

Abstract
Aims
We investigated the role of src family kinases (srcFK) in agonist-mediated Ca2+-sensitization in pulmonary artery and whether this involves interaction with the rho/rho-kinase pathway.
Methods and results
Intra-pulmonary arteries (IPAs) and cultured pulmonary artery smooth muscle cells (PASMC) were obtained from rat. Expression of srcFK was determined at the mRNA and protein levels. Ca2+-sensitization was induced by prostaglandin F2α (PGF2α) in α-toxin-permeabilized IPAs. Phosphorylation of the regulatory subunit of myosin phosphatase (MYPT-1) and of myosin light-chain-20 (MLC20) and translocation of rho-kinase in response to PGF2α were also determined. Nine srcFK were expressed at the mRNA level, including src, fyn, and yes, and PGF2α enhanced phosphorylation of three srcFK proteins at tyr-416. In α-toxin-permeabilized IPAs, PGF2α enhanced the Ca2+-induced contraction (pCa 6.9) approximately three-fold. This enhancement was inhibited by the srcFK blockers SU6656 and PP2 and by the rho-kinase inhibitor Y27632. Y27632, but not SU6656 or PP2, also inhibited the underlying pCa 6.9 contraction. PGF2α enhanced phosphorylation of MYPT-1 at thr-697 and thr-855 and of MLC20 at ser-19. This enhancement, but not the underlying basal phosphorylation, was inhibited by SU6656. Y27632 suppressed both basal and PGF2α-mediated phosphorylation. The effects of SU6656 and Y27632, on both contraction and MYPT-1 and MLC20 phosphorylation, were not additive. PGF2α triggered translocation of rho-kinase in PASMC, and this was inhibited by SU6656.
Conclusions
srcFK are activated by PGF2α in the rat pulmonary artery and may contribute to Ca2+-sensitization and contraction via rho-kinase translocation and phosphorylation of MYPT-1.
Keywords: Pulmonary circulation, Smooth muscle, Vasoconstriction/dilation, Tyrosine, Protein kinases, Prostaglandins, Rho-kinase
1. Introduction
Ca2+-sensitization in smooth muscle is the process by which increased myosin light-chain phosphorylation, and hence contractile force, occurs, generated in response to an external stimulus independently of changes in intracellular Ca2+ concentration ([Ca2+]i).1 In smooth muscle, this is believed to occur principally through phosphorylation of myosin-binding subunit (or MYPT-1) and subsequent inhibition of myosin light-chain phosphatase.2 MYPT-1 is phosphorylated by rho-kinase at two sites (thr-697 and thr-855 in rat), the first of which results in inhibition of phosphatase activity3 and the second results in dissociation of the phosphatase from myosin.4 Rho-kinase is activated by binding to the GTP-bound form of the small G-protein RhoA and subsequent translocation.5 The mechanisms by which agonist stimulation activates RhoA are not fully understood, but the involvement of guanine nucleotide exchange factors (RhoGEF) is a requirement.6
src family kinases (srcFK) are a group of closely related non-receptor tyrosine kinases, the eponymous member of which has been found to be highly expressed in vascular tissues.7srcFK are activated by myristoylation and/or palmitoylation,8 translocation to the plasma membrane,9 and subsequent auto-phosphorylation at tyr-416.10 Tyrosine kinases in general are implicated in vascular smooth muscle contraction, both via actions upon Ca2+ release and/or influx and via Ca2+-sensitization.11–15 Although evidence for the latter is somewhat contradictory,11,14 one study clearly shows that sphingosylphosphorylcholine-mediated contraction and translocation of rho-kinase in pig coronary artery are dependent on srcFK.16srcFK may regulate rho activation via phosphorylation of RhoGEF.6
Ca2+-sensitization and the rho/rho-kinase pathway are important components of contractile function in pulmonary artery, in response to agonists and acute or chronic hypoxia, both in normal and patho-physiological settings,17,18 whereas the role(s) of srcFK are largely unexplored.19 In the present study, therefore, we evaluated the role of srcFK in Ca2+-sensitization-dependent prostaglandin F2α (PGF2α)-mediated contraction in rat small distal pulmonary arteries and srcFK interaction with the rho/rho-kinase pathway.
2. Methods
2.1. Animals, tissue isolation, and cell culture
This study conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). Male Wistar rats (200–250 g) were killed by lethal overdose of pentobarbital (ip). Lungs were excised and placed in cold physiological salt solution (PSS), and first to third-order branches of the intra-pulmonary artery (IPA) were dissected free of surrounding parenchyma. First, second, and third-order branches (∼150–1000 µm diameter) were used for the extraction of mRNA for polymerase chain reaction (PCR) and protein for western blot experiments. Second or third-order branches (∼150–600 µm diameter) were used for the preparation of pulmonary artery smooth muscle cells (PASMC) and measurement of isometric tension.
PASMC were dispersed enzymatically and grown in DMEM with 10% FCS to passage 4 or 5. Cells were then growth-arrested in serum-free media for 24 h and harvested for PCR/western blot or plated on 13 mm coverslips and then growth-arrested for staining and translocation experiments. Identification of each line of cells as smooth muscle was verified by positive staining with anti-smooth muscle α-actin, and anti-calponin antibodies (Santa Cruz Biotechnology, CA, USA).
