

Abstract
Inhibitors of hepadnaviral DNA polymerases are predicted to inhibit both minus and plus strand of viral DNA synthesis and arrest viral DNA replication at the stage of pregenomic (pg) RNA-containing nucleocapsids. However, analyses of the RNA species of human and duck hepatitis B viruses (HBV and DHBV, respectively) in hepatoma cells treated with viral DNA polymerase inhibitors revealed the genesis of novel RNA species migrating slightly faster than the full-length pgRNA. The DNA polymerase inhibitor-induced accumulation of these RNA species were abolished in the presence of alpha-interferon or HBV nucleocapsid assembly inhibitors. Moreover, they were protected from microccocal nuclease digestion and devoid of a poly-A tail. These characteristics suggest that the novel RNA species are most likely generated from RNase H cleavage of encapsidated pgRNA, after primer translocation and synthesis of the 5′ terminal portion of minus strand DNA. In support of this hypothesis, DNA polymerase inhibitor treatment of chicken hepatoma cells transfected with a DHBV genome encoding an RNase H inactive DNA polymerase (E696H) failed to produce such RNA species. Our results thus suggest that the currently available DNA polymerase inhibitors do not efficiently arrest minus strand DNA synthesis at the early stage in hepatocytes. Hence, development of novel antiviral agents that more potently suppress viral DNA synthesis or viral nucleocapsid assembly inhibitors that are mechanistically complementary to the currently available DNA polymerase inhibitors are warranted.
1. Introduction
Despite the availability of effective vaccines for the last three decades, chronic hepatitis B virus (HBV) infection still affects 240 million people worldwide and remains a major public health problem (Ott et al., 2012). Treatment of chronic hepatitis B patients with nucleos(t)ide analog HBV DNA polymerase inhibitors, such as lamivudine, adefovir, entecavir, telbivudine and tenofovir, dramatically reduces the viral load, significantly improves the liver function and lowers the incidence of liver failure and hepatocellular carcinoma (Liaw, 2013b). However, chronic HBV infection-related diseases still account for 600,000 deaths per year worldwide (Ott et al., 2012).
HBV is the prototype member of Hepadnaviridae family and contains a relaxed circular (rc) partially double-stranded, 3.2kb in length, DNA genome with its DNA polymerase covalently linked to the 5′ terminus of minus strand. One of the most characteristic features of hepadnaviruses is that their genomic DNA is replicated via protein-primed reverse transcription of an RNA intermediate called pregenomic (pg) RNA in the cytoplasmic nucleocapsids (Summers and Mason, 1982). HBV DNA polymerase (P) is a multifunctional protein containing four structurally and functionally distinct domains, a terminal protein (TP) domain at the amino-terminus region followed by a spacer domain, a reverse transcriptase (RT) domain and an RNase H domain at the carboxy-terminus region. While the RT and RNase H domains share homologous sequence motifs with retroviral reverse transcriptases, the TP and spacer domains are unique to the hepadnaviral P proteins, with no phylogenetically related structures present in other viral DNA polymerases (Miller and Robinson, 1986). The HBV P protein plays essential roles in pgRNA encapsidation as well as viral DNA synthesis (Wang and Seeger, 1992, 1993). Specifically, once translated from pgRNA, the P protein binds to a stem-loop structure at the 5′ terminal region of pgRNA, termed epsilon, which triggers the recruitment of core proteins to package the pgRNA-polymerase complex into nucleocapsids (Bartenschlager and Schaller, 1992). Meanwhile, the interaction of P protein and epsilon also initiates the synthesis of the minus strand of viral DNA by using the P protein itself as a protein primer for the formation of a phosphodiester linkage between a hydroxyl residue of a tyrosine located in its TP domain and dGMP with the proposed bulge region of epsilon RNA acting as a template (Bartenschlager and Schaller, 1988; Wang and Seeger, 1992; Zoulim and Seeger, 1994). The priming reaction terminates following the synthesis of only 3 to 4 nucleotides, which are subsequently transferred to a complementary sequence motif near the 3′ terminus of pgRNA to continue the elongation of the minus strand of viral DNA toward the 5′ terminus of pgRNA (Tavis et al., 1994; Wang and Seeger, 1993). The pgRNA is simultaneously degraded by the RNase H domain of the P protein, except for its capped 5´ terminal region including 5´ DR1, which is translocated to the DR2 region of the minus strand DNA to prime the synthesis of plus strand DNA (Lien et al., 1987; Lien et al., 1986).
Nucleos(t)ide analogs inhibit HBV DNA synthesis by acting as DNA chain terminators. Structurally, these analogs lack the 3′-hydroxyl group that permits covalent binding to an incoming nucleotide on the elongating DNA chain. Therefore, incorporation of the nucleotide analog into the nascent DNA chain will arrest further chain elongation (Jordheim et al., 2013; Menendez-Arias et al., 2014). In vitro biochemical studies showed that while entecavir-TP (a dGTP analog) inhibits protein priming initiated specifically with dGTP, tenofovir DF-TP as well as PFA, a pyrophosphate analog DNA polymerase inhibitor, strongly inhibit the addition of two dAMP nucleotides to the initial deoxyguanosine nucleotide (Jones et al., 2013; Wang and Seeger, 1992). These findings suggest that treating HBV-infected hepatocytes with viral DNA polymerase inhibitors should arrest viral DNA replication at the priming stage and nucleocapsids should thus contain full-length pgRNA. However, analyses of the RNA species of HBV and duck hepatitis B virus (DHBV) in hepatoma cells treated with viral DNA polymerase inhibitors revealed the existence of additional RNA species migrating slightly faster than the full-length pgRNA (Liu et al., 2013). Our studies reported herein demonstrate that the DNA polymerase inhibitor-induced novel viral RNA species are encapsidated in the nucleocapsids and generated from RNase H cleavage of pgRNA, after the DNA polymerase-primer complex translocation and synthesis of the 5′ terminal portion of the minus strand DNA from the 3′ terminal region of pgRNA. Our finding suggests that DNA polymerase inhibitors do not efficiently arrest hepadnaviral DNA replication in the priming stage in vivo. It is possible that the viral polymerases may adopt a distinct conformations in the early stage of minus strand DNA synthesis, which renders the enzyme less sensitive to nucleos(t)ide analog inhibitors. Nevertheless, considering the potential role of viral nucleocapsid-associated viral DNA in amplification of liver inflammation through activation of pattern recognition receptors in hepatocytes or liver nonparenchymal cells (Chang et al., 2012; Chang et al., 2014; Sato et al., 2015), it is conceivable that treatment of chronic hepatitis B with nucleocapsid assembly inhibitors, which preclude the formation of pgRNA-containing capsids and subsequent synthesis of viral DNA, along or in combination with DNA polymerase inhibitors might be more efficient in attenuating liver inflammation, in addition to more potently suppressing viral replication.
