistochemical staining with Hydroxyprobe-1. (D) Dermal blood flow on the tibialis muscle of SHRs was evaluated using laser Doppler imager. The blood perfusion signal is displayed in colour codes ranging from dark blue (0) to white (1000). Representative laser Doppler images of each group and its blood perfusion percentage, which was normalized to the blood perfusion extent of the normotensive control, Wistar Kyoto rats. *P < 0.05, **P < 0.01 vs. pLacZ-transferred or untreated SHRs (n = 6). Scale bars on CD31- or Hydroxyprobe-1-stained figures are 20 and 10 µm, respectively.
3.4. COMP-Ang-1 induces beneficial effects in spontaneously hypertensive rats via Tie2 receptors on endothelium
To investigate whether the beneficial outcomes observed in pCOMP-Ang-1-transferred SHRs were mediated by COMP-Ang-1 protein systemically secreted from transfected skeletal muscles of SHRs, we evaluated the phosphorylated extent of Tie2, an endothelial-specific receptor for COMP-Ang-1, in the lung of pLacZ- or pCOMP-Ang-1-transferred SHRs. As shown in Figure 5A, Tie2 was found to be highly phosphorylated in the lung of the pCOMP-Ang-1 group compared with that of the pLacZ group. It demonstrated that a significant amount of COMP-Ang-1 protein secreted into the bloodstream from the gene-transferred adductor muscle, bound, and subsequently activated the Tie2 receptor on the vascular endothelium. This result was further verified by the following experiment, where SHRs were pre-treated with adenovirus carrying soluble Tie2 receptor 1 day before gene transfer. The soluble extracellular domain of Tie2 has been found to compete with the native Tie2 receptor on the endothelium in binding with its ligand, thereby inhibiting ligand-induced Tie2 activation.
Figure 5.
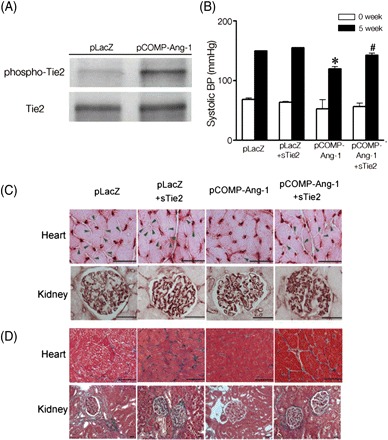
COMP-Ang-1 protein induces beneficial effects in SHRs via Tie2 receptors on endothelium. (A) Tie2 phosphorylation in the lung tissues of pLacZ- or pCOMP-Ang-1-transferred SHRs sacrificed 3 weeks after the gene transfer. One day before the first gene transfer of pLacZ or pCOMP-Ang-1, SHRs were intravenously injected with 1 × 109 pfu of adenoviral soluble Tie2 (sTie2). Then their systolic BP (B) capillary density (C) [arrowheads in (C) indicate the absence of blood vessels], and tissue damage (D) in the heart and kidney were evaluated as described earlier. *P < 0.05 vs. pLacZ or pLacZ+sTie2; #P < 0.05 vs. pCOMP-Ang-1 (n = 3). Scale bar is 20 µm.
Figure 5B demonstrates that adenovirus-mediated overexpression of soluble Tie2 receptor significantly elevated BP to a similar extent to those in pLacZ-transferred SHRs. In addition, it caused the severe microvascular rarefaction and target organ damage in pCOMP-Ang-1-transferred SHRs as observed in pLacZ-transferred SHRs (Figure 5C and D; Supplementary material online, Figure S2). These results suggest that pre-treatment of soluble Tie2 receptor abolished the beneficial effect of COMP-Ang-1 protein in SHRs, indicating that COMP-Ang-1 had antihypertensive effects in SHRs via binding and activating Tie2 receptors on the vascular endothelium.
