

ABSTRACT
The fungi that reside in the human lungs represent an understudied, but medically relevant comm-unity. From the few studies published on the lung mycobiome, we find that there are fungi in both the healthy and diseased respiratory tract, that these fungi vary widely between individuals, and that there is a trend toward lower fungal diversity among individuals with disease. This review discusses the few studies of the lung mycobiome and details the challenges that accompany lung mycobiome studies. These challenges include sample collection and processing, sequence amplification and processing, and a history of multiple names for species. Some challenges may never be solved, but others can be solved with more data and additional studies of the lung mycobiome.
KEYWORDS: fungi, lung, mycobiome, next-generation sequencing, respiratory tract
In less than a decade, we have progressed from the belief that healthy lungs are a sterile environment to studying inter-kingdom interactions between microbial residents of the lung. In part due to the debate about the sterility of the lungs, next-generation sequencing (NGS)-based studies of the lung microbiome have lagged behind those of the gut microbiome, with the first studies of the lung microbiome being published in 2010 and 2011.1–3 These early NGS studies, and many studies since, focused exclusively on the bacteria present in the lungs under health and disease. However, the microbial community that inhabits the lungs also contains viruses, fungi, and other eukaryotes. For comprehensive reviews of other members of the lung microbiome, see ref.4 for bacteria and ref.5 for viruses. Here we focus on the fungal component of the community, also known as the lung mycobiome. We outline what we know about the lung mycobiome, the challenges to studying the lung mycobiome (or why there are so few lung mycobiome studies), and conclude with future directions in the field.
Why is the lung mycobiome important?
In addition to causing clinical fungal infections, the lung mycobiome may have profound inflammatory effects that can cause or worsen lung disease. Fungi contain pathogen-associated molecular patterns (PAMPS) such as glucans, chitin, and mannans present in the fungal cell wall.6,7 These PAMPs are recognized by pattern recognition receptors (PRRs) that then activate immune cells leading to inflammation (Fig. 1). Activation of macrophages, T cells, and B cells leads to cytokine release and immune activation. Both the adaptive and innate immune responses are triggered by fungi, and the respiratory epithelium plays a key role in the response to fungi. Fungi have been linked to such chronic lung diseases as asthma and COPD.8,9 Given the ubiquity of fungi in the environment, the potential respiratory exposure to fungi, and the ability of fungi to trigger inflammation, the mycobiome may play a key role in shaping the respiratory immune response and contribute to lung damage.
Figure 1.
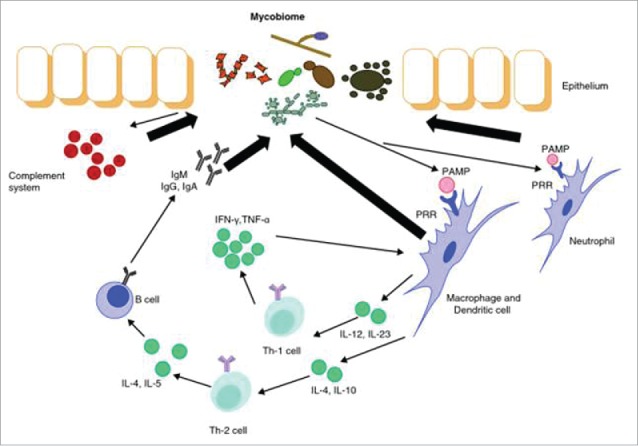
Interaction between the mycobiome and the immune system. When pattern recognition receptors (PRRs) recognize the pathogen-associated molecular patterns (PAMPs) on fungal cell walls macrophages, T cells, and B cells are activated. The fate of the activated T cells is determined by the cytokines were stimulated. INF, interferon; IL, interleukin; TNF, tumor necrosis factor. Figure from ref.37.
What do we know about the lung mycobiome?
There have been fewer than 10 NGS lung mycobiome papers published. Despite the low number of published NGS studies, several themes emerge from the literature: (1) fungi are present in the human respiratory tract, even during health; (2) the fungi present in the respiratory tract are highly variable between individuals; and (3) many diseases are accompanied by decreased diversity of fungi in the lungs.