2.2. Solutions, drugs, and chemicals
PSS contained (mM): NaCl 118; NaHCO3 24; KCl 4; CaCl2 1.8; MgSO4 1; NaH2PO4 0.434, glucose 5.56. Ca2+-free relaxing solution contained (mM): PIPES 30, Mg(Ms)2 5.3, KMs 46.6, K2EGTA 10, Na2ATP 5, Na2 creatinine phosphate 10, and the pH was set at 7.1. Ca2+-containing intracellular solution was identical except for the substitution of CaEGTA for K2EGTA. Free [Ca2+] was adjusted by mixing the two solutions in the appropriate proportion, as calculated by WEBMAXC software (www.stamford.edu). SU6656, PP2, PP3 and Y27632 were all obtained from Calbiochem (Merck Biosciences Nottingham, UK). PGF2α (tromethamine salt) was purchased from Biomol (Exeter, UK). All other reagents were obtained from Sigma (Poole, UK) Calbiochem, Invitrogen (Paisley, UK), or Fisher (Loughborough, UK).
2.3. RNA isolation and reverse transcriptase–polymerase chain reaction
Total RNA was extracted from IPA or PASMC using the Qiagen RNeasy mini kit and TissueLyser (Qiagen, Crawley, UK). RNA was treated with TURBO DNase (Ambion, Austin, TX, USA) to remove any remaining contaminating DNA and then reverse-transcribed in the presence of RNAguard (GE Healthcare, Chalfont St Giles, UK) by using random hexamers and revert-aid reverse transcriptase (Fermentas International, York, UK). MacVector™ (version 7.2) and Ensembl Genome Browser (www.emsembl.org) were used to design RT–PCR primer pairs. Sense and antisense primers on either side of a small intron (<1 kb) were made to allow distinction from amplification of any contaminating DNA as opposed to reverse-transcribed mRNA. Primer pairs are as follows. BLK (accession no. BC098683): sense GGACAATGGAGGCTATTACATCTCG; antisense ATTCTTCGGGGCTGGGTTCACAC. FGR (accession no. BC062025): sense TCTATGCTACTTGCTCACCGCAC; antisense ATAAATGGGTTCCTCTGACACCAC. FRK (accession no. U09583): sense TGTGTGGTCTTTTGGAATCCTGC; antisense TTGGTCGTTGCTTGGGCTCTAC. FYN (accession no. U35365): sense GAAGAGCCCATTTACATTGTCACG; antisense ATGAGTCCGTTCCCCACCAG. HCK (accession no. BC078890): sense CTGGACAGTGGAGGCTTCTACATC; antisense ATGGCTTCTGGGGTTTGGG. LCK (accession no. BC099218): sense TCCCCTCGTATCACTTTTCCCG; antisense CCCTTGCTTCAGACTTTTCACTGC. LYN (accession no. AF000300): sense GACAATCTGAATGACGATGGAG; antisense CGTAGTTGCTGGGGATGAAGC. SRC (accession no. AF157016): sense TTCAAGAAAGGGGAGCGGCTGC; antisense TGTCAAAGTCGGATACAGAGAGGC. YES1 (accession no. BC079403): sense GCAAAATGGGGAGAAAAGATGCTG; antisense TGGTCGTGATGTAGTATCCACCG.
All PCR primers were supplied by MWG Biotech (Ebersberg, Germany). PCR was carried out using 100 ng of reverse-transcribed RNA, 1× PCR II buffer, 4 mM MgCl2, 2 U Amplitaq Gold (Applied Biosystems, Warrington, UK), 0.5 U Perfect Match (Stratagene Europe, The Netherlands), 0.25 mM dNTPs (Fermentas International, York, UK), and 1.25 µM primer pair in a final volume of 40 µL. PCR cycling conditions were 10 min 95°C followed by 4 cycles of 2 min 95°C, 10 min 57°C, 2 min 72°C and then a variable number of cycles of 2 min 95°C, 2 min 57°C, 2 min 72°C (total number of cycles indicated in figure legends). Eighty microlitres of PCR products (reaction equivalent on 20 ng reverse-transcribed RNA) were analysed by electrophoresis on 2.8% agarose gels run in 1× TAE buffer (National Diagnostics, Yorkshire, UK) with PhiX174 DNA/HinfI Marker (Fermentas International, York, UK). Gel-purified PCR fragments were sequenced to confirm identity (Geneservice, Medical Solutions plc, UK).
2.4. Western blot
IPA segments were treated with PGF2α (20 µM), following a 15 min equilibration period in PSS and a 15 min pre-incubation with pharmacological agents where appropriate, gassed with 5% CO2/balance air at 37°C, prior to snap-freezing. Tissue was homogenized and protein extracted in 50 µL of Tris/SDS sample buffer containing phosphatase inhibitor cocktail I and II (Sigma) and protease inhibitor cocktail I (Sigma). Protein was extracted from PASMC by the same method. Protein extracts (12–15 µL, ∼10 µg, per lane) were run on SDS/PAGE gels (4–12% gradient, Invitrogen), transferred to nitrocellulose membrane, blocked with 5% skimmed milk for 1 h, probed with primary antibody (1:1000, in Tris-buffered saline with 0.1% skimmed milk) overnight at 4°C and then with horseradish-peroxidase conjugated anti-IgG secondary antibody (1:5000 in tris-buffered saline with 1% milk) for 1 h at room temperature. For phosphorylation experiments, membranes were first probed with anti-phospho-antibodies, stripped for 1 h (Pierce stripping buffer), re-blocked, and re-probed with corresponding anti-total antibodies. Protein bands were visualized with Supersignal West Femto Maximum Sensitivity Substrate (Pierce, Cramlington, UK) or ECL western blotting detection reagent (Amersham, Bucks, UK) and exposed to photographic film. Rabbit anti-src, rabbit anti-phospho-srcFK (tyr-416), mouse anti-phospho-tyrosine, rabbit anti-myosin light-chain, mouse anti-phospho-myosin light-chain (ser-19), and mouse anti-β-actin antibodies were obtained from Cell Signaling. Sheep anti-MYPT-1, rabbit anti-phospho-MYPT-1 (thr-695: equivalent to thr-697 in rat), and rabbit anti-phospho-MYPT-1 (thr-850: equivalent to thr-855 in rat) antibodies were obtained from Upstate (UK).