3.5. COMP-Ang-1increases nitrite production by activating endothelial nitric oxide synthase
To investigate the mechanisms behind the antihypertensive effects of COMP-Ang-1 in SHRs, a major endothelial homeostatic factor, NO, was chosen to assess in the experiments. Because COMP-Ang-1, a potent variant of Ang-1, strongly phosphorylates Tie2 receptors on the endothelium, it is conceivable that COMP-Ang-1 enhances NO production by activating eNOS, a known Tie2 downstream-signalling molecule. As shown in Figure 6A, pCOMP-Ang-1-transferred SHRs had higher plasma concentrations of nitrite, an oxidative byproduct of NO, compared with other control SHRs that had gradually lower plasma nitrite concentration as the BP increased. To verify that the COMP-Ang-1 protein in blood induces increases in the plasma nitrite concentration via Tie2, HUVECs were treated with serum from SHRs and then their nitrite production was assessed as shown in Figure 6B. As a result, serum obtained from pCOMP-Ang-1-transferred SHRs significantly promoted nitrite production in HUVECs. However, the increase in nitrite disappeared when the serum was pre-treated with soluble Tie2. Moreover, it was found that eNOS was more activated in the lung, heart, and kidney of pCOMP-Ang-1-transferred SHRs than in those of the pLacZ control pre-treated with soluble Tie2 (Figure 6C; Supplementary material online, Figure S3). Consistently, in vitro results shown in Figure 6D and E demonstrate that the COMP-Ang-1 protein was able to phosphorylate eNOS and to increase significantly nitrite production in HUVECs (at 100 ng/mL, which was a much lower concentration than the peak plasma levels in vivo). Given that essential hypertension is characterized by NO deficiency, these data suggest that the eNOS activation and subsequent NO production by expressed COMP-Ang-1 protein may help the endothelium to maintain vascular homeostasis and prevent the elevation of BP in pCOMP-Ang-1-transferred SHRs.
Figure 6.
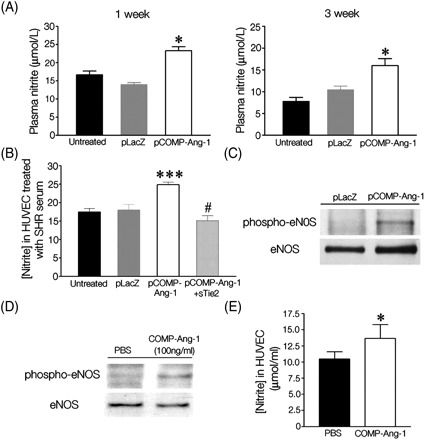
COMP-Ang-1 enhances nitric oxide production by activating endothelial nitric oxide synthase (eNOS). (A) Nitrite levels in peripheral bloods from SHRs. (B) Nitrite levels in human umbilical vein endothelial cells (HUVEC) after the incubation with serum obtained from SHRs. Of note, pre-treatment with soluble Tie2 abolished the increase of nitrite production in HUVEC treated with serum from pCOMP-Ang-1 transferred SHRs. *P < 0.05, ***P < 0.001 vs. untreated or pLacZ; #P < 0.001 vs. pCOMP-Ang-1 (A, n = 5; B, n = 4). (C) eNOS phosphorylation in the lung tissues of pLacZ- or pCOMP-Ang-1-transferred SHRs sacrificed 3 weeks after the gene transfer. (D) eNOS phosphorylation in HUVEC after 60 min incubation with COMP-Ang-1 protein (100 ng/mL). (E) Nitrite production in HUVEC after the treatment with COMP-Ang-1 protein (100 ng/mL). *P < 0.05 vs. PBS (n = 3).