Fungi found in the human respiratory tract cover a range of phylogenies, but are predominantly from the Dikarya sub-kingdom which is composed of the phyla Ascomycota and Basidiomycota. In fact, the most common taxa identified in healthy lung samples were the family Davidiellaceae, and the genera Cladosporium, Eurotium, Penicillium, and Aspergillus.10 Other genera found in healthy individuals include Candida, Neosartorya, Malassezia, Hyphodontia, Kluyveromyces, and Pneumocystis11 (Fig. 2). These eleven taxa cover the range of fungal growth patterns from filamentous, to yeast and yeast-like.
Figure 2.

Distribution of fungal phyla in the sputum of healthy individuals8. The inner ring displays the class of fungi while the outer ring displays the phyla. Any class or phyla that represented over 10% of the reads is labeled with its percentage of reads and classes or phyla less than 0.1% are not represented.
The fungi present in the respiratory tract are also highly variable between individuals. Even in patients with the same disease, different patients have been shown to harbor distinct fungal communities.12 In our experience, the number of “private species,” those present in only one individual, can be greater than the number of species shared across samples. Whether this difference is due to mis-identification of the fungi (perhaps due to a sequencing error) or a patient's unique environmental exposures has yet to be examined. It has been proposed that the macromycetes, or macroscopic fungi commonly known as mushrooms, observed in a subset of samples, represent the outdoor environment that a patient is exposed to as they often contain wood-inhabiting fungi and cereal grain pathogens.11 Even the amount of fungal diversity in the lungs is highly variable between individuals. Compared to bacterial diversity in the lungs, average fungal diversity in the same samples is lower,13 but has a higher coefficient of variation, or ratio of standard deviation to the mean. We see in our own data, a subset of 35 BAL samples from a study of the lung mycobiome in HIV-infected and uninfected individuals for which we have both 16S and ITS sequence data.,9 a coefficient of variation for bacterial diversity, as measured by the Shannon diversity index, of 22.9% and a coefficient of variation for fungal diversity of 73.9%. Because bacterial diversity is relative stable, other factors, including patient health and environmental exposures, appear to have a greater impact on the diversity of the fungi.
Only a limited number of diseases have been examined for their impact on, or association with the lung mycobiome. Most diseases that have been studied, including CF, asthma, lung transplant, and COPD, have been associated with decreases in fungal diversity.10,11,9,14 Across these diseases, lower fungal diversity is correlated with lower respiratory function. The reduced diversity may be caused by an overgrowth of a single fungal species, or by the loss of rare species that comes with a reduction in overall fungal abundance.
CF has received the most attention with studies that range from correlating community characteristics with patient health indicators to comparing NGS and sequencing detection to community stability over time. Delhaes et al examined sputum of 4 CF patients, each sampled twice, and found that both bacterial and fungal community richness was positively correlated with indicators of health and lung function,15 i.e. more fungal species were seen in the patients with the lowest disease severity scores, highest body-mass indices, highest forced vital capacity, and highest forced expiratory volume in 1 second. Harrison et al found that over 82% of the species identified by sequencing were not found by culture-based methods which detected fungi in only 27% of the sputum samples from 55 CF patients compared to a 90% detection rate by sequencing.14 Willger and colleagues sought to compare sputum from 6 CF patients before and after antimicrobial therapy and found that the fungal communities were relatively stable.16 Similarly, a study of 89 sputum samples from 28 CF patients showed that the fungal communities were stable through clinical exacerbation and treatment.12 This study combined NGS of the mycobiome with phenotypic and genotypic analysis of Candida isolates from the samples to identify mutations leading to the filamentous phenotype in the presence of filamentation repressive cues from the bacteria Pseudomonas aeruginosa.17 It is the filamentous phenotype that is considered pathogenic and evading the repressive signals from other members of the microbiome could lead to Candida infection.