2. Materials and Methods
2.1 Cell Lines, IFN-α and Chemicals
Chicken hepatoma cells (LMH) were cultured in DMEM/F12 medium (Mediatech) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. dstet5 is a LMH-derived stable cell line supporting the replication of an envelope protein-deficient DHBV genome in a tetracycline dependent manner (Guo et al., 2003). HepAD38 is a human hepatoma cell (HepG2)-derived stable cell line supporting HBV genome replication in a tetracycline dependent manner (Guo et al., 2007). Both cell lines were maintained in DMEM/F12 medium (Mediatech) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml tetracycline and 200 μg/ml G-418. Chicken alpha-interferon (IFN-α) was produced and titrated as described previously (Guo et al., 2003). Foscarnet (PFA), Lamivudine (LAM), and Adefovir (ADV) were purchased from Sigma. Entecavir (ETV) was provided by Dr. William S. Mason at Fox Chase Cancer Center, Philadelphia. Bay 41-4109 was provided by Dr. Lai Wei at Hepatology Institute of Peking University, China. AT-61 and DVR-23 are synthesized in house as described previously (Campagna et al., 2013; Xu et al., 2010).
2.2 Plasmids
pCMV-DHBV expressing the wild-type DHBV pregenomic (pg) RNA under the transcriptional control of cytomegalovirus (CMV) immediate early (IE) promoter has been described previously (Condreay et al., 1990). A plasmid expressing a mutant DHBV polymerase protein containing E696H mutation in the RNase H domain, designated as pCMV-DHBV/PE696H, was generated by site-directed mutagenesis with primers 5′-GGTCAGAGATATACATGTTCAGCACCTATTGATGTCTTGTTTAG-CC-3′ and 5′-GGCTAAACAAGACATCAATAGGTGCTGAACATGTATATCTC-TTGACC-3′ by using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) and following the manufacturer’s instructions. The desired mutation was verified by DNA sequencing.
2.3 Northern Blot analysis of HBV or DHBV RNA
Total cellular RNA in HepAD38 cells or dstet5 cells was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s direction. Encapsidated DHBV RNA were extracted as described previously (Xu et al., 2010). Briefly, dstet5 cells cultured in 12-well plates were lysed with 300 μl of lysis buffer (10m M Tris-HCl (pH 7.6), 100 mM NaCl, 1 mM EDTA and 0.1% NP-40) per well. Cell debris and nuclei were removed by centrifugation at 10,000 g for 10 min. The supernatants were digested with 20 U/ml of micrococcal nuclease (MNase) at 37°C for 30 min. The core particles were precipitated with 35% Polyethylene glycol (PEG) 8000 dissolved in 1.5 M NaCl in ice for 60 min. After centrifugation at 6,000 rpm for 6 min in an eppendorf centrifuge (5427R), the core particles were dissolved in TNE buffer (10 mM Tris-HCl, pH7.6; 150 mM NaCl and 1 mM EDTA). Encapsidated RNA in core particles was extracted with TRIzol reagent according to the manufacturer’s direction. Six micrograms of total RNA or one half of the encapsidated RNA prepared from one well of a 12-well plate was separated in a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto a Hybond-XL membrane in 20× SSC buffer. For the detection of HBV or DHBV RNA, membranes were probed with an α-32P-UTP (800 Ci/mmol, Perkin Elmer) labeled minus-strand specific full length HBV or DHBV riboprobe as described previously (Guo et al., 2003; Xu et al., 2010). Membranes were exposed to a phosphoimager screen and hybridization signals were quantified using QuantityOne software (Bio-Rad).
2.4 Purification and analysis of poly A+ mRNA from total DHBV RNA
To determine whether these novel RNA species contain a poly A+ tail, poly A+ mRNA was extracted from total RNA preparations with Oligotex mRNA Kit (Qiagen). To ensure maximal extraction of polyA+ RNA from the total RNA preparations, 6 μg of total RNA prepared from dstet5 cells left untreated or treated with viral DNA polymerase inhibitors were incubated with 15 μl of Oligotex suspension that has a capacity to bind 9 μg of polyA mRNA. The total cellular RNA and polyA+ RNA were separated on a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto a Hybond-XL membrane in 20× SSC buffer. The membranes were probed with an α-32P-UTP (800 Ci/mmol, Perkin Elmer) labeled minus-strand specific full length DHBV riboprobe as described previously (Guo et al., 2003; Xu et al., 2010).
2.5 Southern Blot analysis of HBV or DHBV DNA
To determine the antiviral activity of different compounds against HBV or DHBV, HepAD38 or dstet 5 cells were seeded into 12-well plates at a density of 5 × 105 cells per well and cultured in DMEM/F12 media with 1μg/ml tetracycline for 2 days. The cells were then treated with different compounds for 5 days (HepAD38) or 3 days (dstet5) in the absence of tetracycline. Total intracellular viral core-associated DNA were extracted and analyzed by Southern blot hybridization with an α-32P-UTP (800 Ci/mmol, Perkin Elmer) labeled full-length HBV or DHBV riboprobe that specifically hybridizes to minus-strand of viral DNA (Guo et al., 2003; Xu et al., 2010).
2.6 Transfection
LMH cells were seeded into 12-well plates at a density of 8 × 105 cells per well and cultured with DMEM/F12 supplemented with 10% heat-inactivated fetal bovine serum for 12 h. Each well was transfected with 1 μg of pCMV-DHBV or pCMV-DHBV/PE696H with Lipofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. Six hours after transfection, cells were left untreated or treated with lamivudine for two days and core-associated DNA was extracted. For detailed analysis of viral minus and plus strand DNA synthesis, an aliquot of the core-associated DNA from each preparations were denatured in 50% formamide at 95°C for 2 min as previously described (Chen et al., 1996) to convert double-stranded DNA into single-stranded forms. The non-denatured and denatured core-associated DNA were analyzed by Southern blot hybridization as described above.