2.5. Force measurement and α-toxin permeabilization
IPAs were mounted on a Mulvany–Halpern wire myograph (DMT A/S, Aarhus, Denmark), bathed in PSS (gassed with 95% air/5% CO2, pH 7.4), and stretched to 90% of the circumference necessary to create an equivalent transmural pressure of 4.4 kPa (30 mmHg). IPAs were equilibrated with three 3 min contractions to 80 mM K+PSS and then permeabilized with α-toxin as follows. First, arteries were equilibrated in relaxing solution (as mentioned earlier) and then permeabilized by incubation at pCa 6.5 with 60 µg/mL α-toxin until the resulting vasoconstriction reached a plateau. Following re-equilibration with relaxing solution, submaximal vasoconstriction was elicited with pCa 6.9, and then PGF2α (100 µM) was applied, in the presence of 1 µM GTP to support G-protein activation. All contraction experiments were conducted at 26°C and in the presence of 10 µM cyclopiazonic acid to remove the influence of Ca2+ store release on contraction.
2.6. Staining and image analysis
PASMC were treated with 20 µM PGF for 10 min in DMEM at 37°C with or without prior application of 30 µM SU6656 for 10 min. Reactions were terminated by addition of paraformaldehyde fixative. For ROCK-2 translocation, fixed cells were permeabilized with Triton-100 and stained overnight with anti-ROCK-2 primary antibody (1:100, Santa Cruz Biotechnology) at 4°C following incubation with Alexa Fluor 488-labelled secondary antibody (1:1000, Santa Cruz) for 1 h at room temperature. Specificity of anti-ROCK-2 was confirmed by western blot (single band at 160 kDa). Staining was negative with secondary antibody alone.
2.7. Data analysis and statistics
Images of western blot bands of appropriate molecular weights were quantified using Image J software (rsb.info.nih.gov). Band intensity was expressed as a ratio of phospho/total for each protein, and values from each treated sample were normalized to those from control (untreated) samples run on the same gel. Two to three control samples were run on each gel and averaged. Statistical analysis was performed with SigmaStat (Systat software Inc.). Time-dependent drug responses were examined by two-way repeated measures ANOVA with appropriate post-tests. Individual comparisons (e.g. single-dose PGF2α vs. control) were performed by Student’s t-test or Mann–Whitney rank sum test, where appropriate. Comparisons of the effects of different drugs or dose-responses of single drugs against PGF2α were performed by one-way ANOVA with post-tests or by Kruskal–Wallis one-way ANOVA on ranks with post-tests, where appropriate.
3. Results
3.1. Expression of src family kinases
PCR was performed on mRNA extracts from IPA. All nine src family members were expressed, and the three most abundant are shown in Figure 1A (see Supplementary material online, Figure Si for all nine). A similar expression pattern was obtained from cultured rat PASMC (see Supplementary material online, Figure Sii). srcFK were also expressed at the protein level. Three bands were visualized, two at ∼59/60 kDa and one at ∼54 kDa. These correspond to the three most highly expressed at the mRNA level, src, and the closely related fyn and yes, with expected molecular weights in rat of 60, 60, and 54 kDa, respectively (as calculated from the predicted amino acid sequence at www.bioinformatics.org). Representative blots are shown in Figure 1B. Similar protein expression was obtained from cultured rat PASMC (see Supplementary material online, Figure Sii). In order to demonstrate full kinase activity, srcFK require auto-phosphorylation at tyr-416.10 Phospho-srcFK (tyr-416) immunoreactivity was inhibited by the srcFK inhibitor PP2 (at 3 and 30 µM but not at 0.3 µM) but not by the inactive analogue PP3 (30 µM) (Figure 1B), indicating in this preparation that 30 µM PP2 is required to substantially inhibit srcFK auto-phosphorylation, and hence srcFK activity.
Figure 1.

Expression of src family kinases in intra-pulmonary artery. (A) mRNA expression by polymerase chain reaction for src, fyn, and yes. The examples shown are representative of triplicate determinations. Thirty-two polymerase chain reaction cycles were carried out for each cDNA. Values in parentheses indicate expected amplicon sizes in base pairs (bp). (B) Protein expression of src family kinases by western blot analysis of intra-pulmonary artery. Three bands (two at ∼59/60 kDa, and one at 54 kDa) were visualized with anti-src and by anti-phospho-src family kinases (tyr-416). Phospho-src family kinases (tyr-416) immunoreactivity at 60 and 54 kDa was inhibited by PP2 (3 µM, *P < 0.05, n = 8 rats and 30 µM, P < 0.05, n = 8 rats) but not by PP3 (30 µM, n = 8 rats).