Lung transplantation could impact the mycobiome due to the immunosuppression and antibiotics received by recipients as well as structural changes in the lung. In general, lung transplant recipients have reduced fungal richness and increased fungal abundance compared to healthy controls. For example, Charlson et al found that combined bacterial and fungal community richness was reduced in bronchoalveolar lavage (BAL) from 21 lung transplant patients compared to healthy controls and richness was lowest in patients who had a transplant due to CF.10 All transplant patients were receiving antibiotics in addition to immunosuppression at the time of sampling, making it difficult to attribute causality in these changes. In the 4 patients with high fungal amplification from BAL, the dominant species (Candida albicans in 3 samples and Aspergillus fumigatus in one sample) was also found by culture methods, which were only able to identify 4 species: C. albicans, A. fumigatus, Aspergillus flavus, and Paecilomyces lilacinus (also known as Paecilomyces lilacinus). Expanding this dataset to include a total of 149 BAL samples from healthy subjects, HIV-infected subjects, subjects with mixed lung disease, and lung transplant recipients, Bittinger et al showed that fungal abundance increases from healthy subjects to lung transplant recipients with HIV-infected subjects and subjects with mixed lung disease falling in the middle.13 To ensure that they were counting species truly present in the lungs, they used DNA quantification to filter out any species that were likely to have come from contamination before calculating species abundances.
Asthma, COPD, and pneumonia have been less well-studied, with only a single paper published each examining shifts in lung mycobiome communities. For asthma, a case-control study to compare induced sputum samples of 30 subjects with asthma to that of 13 control subjects found 90 species to be more abundant in asthma and 46 species to be more abundant in the controls.8 Species with more than a 5% increase in abundance between the asthma and control sample pools were Psathyrella candolleana, Malassezia pachydermatis, and Termitomyces clypeatus, none of which were seen in the control sample pool. Species with more than a 5% decrease in abundance between the asthma and control sample pools were Eremothecium sinecaudum, Systenostrema alba, Cladosporium clasdosporioides, and Vanderwaltozyma polyspora. The only paper on COPD first compared HIV-infected to HIV-uninfected individuals and then compared HIV-infected individuals with COPD to HIV-infected with normal lung function.9 The authors used an overlap of multiple methods to identify overrepresented species in the BAL of 32 HIV-infected individuals, 10 with and 22 without COPD, and 24 HIV-uninfected controls.9 They found Pneumocystis jirovecii to be the most distinguishing species as it was overrepresented in both HIV and COPD. Finally, in the only published study on pneumonia, which is the largest lung mycobiome study to date, Krause et al compared BAL from 87 healthy controls, 18 patients with extrapulmonary infection on antibiotics, 8 intensive care unit patients without antibiotics, 23 intensive care unit patients with extrapulmonay infection on antibiotics, 34 intensive care unit patients with pneumonia on antibiotics, and 32 patients with candidemia.18 They focused on Candida and found that intensive care unit admission, but not antibiotic therapy, shifted the lung mycobiome to be dominated by Candida. Even this recent study still used culture-based fungal identification as the gold-standard for fungal identification, as this is standard practice in a clinical setting.
Why are there so few lung mycobiome studies?
In addition to the relative newness of the field, studies of the lung mycobiome may be limited because of the numerous challenges that exist at every step. The challenges begin with sampling the lung and continue through sample processing. These are followed by tough choices with regards to amplification and sequencing and more challenges to process the sequencing data. Finally, the historical system of naming fungi that resulted in multiple names for a single species has created difficulties now that NGS is used to define and identify species. Because many of these challenges are applicable to all NGS mycobiome studies, we have included only a brief overview of each one and its relevance to the lung mycobiome.
The human lungs are difficult to access. The two most common means of sampling the lungs are induced sputum (IS) and BAL. Both methods run the risk of contamination from the upper respiratory tract. IS is obtained by having subjects cough after inhalation of hypertonic sputum, potentially introducing mouth microbes during collection, and the bronchoscope may introduce upper respiratory microbes to the lungs during passage through the nose or mouth. However, it has been shown that both IS and BAL mycobiomes are distinct from the oral mycobiome.9 Cui et al showed that there are difference in the communities of IS and BAL, likely because the 2 methods sample the lungs differently. IS samples from a greater anatomic region of the lung, while BAL samples from a subset of the alveoli. Different environmental conditions existing in different portions of the lungs, or microenvironments, will be indistinguishable in an IS sample, but may be missed entirely by a BAL. The choice of sampling method should be selected based on the question under investigation, or, in the case of pre-existing samples, the limits of the sampling method should be addressed to the extent possible.
Once a sample is obtained, DNA needs to be extracted. As with any NGS-based study of fungi, one of the first steps is to break open the cell wall. The fungal cell wall is composed of a combination of glucans and chitin, whose proportions vary by fungal growth patterns.19 The varying composition of the fungal cell wall leads to a range of tensile strengths, and there are a number of methods to break open the cell wall that vary in harshness. For the purposes of extracting DNA from both yeasts and filamentous fungi, mechanical disintegration has proven most effective20; however, this method runs the risk of shearing the DNA and therefore must be carefully calibrated for the given sample composition.