2.7 Mapping the 3′ ends of encapsidated viral RNA
dstet5 cells were seeded into 10-cm dishes at a density of 5.0 × 106 cells per dish and cultured for an additional two days. Cells were treated with 10 μM of LAM or 100 nM of ETV for 3 days in the absence of tetracycline. HepD38 were treated with 10 μM of LAM for 5 days in tetracycline-free medium. The cells were lysed with 2 ml of lysis buffer (10 mM Tris-HCl (pH 7.6), 100 mM NaCl, 1 mM EDTA and 0.1% NP-40) per plate. Cell debris and nuclei were removed by centrifugation at 10,000 g for 10 min. The supernatants were digested with 20 U/ml of MNase at 37°C for 30 min. The core particles were precipitated with 35% PEG 8000 dissolved in 1.5 M NaCl in ice for 60 min. After centrifugation at 6,000 rpm for 6 min in an eppendorf centrifuge (5427R), the core particles were dissolved in TNE buffer (10 mM Tris-HCl, pH7.6; 150 mM NaCl and 1 mM EDTA). Encapsidated RNA was extracted with TRIzol reagent. The poly A+ RNA was removed by using Oligotex mRNA kit (Qiagen) as described in section 2.4. A poly A+ tail was added to the flow-through RNA with a poly (A) Tailing Kit (Thermo Scientific) by following the manufacturer’s direction. The resulting RNA was reverse-transcribed into cDNA by using SuperScript® III Reverse Transcriptase (Invitrogen) with oligo (dT) adaptor 5′-GCTTAGTGACACCTCCTTGGC(dT)25-3′ as primers. The primers for PCR amplification of the DNA complementary to the 3′ terminal fragments of the encapsidated viral RNA are 5′-GCTTAGTGACACCTCCTTGGC-3′ and DHBV specific primer 5′-CATCTCTTCACTACTGCCCTCGG-3′ or HBV specific primer 5′-CTGTGCCAAGTGTTTGCTGACG-3′. The PCR was performed by using Phusion® High-Fidelity DNA Polymerase (New England Biolabs) in the following conditions: 98°C, 5 min; 98°C, 30s, 72 °C 1min, 5 cycles; 98 °C, 30s, 70 °C, 30s, 72 °C, 1 min, 5 cycles; 98 °C, 30s, 68 °C, 30s, 72 °C, 1 min, 30 cycles; 72 °C, 7 min. The PCR products were purified by using Wizard® Smart gel extract kit and cloned into the pGEM-T easy vector (Promega). Approximately 30 viral cDNA clones were sequenced to determine the 3′ terminus of the encapsidated viral RNA.
3. Results
3.1 Novel DHBV RNA species were accumulated in hepatoma cells treated with viral DNA polymerase inhibitors
We previously established a chicken hepatoma-derived stable cell line, known as dstet5, which harbors an integrated transgene for transcription of DHBV pgRNA in a tetracycline (tet) inducible manner (Guo et al., 2003). Upon removal of tet from culture medium, pgRNA is transcribed from the viral transgene, which leads to the sequential occurrence of viral protein translation, nucleocapsid assembly, DNA synthesis and cccDNA formation. As expected, treatment of the cells with LAM, a viral DNA polymerase inhibitor, and IFN-α that inhibits cccDNA transcription and pgRNA encapsidation (Guo et al., 2003; Liu et al., 2013), alone or in combination, for three days after tet removal, dramatically reduced the amounts of viral core-associated DNA replication intermediates (Fig. 1, lower panel). Viral RNA analysis using Northern blot hybridization also demonstrated that both LAM and IFN-α treatment reduced the amounts of viral RNA, presumably due to their inhibition of cccDNA formation and IFN-α suppression of cccDNA transcription (Liu et al., 2013). However, a RNA species migrating slightly faster than the full-length pgRNA was accumulated in cells treated with LAM, but not in cells treated with IFN-α. Interestingly, the LAM-induced accumulation of the novel RNA species was almost abolished in cells treated with both LAM and IFN-α (Fig. 1, upper panel).
Figure 1. A new RNA species was accumulated in dstet5 cells treated with lamivudine.
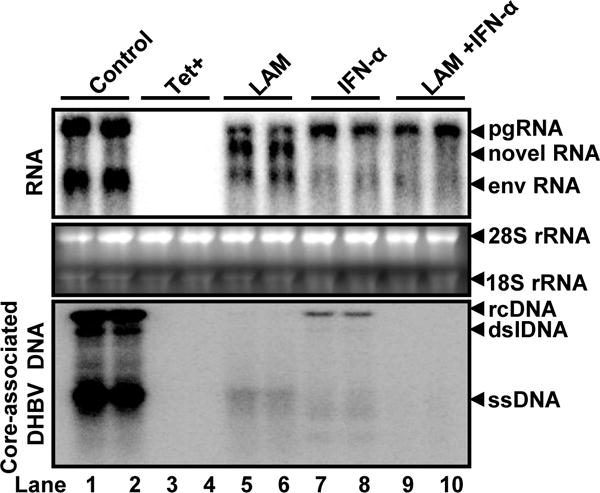
dstet5 cells were left untreated (lanes 1 and 2) or treated with 10 μM lamivudine (LAM) (lanes 5 and 6), 100 U/ml chicken IFN-α (lanes 7 and 8) or combination of 10 μM lamivudine and 100 U/ml chichen IFN-α (lanes 9 and 10) in the absence of tet for 3 days. dstet5 cells cultured in the presence of tet (lanes 3 and 4) served as negative controls. DHBV RNA and core-associated DNA were analyzed by Northern or Southern Blot hybridization assays, respectively. For RNA analysis, ribosomal RNAs (28S and 18S rRNA) served as loading controls. The positions of DHBV pgRNA and subgenomic mRNA encoding viral envelope proteins (envRNA) are indicated. The RNA species accumulated in the cells treated with LAM are indicated as novel RNA. For core-associated DHBV DNA, the forms of relaxed circular (RC), double stranded linear (DSL) and single stranded (SS) DNA are indicated.
To validate and extend this observation, we investigated whether the novel DHBV RNA species accumulation could also be induced upon treatment of dstet5 cells with other viral DNA polymerase inhibitors. As shown in Fig. 2A, treatment of dstet5 cells with the indicated concentrations of PFA, LAM, ADV and ETV for three days after tet removal dramatically reduced the amounts of viral core-associated DNA replication intermediates and induced the accumulation of the novel RNA species. Apparently, the electrophoretic mobility of the novel RNA species differed slightly according to the viral DNA polymerase inhibitors used (Fig. 2A). Moreover, the results presented in Fig. 2B indicated that LAM treatment increased the amounts of the novel RNA species in dstet5 cells in a dose-dependent manner.
Figure 2. Treatment of dstet5 cells with multiple hepadnaviral DNA polymerase inhibitors resulted in the accumulation of the novel sub-genomic RNA species.
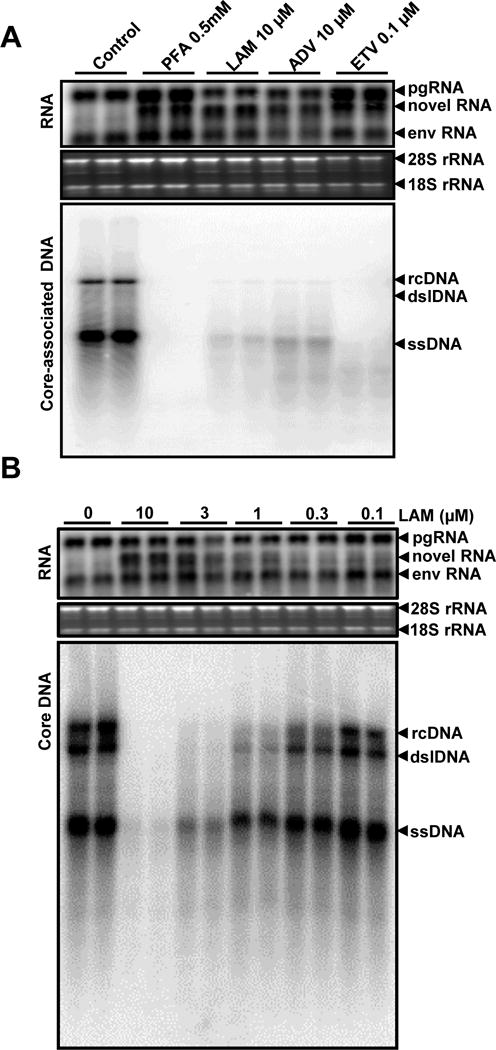
(A) dstet5 cells were treated with the indicated viral DNA polymerase inhibitors for 3 days. DHBV RNA and core-associated DNA were analyzed by Northern and Southern Blot hybridization assays, respectively. (B) Lamivudine induces the accumulation of the novel RNA species in a dose-dependent manner. dstet5 cells were treated with the indicated concentrations of lamivudine for 3 days in the absence of tet. DHBV RNA and core-associated DNA were analyzed as described above. Ribosomal RNAs (28S and 18S rRNA) served as loading controls. The positions of DHBV pgRNA and subgenomic mRNA encoding viral envelope proteins (envRNA) are indicated. The RNA species accumulated in the cells treated with DNA polymerase inhibitors are indicated as novel RNA. For core-associated DHBV DNA, the forms of relaxed circular (RC), double stranded linear (DSL) and single stranded (SS) DNA are indicated.