3.2. src family kinases inhibition antagonizes PGF2α-mediated Ca2+-sensitization
IPAs were permeabilized with α-toxin and submaximally constricted with pCa clamped at 6.9. This constriction was enhanced approximately three-fold by application of PGF2α, reaching a maximum at 40–45 min. At this point, the srcFK inhibitors SU6656 and PP2 or the inactive analogue PP3 was applied. These treatments were compared with parallel time controls in which drug was not applied. DMSO vehicle controls (applied in the place of drug) were indistinguishable from time controls (not shown). SU6656 (Figure 2A) and PP2 (Figure 2B) both caused significant relaxation at 3 and 30 µM, and this was significantly greater than the small degree of spontaneous run-down shown by the time controls. PP3 also caused relaxation at 30 µM, but this was significantly less than for PP2 (Figure 2C). SU6656-mediated relaxation was unaffected by de-endothelialization (n = 6) and was similar when SU6656 was applied prior to the PGF2α (n = 5) as when it was added acutely (see Supplementary material online, Figures Siii and Siv, respectively).
Figure 2.
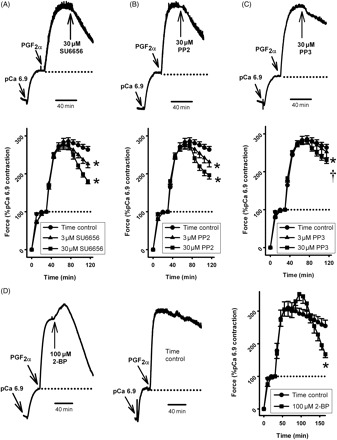
Effect of src family kinase inhibitors on PGF2α-induced contraction in α-toxin-permeabilized intra-pulmonary artery. intra-pulmonary arteries were pre-constricted with pCa6.9 for 30 min, prior to the addition of 100 µM PGF2α for 40 min (A–C) or 90 min (D). Compared with appropriate time controls (n = 12 arteries), this contraction was significantly relaxed by SU6656 [(A) 3 µM, 28 ± 3%, *P < 0.05, n = 8 arteries and 30 µM, 54 ± 4%, *P < 0.01, n = 8 arteries] and by PP2 [(B) 3 µM, 25 ± 5%, *P < 0.05, n = 8 arteries and 30 µM, 46 ± 5%, *P < 0.01, n = 12 arteries]. PP3 also caused relaxation at 30 µM [(C) 28 ± 2%, *P < 0.05, n = 12 arteries] but was significantly less effective than PP2 (†P < 0.05). 2-Bromopalmitate (2-BP) caused a transient contraction (non-significant) followed by significant relaxation [(D) 100 µM, 71 ± 4%, *P < 0.01, n = 7 arteries, compared with time controls, n = 7 arteries].
The srcFK inhibitors SU6656 and PP2 cannot distinguish between different members of the src family. However src may be distinguished by the manner in which it is modified for insertion into the plasma membrane, a process required for full kinase activity.8,20src is simply myristoylated, whereas other family members, including fyn and yes, are also palmitoylated.8,21 We therefore evaluated the effects of the acylation inhibitor 2-bromopalmitate and of ecosapentaenoic acid (EPA, which selectively displaces palmitoylated proteins from the membrane) on PGF2α-induced Ca2+-sensitization.8,20,21 When 100 µM 2-bromopalmitate was applied to a pre-existing PGF2α-induced contraction, a small additional transient contraction was caused, which was followed by a large sustained relaxation (Figure 2D). In contrast to this, 60 µM EPA caused a large sustained additional contraction and no relaxation (288 ± 66% increase, P < 0.01 vs. time controls at 90 min, n = 5).
3.3. PGF2α enhances src family kinases auto-phosphorylation and protein tyrosine phosphorylation
In order to test the hypothesis that PGF2α-mediated contraction was acting via srcFK activation and protein tyrosine phosphorylation, further western blot experiments were performed in PGF2α-treated IPA with anti-phospho-srcFK (tyr-416) and anti-phospho-tyrosine. As shown in Figure 3A, srcFK phosphorylation at tyr-416 was enhanced by PGF2α in a time-dependent manner. The two bands analysed (60 and 54 kDa) showed significant peak enhancements at 2–5 min, whereas at 60 kDa, a significant second-phase-sustained enhancement at 30 min was apparent. Several protein bands were visualized with anti-phospho-tyrosine in IPA: a single intense band at ∼120 kDa, bands at ∼95 and ∼75 kDa, and a close collection of bands centred at ∼65 kDa (Figure 3B). This immunoreactivity was enhanced by PGF2α, significantly inhibited by PP2, but unaffected by PP3 (30 µM). Other fainter bands were present but were not quantified.
Figure 3.