The harsh mechanical treatment to break open the fungal cell walls also creates a challenge by releasing DNA from other cells present in the lung sample, both bacterial or human. The extra DNA released from non-fungal cells, along with any DNA found in the laboratory reagents (a recent study attempted to characterize the bacteria found in DNA extraction kits,21 but no equivalent study has been performed for fungi), necessitates careful primer design for amplification. Common targets for amplification include the gene encoding the 18S ribosomal RNA (rRNA) and the internal transcribed spacer (ITS) region(s) located between the 18S and 26S rRNA genes,22 each with their own benefits and drawbacks. The 18S rRNA gene is conserved across all eukaryotes, so targeting this gene for amplification and sequencing of fungi will include non-fungal microbial eukaryotes including protozoa and helminths, both of which are known to parasitize human airways.23 Because the 18S rRNA gene is conserved across all eukaryotes, amplifying this region of the genome can also amplify any human DNA present in the sample, depending on the specificity of the primers. Due to the low biomass of microbes in the lung, the amount of human DNA in the sample prior to targeted amplification is bound to be higher than the amount of fungal DNA. In contrast, the ITS region is more diverse across eukaryotes and primers have been designed specifically for the amplification of fungal DNA,24 to the exclusion of all other eukaryotes. Some of these primers are narrowly targeted such that they introduce bias toward particular fungal phyla, another issue worthy of careful consideration. The diversity of the ITS region and the specificity of the primers combine to allow a greater depth of taxonomic assignment, often down to the species level. It is this advantage that has led the ITS region to be the “official primary barcoding marker” for fungi.24 However, because it is a non-coding region, ITS sequences cannot be used to determine phylogenetic relationships between unidentified fungi.
One of the greatest of the challenges following sequencing is a lack of data, specifically a lack of reference genomes. The UNITE database of fungal ITS sequences represents the largest collection of fungal sequences and as of version 7, contains more than 64,500 “species hypotheses” at the 1% similarity threshold, where a species hypothesis is any group of sequences that are no more distant than the similarity threshold.25 Compared to the over 203,000 bacterial species hypotheses at the 1% similarity threshold in the May, 2013 release of the GreenGenes 16S rRNA gene database,26 the number of fungal reference species seems small. The sequences within the UNITE database are heavily biased (87% of species hypotheses) toward the Dikarya sub-kingdom25 (Fig. 3). While this bias may accurately represent the distribution of fungal species, or may simply stem from UNITE's history as a database for plant root fungi,27 it certainly explains why the majority of species identified in the lungs belong to this sub-kingdom.
Figure 3.

Distribution of fungal phyla in the UNITE database27. The chart shows the breakdown of phyla of the 64,500 “species hypotheses” at the 1% similarity threshold found in the UNITE ITS database. Phyla that represent over 10% of the species hypotheses are labeled with its percentage of species hypotheses.
Even after the sequences are identified, there are still challenges to be overcome. Despite years of expert mycologists pushing for each fungus to have a single species name, many fungi still have one name for their sexual reproductive stage (or teleomorph) and one name for their asexual reproductive stage (or anamorph).28,29 The problem with this dual naming system in the NGS era is 2-fold. First, it can complicate a search for knowledge prior to the NGS era. Many older studies reference only one name and it can be unclear if the results apply to the opposite morph. There is no way to identify which morph is present in a sample based on its DNA. It can also be that the higher order taxonomic assignments, such as family and order, of the 2 morphs are different, leading to phylogenetic confusion about the placement of the species as a whole. Uncertain phylogenetic placement and phylogenetic restructuring result in taxonomic hierarchies that include incertae sedis (Latin for “of uncertain placement”), as seen in the taxonomies of members of the former phyla Zygomycota.30 The second problem with a dual naming system is that sample sequences may have 2 or more identical matches when a database has reference sequences for both the teleomorph and anamorph causing ambiguous assignments. Curated databases such as Mycobank31 can aid in the reduction of duplicate reference sequences, but similar curation is not readily available for pre-NGS knowledge.
Where do we go from here?