4. Discussion
In hypertension, the vasculature undergoes a variety of structural alterations. For example, pre-capillary resistance vessels increase in wall thickness to maintain constant wall stress in the face of high BP. The microvasculature, such as small arterioles and capillaries, becomes constricted to the point of non-perfusion, and vessels eventually disappear.5 Although these anatomical changes in the vasculature were initially appreciated as a consequence of increased vascular pressure, recent evidence in human hypertensive patients and SHRs suggests that it may not always be the case. In particular, the disappearance of microvasculature, termed microvascular rarefaction, was shown to precede manifest elevation of BP in persons with a familial history of hypertension, which suggested that microvascular rarefaction could initiate the development of hypertension and its related pathogenic changes.13–15 In addition, anti-cancer clinical trials performed with angiogenesis inhibitors concluded that vascular abnormalities would be a contributing factor in the development of hypertension caused by vascular endothelial growth factor (VEGF) antagonists such as BAY 43-9006 or PTK787/ZK222584.16,17 These observations support the concept that the endothelial dysfunction could cause hypertension by reducing the microvascular density in persons prone to hypertension and further suggest that a new therapeutic target for hypertension would be to maintain endothelial integrity and function, thereby inhibiting the progress of microvascular rarefaction and the subsequent development of hypertension and its related pathological sequences.
On the basis of these findings, we performed the pioneering study to investigate the therapeutic potential of endothelial survival factors in preventing not only hypertension, but also its related pathological consequences. We found that a strong endothelial survival factor, COMP-Ang-1, was effective in reducing microvascular rarefaction and tissue damage in the heart and kidney, as well as in preventing the development of hypertension in a genetic hypertension animal model, SHRs. In particular, as the microvasculature functions primarily to provide sufficient nutrients and oxygen to the surrounding tissues in response to tissue demand, the attenuated microvascular rarefaction in COMP-Ang-1-gene-transferred SHRs is thought to directly contribute to preventing the tissue ischaemia and subsequent pathological changes in the heart and kidney. However, these effects of COMP-Ang-1 gene transfer on systolic BP and histopathological sequels in target organs completely disappeared when SHRs were pre-treated with adenoviral soluble Tie2 receptor, which indicates that therapeutic effects of COMP-Ang-1 gene transfer in SHRs occur through the interaction between expressed COMP-Ang-1 protein in blood and the Tie2 receptor in the endothelium.
An important issue to be addressed is the possible mechanism responsible for the beneficial effect of COMP-Ang-1 on SHRs. In cardiovascular diseases including essential hypertension, their common mechanism is the endothelial dysfunction in which the decrease in NO bioavailability is likely to be crucial. As has been reported in many publications, NO plays a number of vasculoprotective roles by regulating endothelial homeostasis, angiogenic growth factor-induced angiogenesis, and vascular remodelling18–20. In particular, NO reduces the recruitment and adhesion of leukocytes on the endothelium by downregulating the expression of adhesion-related molecules and chemokines. In addition, NO is crucial for maintaining normal BP by controlling endothelium-mediated vasorelaxation. In this regard, its decreased production and/or increased consumption leads to an apparent reduction in the bioavailability of NO, resulting in endothelial dysfunction as frequently observed in hypertensive subjects and SHRs as well. Considering this close association of NO with hypertension, the substantially phosphorylated eNOS and high plasma level of nitrite, a metabolite of NO, in pCOMP-Ang-1-transferred SHRs implied that increased NO synthesis may contribute to maintaining endothelium-mediated vasodilation and to preserving endothelial integrity and function in growing SHRs. Indeed, the in vitro experiments done in HUVECs showed that the COMP-Ang-1 protein in blood from SHRs was able to increase nitrite production by activating the Tie2/eNOS-signalling cascade in the endothelium. However, the present result is not sufficient to conclude that NO is the major factor inducing the antihypertensive effect in COMP-Ang-1-transferred SHRs.
Not all endothelial survival or growth factors depend on the NO-signalling cascade for their anti-apoptotic and proliferating effects on endothelial cells. For example, NO production is necessary for VEGF-induced angiogenesis, but not for basic fibroblast growth factor (bFGF)-induced angiogenesis.21 The inhibition of NO synthesis by eNOS inhibitors completely blocks VEGF-induced corneal angiogenesis by suppressing endothelial sprouting, migration, and proliferation, but has no effect on bFGF-mediated actions on endothelial cells both in vivo and in vitro.22 In fact, COMP-Ang-1 seems to have similar NO-independent activity as well. In previous research on dermal wound-healing experiments, COMP-Ang-1 accelerated the wound-repairing process even in eNOS knockout mice as well, indicating that COMP-Ang-1-induced angiogenesis does not depend on the eNOS-mediated NO synthesis alone in the endothelium.23 In this regard, it cannot be ruled out that NO-independent pathway of COMP-Ang-1 in endothelial cells might significantly contribute to preventing the development of hypertension as well as the appearance of target organ damage in SHRs. With the current data, it is not clear which mechanism plays a dominant role in inducing therapeutic effects of COMP-Ang-1 in hypertensive animal model. Answering this question might require the further experiment, in which SHRs need to be pre-treated with NOS inhibitor in order to exclude the NO-mediated effect of COMP-Ang-1.