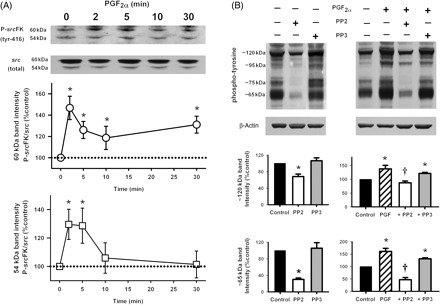
Effects of PGF2α on src family kinases tyr-416 auto-phosphorylation and tyrosine phosphorylation of multiple proteins in intra-pulmonary artery. (A) PGF2α-enhanced phospho-src (tyr-416) immunoreactivity at 60 and 54 kDa in a time-dependent manner (20 µM, *P < 0.05, n = 10–12 rats). (B) PGF2α also enhanced phospho-tyrosine immunoreactivity at multiple protein bands (20 µM, *P < 0.01, n = 10–15 rats). This was inhibited by PP2 (30 µM, †P < 0.05 vs. PGF2α, n = 10–15 rats) but not by PP3 (30 µM, n = 12 rats). Data are expressed as the ratio of phospho-tyrosine/β-actin immunoreactivity.
3.4. PGF2α mediates Ca2+-sensitization via MYPT-1 and MLC20 phosphorylation
Western blot experiments were carried out for MYPT-1 (both phospho-thr-697 and phosho-thr-855) and MLC20 (phospho-ser-19) in PGF-treated IPA. PGF2α significantly enhanced phosphorylation of MYPT-1 at both thr-697 (Figure 4A) and thr-855 (Figure 4B), although this effect was greater at thr-855. As expected, PGF2α also significantly enhanced ser-19 phosphorylation on MLC20 (Figure 4C). With both MYPT-1 at thr-855 and MLC20 at ser-19, the PGF-stimulated increase in phosphorylation peaked at 10 min. This time point was therefore used in subsequent phosphorylation experiments.
Figure 4.

Effects of PGF2α on phosphorylation of MYPT-1 (120–130 kDa) and MLC20 (18 kDa) in intra-pulmonary artery. Phosphorylation was enhanced on MYPT-1 at thr-697 [(A) *P < 0.05, n = 6–8 rats] and thr-855 [(B) *P < 0.05; **P < 0.01, n = 6–10 rats] and on MLC20 at ser-19 [(C) *P < 0.05; **P < 0.01, n = 6–11 rats].
3.5. MYPT-1 and MLC20 phosphorylation are sensitive to inhibition of src family kinases and rho-kinase
Basal phosphorylation of MYPT-1 at thr-697 and thr-855 and MLC20 at ser-19 was not significantly affected by 30 µM SU6656, but was reduced by the rho-kinase inhibitor Y27632 (10 µM) (Figure 5A–C). Y27632 was much more effective against MYPT-1 phosphorylation at thr-855 than at thr-697. After 10 min exposure to PGF2α, phosphorylation of MYPT-1 at thr-697 and thr-855 and MLC20 at ser-19 was enhanced (Figure 5D–F). This PGF2α-induced enhancement of MYPT-1 phosphorylation at thr-855 and MLC20 phosphorylation at ser-19 was nearly abolished by 30 µM SU6656 (Figure 5E and F). Y27632 also greatly inhibited the phosphorylation of these two sites. The effects of the two inhibitors when used in combination were however not additive (Figure 5D–F).
Figure 5.

Effects of inhibition of src family kinases and rho-kinase on MYPT-1 (120–130 kDa) and MLC20 (18 kDa) phosphorylation. [(A)–(C)] In the absence of PGF2α, phosphorylation of all three sites was not affected by SU6656 (30 µM, n = 7 rats), but variably inhibited by Y27632 (10 µM, *P < 0.05, n = 8 rats). [(D)–(F)] In the presence of PGF2α, phosphorylation of all three sites was enhanced (*P < 0.05; **P < 0.01, n = 8–11 rats), and this enhancement was inhibited by SU6656 (30 µM, MYPT-1 at thr-855 and MLC20 at ser-19, †P < 0.01, n = 8 rats), Y27632 (10 µM, †P < 0.01, n = 9 rats), and the two drugs in combination (†P < 0.01, n = 8 rats). No additive effect of SU6656 and Y27632 was apparent.
3.6. Interaction between src family kinases and rho-kinase during PGF2α-mediated Ca2+-sensitization
The results shown in Figure 5 suggest that srcFK activation may be part of the same pathway as PGF2α-induced rho-kinase-mediated phosphorylation of MYPT-1. This was corroborated by further contraction experiments in α-toxin-permeabilized IPA. As expected, 30 µM SU6656 did not have any significant effect on pCa 6.9-induced contraction in the absence of PGF2α (Figure 6A). PP2, PP3, and 2-bromopalmitate were similarly without effect (PP2 and PP3: 30 µM, n = 8, 2-BP: 100 µM, n = 5, not shown). Y27632, however, did relax this contraction, indicative of significant basal rho-kinase activity in the absence of agonist (Figure 6A). Y27632 also greatly relaxed the PGF2α-induced contraction, but this relaxation was not significantly greater when SU6656 was applied simultaneously with Y27632 (Figure 6B). The PKC-δ selective blocker rottlerin was completely without effect on PGF2α contraction (see Supplementary material online, Figure Sv), eliminating the possibility that our results were influenced by inhibition of PKC-δ by Y27632, as shown previously by Wilson et al.22
Figure 6.