Many of the challenges to studying the lung mycobiome are unavoidable. There is likely never going to be easy access to the human lung that avoids the upper respiratory tract, and the microbes in the lung will always be low in biomass. However, improvement is possible in primer design, reference databases, and analytic methods. Going forward, many of the advances made in the study of the lung bacteria will aid in the study of the lung mycobiome. Once the sequencing data is collapsed into a “biom” file or taxa table, a table that displays the abundance of each taxonomic group for every sample, it makes little difference if the taxa are bacterial or fungal. All of the statistical methods to handle the abnormal distributions32 and complex study designs associated with bacterial studies can be used on fungal studies with little or no modifications. Similarly, as bacterial studies shift from cross-sectional to longitudinal, so too should fungal studies. There have already been studies into the daily changes in bacterial communities that occur during CF and its exacerbations33 but fungi were not examined. The tools, including sequencing capacity, that are developed to handle daily sampling of the bacteria can also be put to use to analyze the fungi present during the same time period.
When more studies include both bacterial and fungal amplicon sequences, we can begin to look at cross-kingdom interactions. Interactions between bacteria and fungi are important among oral microbiota34 and identified as an emerging field across biology,35 so they will no doubt be important to the study of the lung microbiota. Looking farther into the future, as amplicon sequencing gives way to whole metagenome and whole metatranscriptome sequencing, these delineations between bacterial and fungal communities will fall away. Both kingdoms will be sequenced simultaneously and their members' abundances and transcriptional activity, relative to each other, will be apparent.
Another avenue for future investigation will be the mechanisms of interactions between the mycobiome and the host. As a part of the mucosal immune system, the lungs, and the microbes within, play an important role in human health and disease. The impacts of inflammation on the development of many lung diseases represent an area of active investigation, one in which the contribution from the lung mycobiome could prove crucial to understanding.
In both the short- and long-term, the critical need for the lung mycobiome is more data, in the forms of reference sequences and additional lung mycobiome studies. Adding sequences to reference databases by sequencing more fungi will help in identifying species that are currently unclassifiable. These sequences can come from culturing some of the estimated 99% of the world's fungi that have yet to be reliably grown in the lab,36 or from assembling genomes present in deeply sequenced metagenomes. The latter makes it possible to obtain sequences from unculturable fungi without the time and manpower required to optimize the culturing conditions of newly cultured organisms. The other and perhaps more important way to contribute to the knowledge of the lung mycobiome is to perform more lung mycobiome studies. Additional lung mycobiome studies will provide more information about the changes in the fungi present under health and disease conditions and will help to explain the role of fungi in the respiratory immune response.
Abbreviations
- BAL
bronchoalveolar lavage
- CF
cystic fibrosis
- COPD
chronic obstructive pulmonary disease
- HIV
human immunodeficiency virus
- IFN
interferon
- IL
interleukin
- IS
induced sputum
- ITS
internal transcribed spacer
- NGS
next generation sequencing
- PAMP
pathogen-associated molecular patterns
- PRR
pattern recognition receptors
- rRNA
ribosomal RNA
- TNF
tumor necrosis factor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
A.M. was supported by K24 HL123342 from the NHLBI for mentoring in studies of the lung microbiome.