3.2 DNA polymerase inhibitors also induced the accumulation of novel HBV RNA species in human hepatocyte-derived cells
To test whether inhibition of HBV DNA replication by DNA polymerase inhibitors could also induce the accumulation of the novel RNA species, a human hepatoma HepG2-derived stable cell line harboring an integrated transgene for transcription of HBV pgRNA in a tetracycline (tet) inducible manner, designated as HepAD38 (Ladner et al., 1997), were treated with a serial dilution of LAM for 5 days after tet removal. As shown in Fig. 3, LAM dose-dependently inhibited HBV DNA replication and induced the accumulation of the novel HBV RNA species in HepAD38 cells.
Figure 3. DNA polymerase inhibitor also induced the accumulation of a novel HBV RNA species in HepAD38 cells.
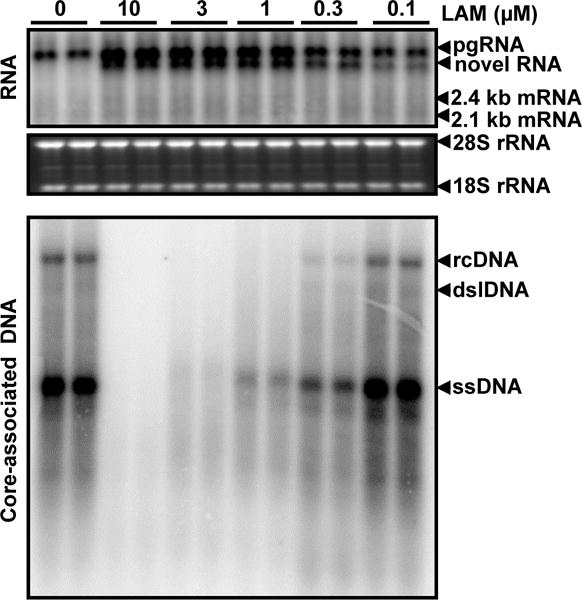
HepAD38 cells were treated with the indicated concentrations of lamivudine for 5 days in the absence of tet. Intracellular HBV RNA and core-associated DNA were analyzed by Northern and Southern Blot hybridization assays, respectively. For RNA analysis, ribosomal RNAs (28S and 18S rRNA) served as loading controls. The positions of HBV pgRNA and 2.4 and 2.1 kb mRNA encoding viral envelope proteins are indicated. The HBV RNA species accumulated in the cells treated with LAM was indicated as novel RNA. For core-associated HBV DNA, the forms of relaxed circular (RC), double stranded linear (DSL) and single stranded (SS) DNA are indicated.
Taken together, the results presented in the above sections indicate that inhibition of either HBV or DHBV replication with DNA polymerase inhibitors results in accumulation of the RNA species migrating slightly faster than full-length pgRNA in chicken or human hepatocyte-derived cell lines.
3.3 HBV capsid assembly inhibitors prevent LAM-induced accumulation of the novel RNA species
The two plausible mechanisms underlying the accumulation of these novel RNA species are alternative splicing or cleavage of pre-C-mRNA or pgRNA. Because IFN-α inhibited DHBV pgRNA encapsidation (Guo et al., 2003; Schultz et al., 1999) and efficiently abolished the LAM-induced accumulation of the novel RNA species in dstet5 cells (Fig. 1), we favored the hypothesis that the precursor of the novel RNA species is most likely the encapsidated pgRNA. To further investigate this hypothesis, we took advantage of the small molecules that specifically inhibit HBV capsid assembly or pgRNA encapsidation, such as Bay 41-4109 (Deres et al., 2003; Stray et al., 2005; Stray and Zlotnick, 2006), AT-61 (Feld et al., 2007; King et al., 1998) and DVR-23 (Campagna et al., 2013), and determined if inhibition of capsid assembly or pgRNA encapsidation would prevent the accumulation of LAM-induced novel RNA species in HepAD38 cells. As expected, treatment of HepAD38 cells with LAM and each of the three HBV capsid/nucleocapsid assembly inhibitors, alone or in combination, efficiently suppressed HBV DNA synthesis, but did not apparently affect the levels of HBV pgRNA (Fig. 4). However, as predicted by our hypothesis, combinational treatment of HepAD38 cells with LAM and each of the three capsid assembly or pgRNA encapsidation inhibitors completely prevented the accumulation of the novel RNA species (Fig. 4, upper panel).
Figure 4. HBV capsid assembly inhibitors prevent lamivudine-induced accumulation of the novel HBV RNA species.
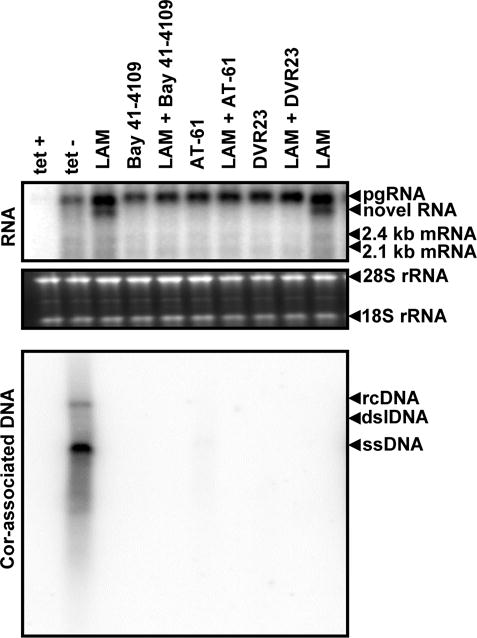
HepAD38 cells were left untreated (tet-) or treated with 10 μM LAM, 2 μM Bay 41-4109, 20 μM AT-61, 5 μM DVR-23, alone or in the indicated combinations for 5 days in the absence of tet. HBV RNA and core-associated DNA were analyzed by Northern and Southern Blot hybridization assays, respectively. For RNA analysis, ribosomal RNAs (28S and 18S rRNA) served as loading controls. The positions of HBV pgRNA and 2.4 and 2.1 kb mRNA encoding viral envelope proteins are indicated. The HBV RNA species accumulated in the cells treated with LAM was indicated as novel RNA. For core-associated HBV DNA, the forms of relaxed circular (RC), double stranded linear (DSL) and single stranded (SS) DNA are indicated.