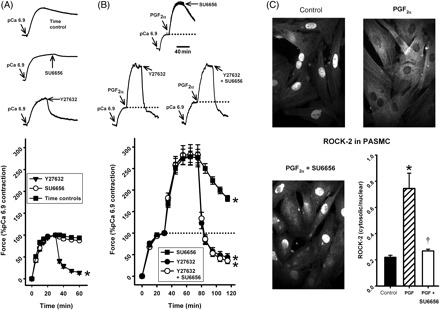
Interaction between src family kinases and rho-kinase activity in PGF2α-stimulated intra-pulmonary artery and pulmonary artery smooth muscle cells. (A) In intra-pulmonary artery, in the absence of PGF2α, the pCa6.9 contraction was inhibited by Y27632 (10 µM, *P < 0.001, n = 8, compared with time controls, n = 8 arteries) but not by SU6656 (30 µM, n = 8 arteries). (B) Y27632 also inhibited contraction in the presence of PGF2α (10 µM, 84 ± 2%, *P < 0.001, n = 8 arteries) and this relaxation was unaltered by the additional presence of SU6656 (30 µM, 87 ± 2%, *P < 0.001, n = 8 arteries). The effect of SU6656 alone (from Figure 2) is included as a comparison. (C) In pulmonary artery smooth muscle cells, PGF2α triggered translocation of rho-kinase (ROCK-2) immunoreactivity from the nucleus to the cytoskeleton (20 µM, *P < 0.05, n = 6 determinations) and this was prevented by SU6656 (30 µM; *P < 0.05, n = 6 determinations).
To examine more closely the interaction between srcFK and rho-kinase during stimulation with PGF2α, we examined in PASMC the subcellular translocation of rho-kinase, a process that is required for its activation and therefore precedes phosphorylation of MYPT-1.5 In control cells, the majority of rho-kinase (ROCK-2, the isoform activated by RhoA) antibody staining was localized to the nucleus; with some cytoplasmic staining, but upon stimulation with PGF2α, most of this translocated to the cytoskeleton. This movement was almost completely reversed by pre-treatment with SU6656 (Figure 6C). In the absence of PGF2α, SU6656 was without effect (not shown). No ROCK-2 was detected in plasma membrane.
4. Discussion
The results of this study clearly demonstrate the involvement of srcFKs in agonist-induced contraction of the pulmonary artery. Most previous works have relied on the use of non-selective tyrosine kinase inhibitors such as genistein and tyrphostins, which do not distinguish between receptor and non-receptor tyrosine kinases, and have focused on the role of these kinases in Ca2+ homeostasis (reviewed in Ward et al.15). The few that have addressed the question of Ca2+-sensitization have drawn opposing conclusions. For example, in α-toxin-permeabilized rat mesenteric arteries, ET-1-induced contractions were inhibited by tyrphostin-A23,14 whereas in β-escin-permeabilized guinea-pig mesenteric arteries, direct Ca2+-mediated contraction was unaffected by tyrphostin.11 This discrepancy may be explained by our current findings, since we showed that the srcFK-selective inhibitors SU665623 and PP224 both selectively inhibited PGF2α-induced but not direct Ca2+-induced contraction in α-toxin-permeabilized rat pulmonary artery and inhibited PGF2α-mediated but not basal MLC20 phosphorylation at ser-19, suggesting the involvement of srcFK only during stimulation of G-protein-coupled receptors.
MYPT-1, the regulatory subunit of myosin phosphatase, is an important mediator of changes in contractile force independent of a change in [Ca2+]i (Ca2+-sensitization).2 As has been previously shown for PGF2α and other agonists in other vascular preparations,22,25,26 we observed that PGF2α enhanced MYPT-1 phosphorylation at two sites, thr-697 and thr-855 (the rat homologues of thr-696 and thr-850 in humans), but it was principally phosphorylation at thr-855 which was most sensitive to rho-kinase inhibition by Y27632. Although both threonine residues were previously shown to be targets of rho-kinase,3,4 our results agree with several other reports that thr-855 or its equivalent in other vascular preparations is the principal target of rho-kinase and that thr-697 is phosphorylated basally by another as-yet-unspecified serine/threonine kinase.22,25,26 As with phosphorylation of MLC20, the enhancement of MYPT-1 phosphorylation at thr-855 by PGF2α was inhibited by SU6656 although basal phosphorylation was unaffected. Interestingly, Y27632 nearly abolished the pCa 6.9-induced contractions in IPA as well as the basal MYPT-1 phosphorylation at thr-855 and significantly inhibited MLC20 phosphorylation, suggesting that there is considerable basal rho-kinase activity in the rat pulmonary artery. In any case, it is stimulated further by the addition of agonist. A similar observation was made in U46619-stimulated rat caudal artery.22
The src family members most likely to be required for the responses described in this study are the highly expressed src and fyn, which have both been previously implicated in smooth muscle contraction,7,16 and possibly yes, which is also highly expressed. Unfortunately, existing kinase inhibitors cannot easily distinguish between src, fyn, or yes. However, these kinases do differ in the way that they are post-translationally modified; src is dual-myristoylated, whereas both fyn and yes are also palmitoylated.8 Previously, using inhibitors of fatty acid acylation and palmitoylation, Nakao et al.16 suggested that sphingosylphosphorylcholine-induced Ca2+-sensitization in porcine coronary artery was mediated specifically by fyn, not src. We, however, found that the non-selective acylation inhibitor 2-bromopalmitate, but not the selective palmitoylation inhibitor EPA,21 inhibited PGF2α-mediated Ca2+-sensitization. Accordingly, but bearing in mind that other important membrane-signalling proteins are also myristoylated and palmitoylated,27 we may tentatively suggest that it is specifically src and not fyn or yes which is the srcFK involved in PGF2α-mediated Ca2+-sensitization in rat IPA. The additional contractions caused by 2-bromopalmitate and EPA are presumably due to further actions of these drugs, independent of PGF2α stimulation or srcFK activity.