References
- [1].Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, Davies J, Ervine A, Poulter L, Pachter L, et al.. Disordered Microbial Communities in Asthmatic Airways. PLoS One [Internet] 2010. [cited 2016June30]; 5:e8578. Available from: http://dx.plos.org/10.1371/journal.pone.0008578; PMID:20052417; http://dx.doi.org/ 10.1371/journal.pone.0008578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, Young VB, Toews GB, Curtis JL, Sundaram B, et al.. Analysis of the Lung Microbiome in the “Healthy” Smoker and in COPD. PLoS One [Internet] 2011. [cited 2016June30]; 6:e16384. Available from: dx.plos.org/10.1371/journal.pone.0016384; PMID:21364979; http://dx.doi.org/ 10.1371/journal.pone.0016384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Huang YJ, Nelson CE, Brodie EL, DeSantis TZ, Baek MS, Liu J, Woyke T, Allgaier M, Bristow J, Wiener-Kronish JP, et al.. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J Allergy Clin Immunol 2011; 127:372–81.e3; PMID:21194740; http://dx.doi.org/ 10.1016/j.jaci.2010.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The Microbiome and the Respiratory Tract. Annu Rev Physiol [Internet] 2016. [cited 2016June15]; 78:481–504. Available from: http://www.annualreviews.org/doi/10.1146/annurev-physiol-021115-105238; PMID:26527-186; http://dx.doi.org/ 10.1146/annurev-physiol-021115-105238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zou S, Caler L, Colombini-Hatch S, Glynn S, Srinivas P. Research on the human virome: where are we and what is next. Microbiome [Internet] 2016. [cited 2016June30]; 4:32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27341799; PMID:27341799; http://dx.doi.org/ 10.1186/s40168-016-0177-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Romani L. Immunity to fungal infections. Nat Rev Immunol [Internet] 2011. [cited 2016June15]; 11:275–88. Available from: http://www.nature.com/doifinder/10.1038/nri2939; PMID:21394104; http://dx.doi.org/ 10.1038/nri2939 [DOI] [PubMed] [Google Scholar]
- [7].Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol [Internet] 2015. [cited 2016June15]; 16:343–53. Available from: http://www.nature.com/doifinder/10.1038/ni.3123; PMID:25789684; http://dx.doi.org/ 10.1038/ni.3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van Woerden HC, Gregory C, Brown R, Marchesi JR, Hoogendoorn B, Matthews IP, Hasleton P, Bartsch P, Collignon A, Weber G, et al.. Differences in fungi present in induced sputum samples from asthma patients and non-atopic controls: a community based case control study. BMC Infect Dis [Internet] 2013. [cited 2016June29]; 13:69. Available from: http://bmcinfectdis.biomedcentral.com/articles/10.1186/1471-2334-13-69; PMID:23384395; http://dx.doi.org/ 10.1186/1471-2334-13-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cui L, Lucht L, Tipton L, Rogers MB, Fitch A, Kessinger C, Camp D, Kingsley L, Leo N, Greenblatt RM, et al.. Topographic Diversity of the Respiratory Tract Mycobiome and Alteration in HIV and Lung Disease. Am J Respir Crit Care Med [Internet] 2015; 191:932–42. Available from: http://dx.doi.org/10.1164/rccm.201409-1583OC; PMID:25603113; http://dx.doi.org/ 10.1164/rccm.201409-1583OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Charlson ES, Diamond JM, Bittinger K, Fitzgerald AS, Yadav A, Haas AR, Bushman FD, Collman RG. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med [Internet] 2012. [cited 2016June29]; 186:536–45. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22798321; PMID:22798321; http://dx.doi.org/ 10.1164/rccm.201204-0693OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E, et al.. The Airway Microbiota in Cystic Fibrosis: A Complex Fungal and Bacterial Community—Implications for Therapeutic Management. PLOS One [Internet] 2012. [cited 2016June29]; 7:e36313. Available from: http://dx.plos.org/10.1371/journal.pone.0036313; PMID:22558432; http://dx.doi.org/ 10.1371/journal.pone.0036313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kim SH, Clark ST, Surendra A, Copeland JK, Wang PW, Ammar R, Collins C, Tullis DE, Nislow C, Hwang DM, et al.. Global Analysis of the Fungal Microbiome in Cystic Fibrosis Patients Reveals Loss of Function of the Transcriptional Repressor Nrg1 as a Mechanism of Pathogen Adaptation. PLOS Pathog [Internet] 2015. [cited 2016June26]; 11:e1005308. Available from: http://dx.plos.org/10.1371/journal.ppat.1005308; PMID:26588216; http://dx.doi.org/ 