3.4 The novel RNA species is protected from MNase digestion and devoid of poly-A tail
The above results strongly support the notion that the novel RNA species accumulated in hepadnaviral DNA polymerase inhibitor-treated cells are derived from encapsidated pgRNA. Accordingly, it can be predicted that the RNA species should be protected from nuclease digestion (Summers and Mason, 1982). Indeed, as demonstrated in Fig. 5, comparing to viral RNA profile presented in total cellular RNA (upper panel), MNase treatment of cell lysates from dstet5 cells treated with DNA polymerase inhibitors efficiently digested subgenomic RNA specifying viral envelope proteins as well as the majority of the pgRNA. However, the polymerase inhibitor-induced RNA species as well as a fraction of the pgRNA were protected from the nuclease digestion (Fig. 5, lower panel). Moreover, a poly-A selection of the total RNA prepared from the cells treated with LAM or ETV indicated that unlike pgRNA and envelope protein RNAs, the novel RNA species cannot bind to oligo(dT) and are thus devoid of a poly-A tail (Fig. 6). Hence, our results imply that the DNA polymerase inhibitor-induced novel viral RNA species were conceivably generated by the cleavage of encapsidated pgRNA at its 3′ terminal region.
Figure 5. The novel DHBV RNA species is protected from MNase digestion.
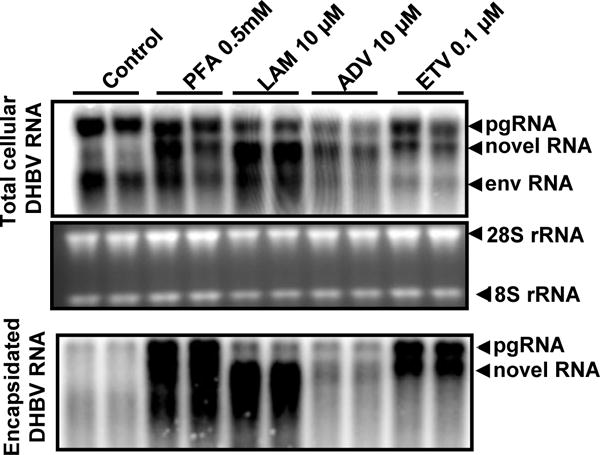
dstet5 cells were left untreated (Control) or treated with the indicated DNA polymerase inhibitors for 3 days in the absence of tet. Cell lysates were centrifugated at 10,000 g for 10 min to remove cell debris and nuclei. The supernatants were digested with 20 U/ml of MNase at 37°C for 30 min. The core particles were precipitated with 35% PEG 8000 in ice for 60 min. Encapsidated RNA was extracted with TRIzol reagent. Total cellular RNA (upper panel) and encapsidated DHBV RNA (lower panel) were analyzed by Northern Blot hybridization. Ribosomal RNAs (28S and 18S rRNA) served as loading controls. The two lanes under each treatment condition are the results of experimental duplicates. The positions of DHBV pgRNA and subgenomic mRNA encoding viral envelope proteins (envRNA) are indicated. The RNA species accumulated in the cells treated with DNA polymerase inhibitors are indicated as novel RNA.
Figure 6. The novel RNA species is devoid of a poly-A tail.
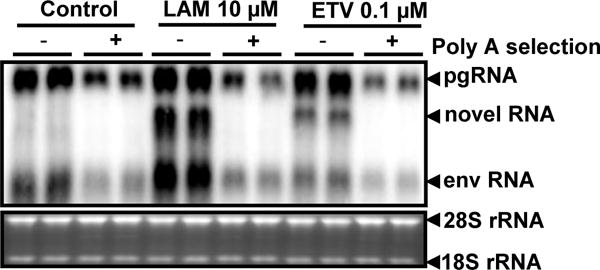
dstet5 cells were treated with 10 μM LAM or 0.1 μM ETV for 3 days in the absence of tet. Total cellular RNA were extracted with TRIzol reagents and polyA+ mRNA were extracted with an Oligotex mRNA mini kit from total RNA preparations. Each of the lanes was loaded with 6 μg of total cellular RNA or polyA-selected RNA from 6 μg of total cellular RNA from dstet5 cells left untreated or treated with the indicated concentrations of viral DNA polymerase inhibitors. To ensure each of the lanes are loaded with equal amount of RNA, each of the polyA-selected RNA samples was mixed with 6 μg of total cellular RNA prepared from parental LMH cells. DHBV RNA were detected by Northern blot hybridization. Ribosomal RNAs (28S and 18S rRNA) served as loading controls. The positions of DHBV pgRNA and subgenomic mRNA encoding viral envelope proteins (envRNA) are indicated. The RNA species accumulated in the cells treated with DNA polymerase inhibitors are indicated as novel RNA.
3.5 Generation of the novel RNA species requires RNase H activity
Reverse transcription replication of HBV DNA begins with binding of the viral P protein to a stem–loop structure epsilon (ε) located in the 5′ terminal redundancy of pgRNA, which facilitates packaging of the P protein and pgRNA into viral nucleocapsids. After the 3 nucleotides from a bulge in the side of epsilon are copied, the P protein and the 3 nucleotides translocate and anneal to the copy of the 11nt sequence, termed DR1, located in the 3′ terminal redundancy sequence of pgRNA to resume the elongation of minus stranded viral DNA (Tavis et al., 1994; Wang and Seeger, 1993). Importantly, while the reverse transcriptase of the P protein copies the pgRNA into minus strand DNA, the RNase H of the P protein cleaves the pgRNA after it has been copied (Tavis et al., 2013). This unique replication strategy as well as the characteristics of the novel RNA species suggests that viral DNA polymerase inhibitors may not arrest viral DNA replication at priming stage. Instead, the DNA polymerase inhibitor-induced novel viral RNA species are most likely generated from RNase H cleavage of pgRNA, after the P protein-primer complex translocation and synthesis of 5′ terminal portion of minus strand DNA from the 3′ terminal region of pgRNA. To test this hypothesis, LMH cells were transfected with a plasmid pCMV-DHBV expressing the wild-type DHBV pgRNA under the control of CMV IE promoter or pCMV-DHBV/PE696H expressing a pgRNA encoding a polymerase protein with an E696H mutation in the RNase H domain. The transfected cells were left untreated or treated with 10 μM LAM for 48 h. As shown in Fig. 7A, while both single- and double-stranded DHBV DNA were synthesized in pCMV-DHBV transfected cells, only incomplete single-stranded DHBV DNA were synthesized in cells transfected with pCMV-DHBV/PE696H. This observation is consistent with the reported RNase H-deficient phenotype of the P protein mutation (Chen and Marion, 1996; Chen et al., 1994). Moreover, in support of our hypothesis, LAM treatment only induced the accumulation of the novel RNA species in the cells transfected with pCMV-DHBV, but not pCMV-DHBV/PE696H (Fig. 7B). The results thus are consistent with the notion that generation of the novel DHBV RNA species in DNA polymerase inhibitor-treated hepatoma cells requires RNase H activity.