In conclusion, on the basis of the recent understanding that microvascular abnormality resulting from endothelial apoptosis and dysfunction is one of the mechanisms underlying hypertension and target organ damage, our study provides a new perspective in the treatment of hypertension by showing that one of the endothelial survival factors, COMP-Ang-1, effectively prevents hypertension as well as target organ damage in hypertensive animal model.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by NRL Program of Korea Science and Engineering Foundation (M1-0412-00-0048), Science Research Center Grant (Molecular Therapy Research Center), Samsung Biomedical Research Institute Grant (SBRI; CA7-404-1), Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2006-311-E00293), and Basic Research Program of the Korea Science and Engineering Foundation (R01-2007-000-11246-0).
Supplementary Material
Acknowledgements
The authors thank Jeong-Bae Park (Department of Medicine, Kwandong University) for helpful discussions.
Conflict of interest: none declared.
References
- 1.Cohuet G, Struijker-Boudier H. Mechanisms of target organ damage caused by hypertension: therapeutic potential. Pharmacol Ther. 2006;111:81–98. doi: 10.1016/j.pharmthera.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Redon J. Antihypertensive treatment: should it be titrated to blood pressure reduction or to target organ damage regression? Curr Opin Nephrol Hypertens. 2005;14:448–452. doi: 10.1097/01.mnh.0000168935.95527.0a. [DOI] [PubMed] [Google Scholar]
- 3.Grimm RH, Jr, Flack JM, Byington R, Bond G, Brugger S. A comparison of antihypertensive drug effects on the progression of extracranial carotid atherosclerosis. The Multicenter Isradipine Diuretic Atherosclerosis Study (MIDAS) Drugs. 1990;40(Suppl. 2):38–43. doi: 10.2165/00003495-199000402-00011. [DOI] [PubMed] [Google Scholar]
- 4.Samuelsson OG, Wilhelmsen LW, Svardsudd KF, Pennert KM, Wedel H, Berglund GL. Mortality and morbidity in relation to systolic blood pressure in two populations with different management of hypertension: The Study of Men Born in 1913 and the Multifactorial Primary Prevention Trial. J Hypertens. 1987;5:57–66. doi: 10.1097/00004872-198702000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Levy BI, Ambrosio G, Pries AR, Struijker-Boudier HA. Microcirculation in hypertension: a new target for treatment? Circulation. 2001;104:735–740. doi: 10.1161/hc3101.091158. [DOI] [PubMed] [Google Scholar]
- 6.Rizzoni D, Rodella L, Porteri E, Rezzani R, Guelfi D, Piccoli A, et al. Time course of apoptosis in small resistance arteries of spontaneously hypertensive rats. J Hypertens. 2000;18:885–891. doi: 10.1097/00004872-200018070-00010. [DOI] [PubMed] [Google Scholar]
- 7.Gobe G, Browning J, Howard T, Hogg N, Winterford C, Cross R. Apoptosis occurs in endothelial cells during hypertension-induced microvascular rarefaction. J Struct Biol. 1997;118:63–72. doi: 10.1006/jsbi.1996.3835. [DOI] [PubMed] [Google Scholar]