10.1371/journal.ppat.1005308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bittinger K, Charlson ES, Loy E, Shirley DJ, Haas AR, Laughlin A, Yi Y, Wu GD, Lewis JD, Frank I, et al.. Improved characterization of medically relevant fungi in the human respiratory tract using next-generation sequencing. Genome Biol [Internet] 2014. [cited 2016June27]; 15:487. Available from: http://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0487-y; PMID:25344286; http://dx.doi.org/ 10.1186/s13059-014-0487-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Harrison M, Twomey K, McCarthy Y, O'Connell O, Febrer M, Alston M, Ryan R, Plant B. LSC 2013 abstract - The role of second-generation sequencing to characterize the fungal microbiota in the adult cystic fibrosis airway, and its correlation with standard culture-based methods and clinical phenotype. Eur Respir J 2013; 42:OP02. [Google Scholar]
- [15].Delhaes L, Monchy S, Fréalle E, Hubans C, Salleron J, Leroy S, Prevotat A, Wallet F, Wallaert B, Dei-Cas E, et al.. The Airway Microbiota in Cystic Fibrosis: A Complex Fungal and Bacterial Community—Implications for Therapeutic Management. PLOS One [Internet] 2012. [cited 2016June30]; 7:e36313. Available from: http://dx.plos.org/10.1371/journal.pone.0036313; PMID:22558432; http://dx.doi.org/ 10.1371/journal.pone.0036313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Willger SD, Grim SL, Dolben EL, Shipunova A, Hampton TH, Morrison HG, Filkins LM, O‘Toole GA, Moulton LA, Ashare A, et al.. Characterization and quantification of the fungal microbiome in serial samples from individuals with cystic fibrosis. Microbiome [Internet] 2014. [cited 2016June29]; 2:40. Available from: http://www.microbiomejournal.com/content/2/1/40; PMID:25408892; http://dx.doi.org/ 10.1186/2049-2618-2-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim SH, Clark ST, Surendra A, Copeland JK, Wang PW, Ammar R, Collins C, Tullis DE, Nislow C, Hwang DM, et al.. Global Analysis of the Fungal Microbiome in Cystic Fibrosis Patients Reveals Loss of Function of the Transcriptional Repressor Nrg1 as a Mechanism of Pathogen Adaptation. PLOS Pathog [Internet] 2015. [cited 2016June29]; 11:e1005308. Available from: http://dx.plos.org/10.1371/journal.ppat.1005308; PMID:26588216; http://dx.doi.org/ 10.1371/journal.ppat.1005308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Krause R, Halwachs B, Thallinger GG, Klymiuk I, Gorkiewicz G, Hoenigl M, Prattes J, Valentin T, Heidrich K, Buzina W, et al.. Characterisation of Candida within the Mycobiome/Microbiome of the Lower Respiratory Tract of ICU Patients. PLOS One [Internet] 2016. [cited 2016June30]; 11:e0155033. Available from: http://dx.plos.org/10.1371/journal.pone.0155033; PMID:27206014; http://dx.doi.org/ 10.1371/journal.pone.0155033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bowman SM, Free SJ. The structure and synthesis of the fungal cell wall. BioEssays [Internet] 2006. [cited 2016June29]; 28:799–808. Available from: http://doi.wiley.com/10.1002/bies.20441; PMID:16927300; http://dx.doi.org/ 10.1002/bies.20441 [DOI] [PubMed] [Google Scholar]
- [20].Klimek-Ochab M, Brzezińska-Rodak M, Zymańczyk-Duda E, Lejczak B, Kafarski P. Comparative study of fungal cell disruption–scope and limitations of the methods. Folia Microbiol (Praha) [Internet] 2011. [cited 2016June29]; 56:469–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21901292; PMID:21901292; http://dx.doi.org/ 10.1007/s12223-011-0069-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW, et al.. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol [Internet] 2014. [cited 2016June29]; 12:87. Available from: http://bmcbiol.biomedcentral.com/articles/10.1186/s12915-014-0087-z; PMID:25387460; http://dx.doi.org/ 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dollive S, Peterfreund GL, Sherrill-Mix S, Bittinger K, Sinha R, Hoffmann C, Nabel CS, Hill DA, Artis D, Bachman MA, et al.. A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol [Internet] 2012; 13:R60–R60. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4053730/; PMID:2275-9449; http://dx.doi.org/ 10.1186/gb-2012-13-7-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Khemasuwan D, Farver CF, Mehta AC. Parasites of the Air Passages. Chest 2014; 145:883–95; PMID:24687710; http://dx.doi.org/ 10.1378/chest.13-2072 [DOI] [PubMed] [Google Scholar]