Figure 7. Accumulation of the novel RNA species requires RNase H activity.
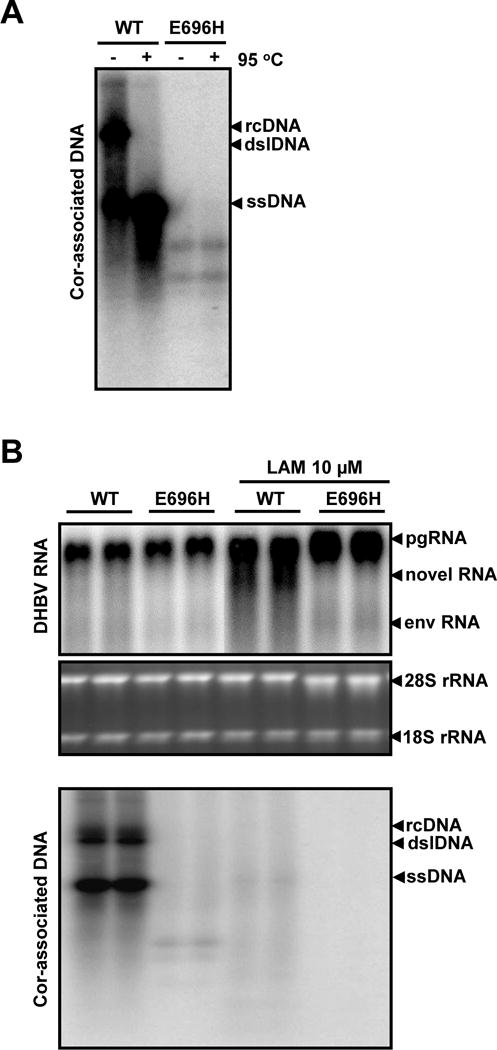
Chicken hepatoma cells (LMH) were transfected with plasmid pCMV-DHBV (WT) or pCMV-DHBV/PE696H (E696H). (A) Forty-eight hours post transfection, the core-associated viral DNA were extracted, resolved in agarose gel, without or with denaturation in 50% formamide at 95°C before loading as indicated on the top of the figure, and detected by hybridization with a full-length riboprobe-specific for minus-stranded viral DNA. (B) The transfected cells were left untreated or treated with 10 μM of lamivudine for 48h. Total cellular RNA and core-associated DNA were extracted and analyzed by Northern (upper panel) and Southern (lower panel) hybridization, respectively. For RNA analysis, ribosomal RNAs (28S and 18S rRNA) served as loading controls. The positions of DHBV pgRNA and subgenomic mRNA encoding viral envelope proteins (envRNA) are indicated. The RNA species accumulated in the cells treated with LAM are indicated as novel RNA. For core-associated DHBV DNA, the forms of relaxed circular (RC), double stranded linear (DSL) and single stranded (SS) DNA are indicated.
3.6 Mapping the 3′ terminus of the novel RNA species
To map the exact 3′ termini of the novel RNA species, dstet5 cells were treated with LAM or ETV for 3 days after tet removal. The cytoplasmic encapsidated viral RNA was prepared. A poly-A tail was then artificially added to the encapsidated RNA and cDNA was synthesized by using an oligo (dT)-containing adaptor as primer. The cDNA were then amplified and cloned into pGEM-T easy vector. Approximately 30 viral cDNA clones were sequenced. To our surprise, the 3′ termini of the encapsidated DHBV RNA did not cluster within a narrow region of pgRNA, but were mapped to a region between nt 1909 to 2522 in DHBV pgRNA (Fig. 8A). However, careful analyses revealed that the 3′ termini of the majority of encapsidated DHBV RNA, i.e. 17 out of 23 in LAM-treated and 16 out of 25 in ETV-treated dstet5 cells, are mapped to the pgRNA sequence between nt 2151 to 2488 and nt 2130 to 2306, respectively. Those results are consistent with the observation that the Northern blot band of the novel RNA species in LAM-treated cells is more defused and migrates slightly faster that in ETV-treated cells (Figs. 2A and 5). Similarly, the 3′ termini of encapsidated HBV RNA in lamivudine-treated HepAD38 cells were mapped to a region between nt 1373 to 1802 in HBV genome, with a higher frequency to end between nt 1726 to 1803 (Fig. 8B).
Figure 8. Mapping the 3′ ends of encapsidated viral RNA.
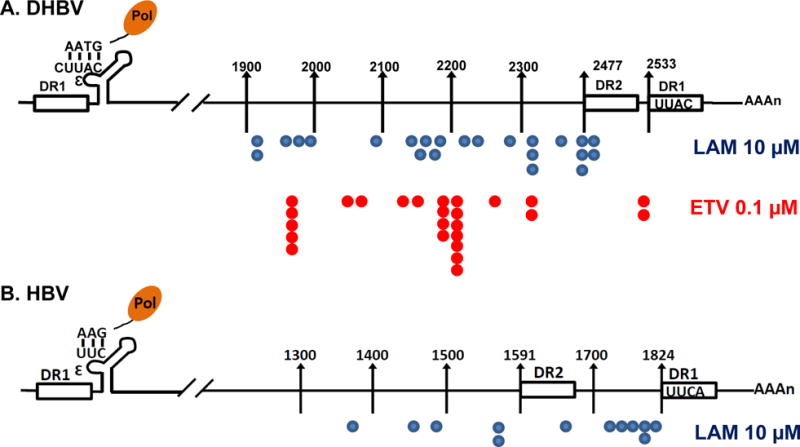
(A) dstet5 cells were treated with 10 μM LAM or 0.1 μM ETV for 3 days in the absence of tet. (B) HepAD38 cells were treated with 10 μM LAM for 5 days in the absence of tet. Encapsidated polyA-free viral RNAs were extracted and their 3′ termini were determined by a 3′-RACE procedure as described in Materials and Methods. The position of 3′ terminus of each of the sequenced DHBV or HBV cDNA was depicted in the diagram of viral pgRNA.