- 8.Cho CH, Kammerer RA, Lee HJ, Steinmetz MO, Ryu YS, Lee SH, et al. COMP-Ang1: a designed angiopoietin-1 variant with nonleaky angiogenic activity. Proc Natl Acad Sci USA. 2004;101:5547–5552. doi: 10.1073/pnas.0307574101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho CH, Kammerer RA, Lee HJ, Yasunaga K, Kim KT, Choi HH, et al. Designed angiopoietin-1 variant, COMP-Ang1, protects against radiation-induced endothelial cell apoptosis. Proc Natl Acad Sci USA. 2004;101:5553–5558. doi: 10.1073/pnas.0307575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim W, Moon SO, Lee SY, Jang KY, Cho CH, Koh GY, et al. COMP-angiopoietin-1 ameliorates renal fibrosis in a unilateral ureteral obstruction model. J Am Soc Nephrol. 2006;17:2474–2483. doi: 10.1681/ASN.2006020109. [DOI] [PubMed] [Google Scholar]
- 11.Cho CH, Kim KE, Byun J, Jang HS, Kim DK, Baluk P, et al. Long-term and sustained COMP-Ang1 induces long-lasting vascular enlargement and enhanced blood flow. Circ Res. 2005;97:86–94. doi: 10.1161/01.RES.0000174093.64855.a6. [DOI] [PubMed] [Google Scholar]
- 12.Lee JS, Byun J, Kim JM, Kim CY, Kim BM, Chung JH, et al. Cardiac expression profiles of the naked DNA vectors encoding vascular endothelial growth factor and basic fibroblast growth factor. Exp Mol Med. 2005;37:447–456. doi: 10.1038/emm.2005.55. [DOI] [PubMed] [Google Scholar]
- 13.Noon JP, Walker BR, Webb DJ, Shore AC, Holton DW, Edwards HV, et al. Impaired microvascular dilatation and capillary rarefaction in young adults with a predisposition to high blood pressure. J Clin Invest. 1997;99:1873–1879. doi: 10.1172/JCI119354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonios TF, Rattray FM, Singer DR, Markandu ND, Mortimer PS, MacGregor GA. Rarefaction of skin capillaries in normotensive offspring of individuals with essential hypertension. Heart. 2003;89:175–178. doi: 10.1136/heart.89.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antonios TF, Singer DR, Markandu ND, Mortimer PS, MacGregor GA. Rarefaction of skin capillaries in borderline essential hypertension suggests an early structural abnormality. Hypertension. 1999;34:655–658. doi: 10.1161/01.hyp.34.4.655. [DOI] [PubMed] [Google Scholar]
- 16.Veronese ML, Mosenkis A, Flaherty KT, Gallagher M, Stevenson JP, Townsend RR, et al. Mechanisms of hypertension associated with BAY 43-9006. J Clin Oncol. 2006;24:1363–1369. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 17.Thomas AL, Morgan B, Drevs J, Unger C, Wiedenmann B, Vanhoefer U, et al. Vascular endothelial growth factor receptor tyrosine kinase inhibitors: PTK787/ZK 222584. Semin Oncol. 2003;30:32–38. doi: 10.1016/s0093-7754(03)00123-4. [DOI] [PubMed] [Google Scholar]
- 18.Kiefer FN, Neysari S, Humar R, Li W, Munk VC, Battegay EJ. Hypertension and angiogenesis. Curr Pharm Des. 2003;9:1733–1744. doi: 10.2174/1381612033454540. [DOI] [PubMed] [Google Scholar]
- 19.Naseem KM. The role of nitric oxide in cardiovascular diseases. Mol Aspects Med. 2005;26:33–65. doi: 10.1016/j.mam.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol. 2006;291:H985–H1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- 21.Sane DC, Anton L, Brosnihan KB. Angiogenic growth factors and hypertension. Angiogenesis. 2004;7:193–201. doi: 10.1007/s10456-004-2699-3. [DOI] [PubMed] [Google Scholar]
- 22.Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, et al. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho CH, Sung HK, Kim KT, Cheon HG, Oh GT, Hong HJ, et al. COMP-angiopoietin-1 promotes wound healing through enhanced angiogenesis, lymphangiogenesis, and blood flow in a diabetic mouse model. Proc Natl Acad Sci USA. 2006;103:4946–4951. doi: 10.1073/pnas.0506352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.