The question arises as to whether srcFK activation in response to PGF2α is upstream of rho/rho-kinase or enhances Ca2+-sensitivity through a separate pathway. The actions of SU6656 and Y27632 were not additive with respect to both inhibition of PGF2α-induced MYPT-1 and MLC20 phosphorylation and inhibition of the associated PGF2α-induced contraction, suggesting that srcFK and rho-kinase may be part of the same pathway. In order to test this hypothesis, we examined translocation of rho-kinase in PASMC, a process that is important for the activation of the kinase.5 In control cells, rho-kinase staining was concentrated in the nucleus, as has been shown previously,28 but upon PGF2α treatment, it translocated to the cytoskeleton. Importantly, this movement was reversed by pre-treatment with SU6656, indicating that the movement of rho-kinase in response to stimulation by PGF2α was likely to be mediated by srcFK. In contrast to the study of Nakao et al.,16 we found no evidence for the presence of rho-kinase at the plasma membrane, either in control or in stimulated cells.
PGF2α mediates Ca2+-sensitization via the TP receptor,29 which is coupled to Gαq/11.30 How this coupling results in activation of srcFK and/or rho/rho-kinase is unclear. For full kinase activity, srcFK require auto-phosphorylation at tyr-41610 and we showed that this is enhanced by PGF2α, indicating that srcFK activity was increased. Src activation often goes hand in hand with focal adhesion kinase (FAK) which is activated by src.31,32 FAK may be the ∼120 kDa protein whose tyrosine phosphorylation in the current study was enhanced by PGF2α and inhibited by PP2. A well-established connection between G-protein-linked receptors such as the TP prostenoid receptor (which is activated by PGF2α29) and mitogenic signalling involves src-dependent transactivation of the EGF-receptor tyrosine kinase.33 This mechanism also involves Gαi, which is known to activate src by direct coupling.34 Studies with the tyrosine phosphatise inhibitor vanadate have suggested that RhoA translocation is stimulated by as-yet-unspecified tyrosine phosphorylation.35 Furthermore, a pathway for tyrosine kinase-dependent modulation of rho activation has been suggested: active rho requires GTP, and it acquires this from rho-specific RhoGEF.6 Two RhoGEFs in native smooth muscle that may enhance RhoA activation by Gαq or Gα12/13 are leukaemia-associated RhoGEF (LARG) and PDZ-RhoGEF,26,36 and LARG is indeed activated by tyrosine phosphorylation.37
In conclusion, we have shown for the first time that agonist-mediated, rho-kinase-dependent Ca2+-sensitization but not basal rho-kinase activity in rat pulmonary artery requires srcFK activity. This has implications for the regulation of rho and rho-kinase-dependent vascular tone and vascular remodelling in health and disease. The precise mechanism(s) by which G-protein-linked receptors couple to srcFK and subsequent rho-kinase activation remain to be determined.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
British Heart Foundation (FS/06/003 to G.K., PG/06/151/21995 to S.D.); Wellcome Trust (078075).
Supplementary Material
References
- 1.Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- 2.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 3.Ichikawa K, Ito M, Hartshorne DJ. Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J Biol Chem. 1996;271:4733–4740. doi: 10.1074/jbc.271.9.4733. [DOI] [PubMed] [Google Scholar]
- 4.Velasco G, Armstrong C, Morrice N, Frame S, Cohen P. Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr-850 induces its dissociation from myosin. FEBS Lett. 2002;527:101–104. doi: 10.1016/s0014-5793(02)03175-7. [DOI] [PubMed] [Google Scholar]
- 5.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- 6.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 7.Oda Y, Renaux B, Bjorge J, Saifeddine M, Fujita DJ, Hollenberg MD. cSrc is a major cytosolic tyrosine kinase in vascular tissue. Can J Physiol Pharmacol. 1999;77:606–617. [PubMed] [Google Scholar]
- 8.Alland L, Peseckis SM, Atherton RE, Berthiaume L, Resh MD. Dual myristylation and palmitylation of Src family member p59-fyn affects subcellular localization. J Biol Chem. 1994;269:16701–16705. [PubMed] [Google Scholar]
- 9.Sandilands E, Brunton VG, Frame MC. The membrane targeting and spatial activation of Src, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. J Cell Sci. 2007;120:2555–2564. doi: 10.1242/jcs.003657. [DOI] [PubMed] [Google Scholar]
- 10.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 11.Di Salvo J, Steusloff A, Semenchuk L, Satoh S, Kolquist K, Pfitzer G. Tyrosine kinase inhibitors suppress agonist-induced contraction in smooth muscle. Biochem Biophys Res Commun. 1993;190:968–974. doi: 10.1006/bbrc.1993.1144. [DOI] [PubMed] [Google Scholar]
- 12.Janssen LJ, Lu-Chao H, Netherton S. Excitation–contraction coupling in pulmonary vascular smooth muscle involves tyrosine kinase and Rho kinase. Am J Physiol Lung Cell Mol Physiol. 2001;280:L666–L674. doi: 10.1152/ajplung.2001.280.4.L666. [DOI] [PubMed] [Google Scholar]