4. Discussion
The nucleos(t)ide analog DNA polymerase inhibitors are the only class of direct acting antiviral agents approved for the management of chronic hepatitis B. They can reduce the viral load in plasma by more than 4 logs in the vast majority of treated patients after one year of treatment. This remarkable success for a monotherapy has made chronic hepatitis B a treatable disease with major health benefits for the treated individuals (Liaw, 2013a). However, analyses of intrahepatic viral DNA indicate that the antiviral therapy is not efficient in reducing HBV core DNA and in particular, the nuclear cccDNA (Cheng et al., 2011; Werle-Lapostolle et al., 2004; Wong et al., 2013). As a consequence, chronic HBV infections are rarely cleared in patients receiving nucleos(t)ide analog therapies and viral replication resurges after treatment cessation, probably due to the presence of cccDNA. While the intricate stability of cccDNA certainly plays a fundamental role in its persistence, the existence of residual intrahepatic viral DNA replicative intermediates under long-term nucleos(t)ide analog therapies (Cheng et al., 2011; Werle-Lapostolle et al., 2004; Wong et al., 2013) and the sequential emergence of nuceos(t)ide analog-resistant mutations in treated patients (Zoulim et al., 2009) strongly suggest that the ongoing replication of HBV DNA, albeit at a very low level, does occur in vivo, and can potentially help replenish the cccDNA pool (Chang et al., 2014; Guo and Guo, 2015).
Interestingly, our work reported herein provides strong evidence suggesting that unlike in the in vitro priming reactions, all the tested HBV DNA polymerase inhibitors failed to completely arrest DHBV or HBV DNA replication at the initial steps in hepatocyte-derived cell lines supporting hepadnaviral replication (Figs. 1 to 4). Instead, the viral DNA replication is more efficiently arrested after the synthesis of approximately 200 to 400 nt of minus-strand DNA. Considering the fact that only a single copy of viral P protein is encapsidated into a nucleocapsid (Zhang and Tavis, 2006), the synthesis of viral minus- and plus-strand DNA is catalyzed by the P protein that always anchors its N-terminal TP domain to the 5′-terminus of minus strand DNA. Such a unique structural relationship between P protein and its catalytic product may require the protein to adopt different conformation during different stages of viral DNA synthesis to overcome the physical constraints in the interactions with its RNA and DNA templates. It is, therefore, reasonable to speculate that changes of the P protein structure may render the DNA polymerase less sensitive to the inhibitors during synthesis of certain regions of viral DNA. Apparently, our results indicate that priming and synthesis of the first few hundred nucleotides of the minus strand DNA is less sensitive to the currently available viral DNA polymerase inhibitors.
One of the general consensus in hepatitis B pathogenesis is that HBV is a non-cytopathic virus and the outcome of HBV infection and severity of associated liver diseases are determined by the nature and strength of the host immune response against the virus (Bertoletti and Ferrari, 2011; Chisari and Ferrari, 1995; Guidotti and Chisari, 2006). Serum alanine aminotransferase (ALT) is a highly sensitive indicator of hepatocellular damage. Elevated levels of serum ALT in HBV-infected patients indicate antiviral immune response-induced hepatocyte injury and are generally correlated well with the severity of hepatic necrotic inflammation. Interestingly, ALT levels gradually decrease during nucleos(t)ide analog therapies and became normalized in approximately 40% and 60% of the patients at 12 and 24 week of treatment, respectively (Liaw et al., 2000; Yoo et al., 2007; Zeng et al., 2006). During this initial period of antiviral therapy, the most significant virologic changes in the livers of treated patients are reduction of viral DNA, but not viral antigens (Liaw et al., 2000). It is therefore possible that viral DNA may play a role in the immune-induced liver damage (Chang et al., 2012; Chang et al., 2014). A likely scenario is that HBV DNA may stimulate cytokine responses via activation of DNA recognition receptors, such as TLR9 and cGAS, in hepatocytes or most likely in intrahepatic macrophages and/or dendritic cells upon their up-taking of damaged HBV-infected hepatocytes (Krug et al., 2004; Sun et al., 2013). The viral DNA induced innate immune responses enhance liver inflammation and hepatocyte damages induced by adaptive antiviral immune response.
Accordingly, the failure to completely suppress intrahepatic HBV DNA replication by the nucleos(t)ide analog therapies strongly argues the need for the development of novel antiviral agents, particularly the inhibitors of viral nucleocapsid assembly and pgRNA encapsidation or novel DNA polymerase inhibitors that preclude or more efficiently inhibit the priming and early steps of viral DNA synthesis (Clark and Hu, 2015; Zlotnick et al., 2015). Those novel antiviral agents can be used as monotherapy or in combination with the currently available DNA polymerase inhibitors to more potently suppress viral DNA synthesis. It can be speculated that such therapeutic regimes should more efficiently reduce liver damage, normalize the levels of serum ALT and achieve a functional cure of chronic hepatitis B (Block et al., 2013; Liang et al., 2015).
Highlights.
Novel viral RNA species shorter than full-length pgRNA accumulate in the cells treated with HBV DNA polymerase inhibitors
The novel RNA species are devoid of poly-A tail and exist in nucleocapsids
The novel RNA species are products of RNase H digestion of viral pgRNA during minus-strand DNA synthesis
HBV DNA polymerase inhibitors do not efficiently arrest minus strand viral DNA synthesis at the early stage in hepatocytes
Acknowledgments
We thank Mohit Sehgal and Julia Ma for their critical reading of the manuscript. This work was supported in part by the NIH grant (R01 AI113267) and the Hepatitis B Foundation through an appropriation from the Commonwealth of Pennsylvania. Dr. Pinghu Zhang is supported by Jiangsu Overseas research& training program for University Prominent Young & Middle-aged Teachers and Presidents, and Program for New Century Excellent Talents in University (Program No. NCET-12-0975), and the Fundamental Research Funds for the Central Universities (Program No. ZJ11206).