- [24].Bellemain E, Carlsen T, Brochmann C, Coissac E, Taberlet P, Kauserud H, Anderson I, Cairney J, Chase M, Fay M, et al.. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol [Internet] 2010. [cited 2016June29]; 10:189. Available from: http://bmcmicrobiol.biomedcentral.com/articles/10.1186/1471-2180-10-189; PMID:20618939; http://dx.doi.org/ 10.1186/1471-2180-10-189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AFS, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, et al.. Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol [Internet] 2013. [cited 2016June29]; 22:5271–7. Available from: http://doi.wiley.com/10.1111/mec.12481; PMID:241124-09; http://dx.doi.org/ 10.1111/mec.12481 [DOI] [PubMed] [Google Scholar]
- [26].McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J [Internet] 2012. [cited 2016June30]; 6:610–8. Available from: http://www.nature.com/doifinder/10.1038/ismej.2011.139; PMID:22134646; http://dx.doi.org/ 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kõljalg U, Larsson K-H, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, et al.. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytologist [Internet] 2005. [cited 2016June29]; 166:1063–8. Available from: http://doi.wiley.com/10.1111/j.1469-8137.2005.01376.x; PMID:15869663; http://dx.doi.org/ 10.1111/j.1469-8137.2005.01376.x [DOI] [PubMed] [Google Scholar]
- [28].de Hoog GS, Chaturvedi V, Denning DW, Dyer PS, Frisvad JC, Geiser D, Gräser Y, Guarro J, Haase G, Kwon-Chung K-J, et al.. Name Changes in Medically Important Fungi and Their Implications for Clinical Practice. J Clin Microbiol [Internet] 2015. [cited 2016June29]; 53:1056–62. Available from: http://jcm.asm.org/lookup/doi/10.1128/JCM.02016-14; PMID:25297326; http://dx.doi.org/ 10.1128/JCM.02016-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Taylor JW. One Fungus = One Name: DNA and fungal nomenclature twenty years after PCR. IMA Fungus [Internet] 2011. [cited 2016June29]; 2:113–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22679595; PMID:22679595; http://dx.doi.org/ 10.5598/imafungus.2011.02.02.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R, et al.. A higher-level phylogenetic classification of the Fungi. Mycological Res 2007; 111:509–47; PMID:17572334; http://dx.doi.org/ 10.1016/j.mycres.2007.03.004 [DOI] [PubMed] [Google Scholar]
- [31].Robert V, Vu D, Amor ABH, van de Wiele N, Brouwer C, Jabas B, Szoke S, Dridi A, Triki M, Ben Daoud S, et al.. MycoBank gearing up for new horizons. IMA Fungus [Internet] 2013. [cited 2016June30]; 4:371–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24563843; PMID:24563843; http://dx.doi.org/ 10.5598/imafungus.2013.04.02.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].White JR, Nagarajan N, Pop M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLOS Comput Biol [Internet] 2009; 5:e1000352. Available from: http://dx.doi.org/10.1371%2Fjournal.pcbi.1000352; PMID:19360128; http://dx.doi.org/ 10.1371/journal.pcbi.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Carmody LA, Zhao J, Kalikin LM, LeBar W, Simon RH, Venkataraman A, Schmidt TM, Abdo Z, Schloss PD, LiPuma JJ, et al.. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome [Internet] 2015. [cited 2016June29]; 3:12. Available from: http://www.microbiomejournal.com/content/3/1/12; PMID:25834733; http://dx.doi.org/ 10.1186/s40168-015-0074-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Diaz PI, Strausbaugh LD, Dongari-Bagtzoglou A. Fungal-bacterial interactions and their relevance to oral health: linking the clinic and the bench. Front Cell Infect Microbiol [Internet] 2014. [cited 2016June30]; 4:101. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25120959; PMID:25120959; http://dx.doi.org/ 10.3389/fcimb.2014.00101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tarkka M, Deveau AL. An Emerging Interdisciplinary Field: Fungal–Bacterial Interactions In: Druzhinina Irina S., Kubicek Chrisitan P, editors. The Mycota IV. Springer; 2016. pp. 162–78. [Google Scholar]
- [36].Hawksworth DL. The fungal dimension of biodiversity: magnitude, significance, and conservation. Mycological Res [Internet] 1991; 95:641–55. Available from: http://www.sciencedirect.com/science/article/pii/S0953756209808101; PMID:18173751817375 [Google Scholar]
- [37].Cui L, Morris A, Ghedin E. The human mycobiome in health and disease. Genome Med [Internet] 2013. [cited 2015June2]; 5:63. Available from: http://genomemedicine.com/content/5/7/63; PMID:23899327; http://dx.doi.org/ 10.1186/gm467 [DOI] [PMC free article] [PubMed] [Google Scholar]