References
- Bartenschlager R, Schaller H. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked protein) believed to prime reverse transcription. Embo J. 1988;7:4185–4192. doi: 10.1002/j.1460-2075.1988.tb03315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartenschlager R, Schaller H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. Embo J. 1992;11:3413–3420. doi: 10.1002/j.1460-2075.1992.tb05420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut. 2011 doi: 10.1136/gutjnl-2011-301073. [DOI] [PubMed] [Google Scholar]
- Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res. 2013;98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. Journal of virology. 2013;87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Block TM, Guo JT. The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antiviral Res. 2012;96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Chang J, Guo F, Zhao X, Guo JT. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta pharmaceutica Sinica B. 2014;4:248–257. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Marion PL. Amino acids essential for RNase H activity of hepadnaviruses are also required for efficient elongation of minus-strand viral DNA. Journal of virology. 1996;70:6151–6156. doi: 10.1128/jvi.70.9.6151-6156.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Robinson WS, Marion PL. Selected mutations of the duck hepatitis B virus P gene RNase H domain affect both RNA packaging and priming of minus-strand DNA synthesis. Journal of Virology. 1994;68:5232–5238. doi: 10.1128/jvi.68.8.5232-5238.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PN, Liu WC, Tsai HW, Wu IC, Chang TT, Young KC. Association of intrahepatic cccDNA reduction with the improvement of liver histology in chronic hepatitis B patients receiving oral antiviral agents. J Med Virol. 2011;83:602–607. doi: 10.1002/jmv.22014. [DOI] [PubMed] [Google Scholar]
- Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- Clark DN, Hu J. Hepatitis B virus reverse transcriptase - Target of current antiviral therapy and future drug development. Antiviral Res. 2015;123:132–137. doi: 10.1016/j.antiviral.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condreay LD, Aldrich CE, Coates L, Mason WS, Wu TT. Efficient duck hepatitis B virus production by an avian liver tumor cell line. Journal of Virology. 1990;64:3249–3258. doi: 10.1128/jvi.64.7.3249-3258.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- Feld JJ, Colledge D, Sozzi V, Edwards R, Littlejohn M, Locarnini SA. The phenylpropenamide derivative AT-130 blocks HBV replication at the level of viral RNA packaging. Antiviral Res. 2007;76:168–177. doi: 10.1016/j.antiviral.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol. 2007;81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Guo H. Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res. 2015;122:91–100. doi: 10.1016/j.antiviral.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol. 2003;77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Murakami E, Delaney W, Furman P, Hu J. Noncompetitive inhibition of hepatitis B virus reverse transcriptase protein priming and DNA synthesis by the nucleoside analog clevudine. Antimicrobial agents and chemotherapy. 2013;57:4181–4189. doi: 10.1128/AAC.00599-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-L- 2′,3′-dideoxy-3′-thiacytidine [published erratum appears in Antimicrob Agents Chemother 1999 Mar;43(3):726] Antimicrob Agents Chemother. 1998;42:3179–3186. doi: 10.1128/aac.42.12.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715–1720. doi: 10.1128/aac.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS. Present and future therapies of hepatitis B: From discovery to cure. Hepatology. 2015;62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw YF. Impact of therapy on the long-term outcome of chronic hepatitis B. Clinics in liver disease. 2013a;17:413–423. doi: 10.1016/j.cld.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Liaw YF. Impact of therapy on the outcome of chronic hepatitis B. Liver international: official journal of the International Association for the Study of the Liver. 2013b;33(Suppl 1):111–115. doi: 10.1111/liv.12057. [DOI] [PubMed] [Google Scholar]
- Liaw YF, Leung NW, Chang TT, Guan R, Tai DI, Ng KY, Chien RN, Dent J, Roman L, Edmundson S, Lai CL. Effects of extended lamivudine therapy in Asian patients with chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. Gastroenterology. 2000;119:172–180. doi: 10.1053/gast.2000.8559. [DOI] [PubMed] [Google Scholar]
- Lien J, Petcu DJ, Aldrich CE, Mason WS. Initiation and termination of duck hepatitis B virus DNA synthesis during virus maturation. J Virol. 1987;61:3832–3840. doi: 10.1128/jvi.61.12.3832-3840.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien JM, Aldrich CE, Mason WS. Evidence that a capped oligoribonucleotide is the primer for duck hepatitis B virus plus-strand DNA synthesis. Journal of Virology. 1986;57:229–236. doi: 10.1128/jvi.57.1.229-236.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Campagna M, Qi Y, Zhao X, Guo F, Xu C, Li S, Li W, Block TM, Chang J, Guo JT. Alpha-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog. 2013;9:e1003613. doi: 10.1371/journal.ppat.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez-Arias L, Alvarez M, Pacheco B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: mechanism of action and resistance. Current opinion in virology. 2014;8:1–9. doi: 10.1016/j.coviro.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Miller RH, Robinson WS. Common evolutionary origin of hepatitis B virus and retroviruses. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:2531–2535. doi: 10.1073/pnas.83.8.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, Watanabe T, Iijima S, Sakurai Y, Watashi K, Tsutsumi S, Sato Y, Akita H, Wakita T, Rice CM, Harashima H, Kohara M, Tanaka Y, Takaoka A. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42:123–132. doi: 10.1016/j.immuni.2014.12.016. [DOI] [PubMed] [Google Scholar]
- Schultz U, Summers J, Staeheli P, Chisari FV. Elimination of duck hepatitis B virus RNA-containing capsids in duck interferon-alpha-treated hepatocytes. J Virol. 1999;73:5459–5465. doi: 10.1128/jvi.73.7.5459-5465.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A. 2005;102:8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray SJ, Zlotnick A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J Mol Recognit. 2006;19:542–548. doi: 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- Summers J, Mason WS. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog. 2013;9:e1003125. doi: 10.1371/journal.ppat.1003125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavis JE, Perri S, Ganem D. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. Journal of Virology. 1994;68:3536–3543. doi: 10.1128/jvi.68.6.3536-3543.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GH, Seeger C. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell. 1992;71:663–670. doi: 10.1016/0092-8674(92)90599-8. [DOI] [PubMed] [Google Scholar]
- Wang GH, Seeger C. Novel mechanism for reverse transcription in hepatitis B viruses. J Virol. 1993;67:6507–6512. doi: 10.1128/jvi.67.11.6507-6512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle-Lapostolle B, Bowden S, Locarnini S, Wursthorn K, Petersen J, Lau G, Trepo C, Marcellin P, Goodman Z, Delaney WET, Xiong S, Brosgart CL, Chen SS, Gibbs CS, Zoulim F. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Wong DK, Seto WK, Fung J, Ip P, Huang FY, Lai CL, Yuen MF. Reduction of hepatitis B surface antigen and covalently closed circular DNA by nucleos(t)ide analogues of different potency. Clin Gastroenterol Hepatol. 2013;11:1004–1010 e1001. doi: 10.1016/j.cgh.2013.01.026. [DOI] [PubMed] [Google Scholar]
- Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol. 2010;84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BC, Kim JH, Chung YH, Lee KS, Paik SW, Ryu SH, Han BH, Han JY, Byun KS, Cho M, Lee HJ, Kim TH, Cho SH, Park JW, Um SH, Hwang SG, Kim YS, Lee YJ, Chon CY, Kim BI, Lee YS, Yang JM, Kim HC, Hwang JS, Choi SK, Kweon YO, Jeong SH, Lee MS, Choi JY, Kim DG, Kim YS, Lee HY, Yoo K, Yoo HW, Lee HS. Twenty-four-week clevudine therapy showed potent and sustained antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology. 2007;45:1172–1178. doi: 10.1002/hep.21629. [DOI] [PubMed] [Google Scholar]
- Zeng M, Mao Y, Yao G, Wang H, Hou J, Wang Y, Ji BN, Chang CN, Barker KF. A double-blind randomized trial of adefovir dipivoxil in Chinese subjects with HBeAg-positive chronic hepatitis B. Hepatology. 2006;44:108–116. doi: 10.1002/hep.21225. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Tavis JE. The duck hepatitis B virus reverse transcriptase functions as a full-length monomer. The Journal of biological chemistry. 2006;281:35794–35801. doi: 10.1074/jbc.M608031200. [DOI] [PubMed] [Google Scholar]
- Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res. 2015;121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F, Durantel D, Deny P. Management and prevention of drug resistance in chronic hepatitis B. Liver Int. 2009;29(Suppl 1):108–115. doi: 10.1111/j.1478-3231.2008.01939.x. [DOI] [PubMed] [Google Scholar]
- Zoulim F, Seeger C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J Virol. 1994;68:6–13. doi: 10.1128/jvi.68.1.6-13.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]