- 13.Jin N, Siddiqui RA, English D, Rhoades RA. Communication between tyrosine kinase pathway and myosin light chain kinase pathway in smooth muscle. Am J Physiol. 1996;271:H1348–H1355. doi: 10.1152/ajpheart.1996.271.4.H1348. [DOI] [PubMed] [Google Scholar]
- 14.Ohanian J, Ohanian V, Shaw L, Bruce C, Heagerty AM. Involvement of tyrosine phosphorylation in endothelin-1-induced calcium-sensitization in rat small mesenteric arteries. Br J Pharmacol. 1997;120:653–661. doi: 10.1038/sj.bjp.0700950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward JP, Knock GA, Snetkov VA, Aaronson PI. Protein kinases in vascular smooth muscle tone—role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction. Pharmacol Ther. 2004;104:207–231. doi: 10.1016/j.pharmthera.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Nakao F, Kobayashi S, Mogami K, Mizukami Y, Shirao S, Miwa S, et al. Involvement of Src family protein tyrosine kinases in Ca2+ sensitization of coronary artery contraction mediated by a sphingosylphosphorylcholine-Rho-kinase pathway. Circ Res. 2002;91:953–960. doi: 10.1161/01.res.0000042702.04920.bf. [DOI] [PubMed] [Google Scholar]
- 17.Fukumoto Y, Tawara S, Shimokawa H. Recent progress in the treatment of pulmonary arterial hypertension: expectation for rho-kinase inhibitors. Tohoku J Exp Med. 2007;211:309–320. doi: 10.1620/tjem.211.309. [DOI] [PubMed] [Google Scholar]
- 18.Robertson TP, Dipp M, Ward JP, Aaronson PI, Evans AM. Inhibition of sustained hypoxic vasoconstriction by Y-27632 in isolated intrapulmonary arteries and perfused lung of the rat. Br J Pharmacol. 2000;131:5–9. doi: 10.1038/sj.bjp.0703537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong WK, Knowles JA, Morse JH. Bone morphogenetic protein receptor type II C-terminus interacts with c-Src: implication for a role in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2005;33:438–446. doi: 10.1165/rcmb.2005-0103OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yurchak LK, Sefton BM. Palmitoylation of either Cys-3 or Cys-5 is required for the biological activity of the Lck tyrosine protein kinase. Mol Cell Biol. 1995;15:6914–6922. doi: 10.1128/mcb.15.12.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb Y, Hermida-Matsumoto L, Resh MD. Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J Biol Chem. 2000;275:261–270. doi: 10.1074/jbc.275.1.261. [DOI] [PubMed] [Google Scholar]
- 22.Wilson DP, Susnjar M, Kiss E, Sutherland C, Walsh MP. Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of MYPT-1 at Thr-855, but not Thr-697. Biochem J. 2005;389:763–774. doi: 10.1042/BJ20050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, et al. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and Fyn-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 25.Ito K, Shimomura E, Iwanaga T, Shiraishi M, Shindo K, Nakamura J, et al. Essential role of rho kinase in the Ca2+ sensitization of prostaglandin F2a-induced contraction of rabbit aortae. J Physiol. 2003;546:823–836. doi: 10.1113/jphysiol.2002.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevenson AS, Matthew JD, Eto M, Luo S, Somlyo AP, Somlyo AV. Uncoupling of GPCR and RhoA-induced Ca2+-sensitization of chicken amnion smooth muscle lacking CPI-17. FEBS Lett. 2004;578:73–79. doi: 10.1016/j.febslet.2004.10.072. [DOI] [PubMed] [Google Scholar]
- 27.Duncan JA, Gilman AG. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS) J Biol Chem. 1998;273:15830–15837. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka T, Nishimura D, Wu RC, Amano M, Iso T, Kedes L, et al. Nuclear Rho kinase, ROCK-2, targets p300 acetyltransferase. J Biol Chem. 2006;281:15320–15329. doi: 10.1074/jbc.M510954200. [DOI] [PubMed] [Google Scholar]
- 29.Snetkov VA, Knock GA, Baxter L, Thomas GD, Ward JP, Aaronson PI. Mechanisms of the prostaglandin F2α-induced rise in [Ca2+]i in rat intrapulmonary arteries. J Physiol. 2006;571:147–163. doi: 10.1113/jphysiol.2005.101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dhein S, Giessler C, Becker K, Ponicke K, Brodde OE. Inositolphosphate formation in thoracic and abdominal rat aorta following Gq/11-coupled receptor stimulation. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:322–329. doi: 10.1007/s002100000344. [DOI] [PubMed] [Google Scholar]
- 31.Minuz P, Fumagalli L, Gaino S, Tommasoli RM, Degan M, Cavallini C, et al. Rapid stimulation of tyrosine phosphorylation signals downstream of G-protein-coupled receptors for thromboxane A2 in human platelets. Biochem J. 2006;400:127–134. doi: 10.1042/BJ20061015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, et al. Ephrin-A1 activates a Src/focal adhesion kinase-mediated motility response leading to Rho-dependent actino/myosin contractility. J Biol Chem. 2007;282:19619–19628. doi: 10.1074/jbc.M701319200. [DOI] [PubMed] [Google Scholar]
- 33.Gao Y, Tang S, Zhou S, Ware JA. The thromboxane A2 receptor activates mitogen-activated protein kinase via protein kinase C-dependent Gi coupling and Src-dependent phosphorylation of the epidermal growth factor receptor. J Pharmacol Exp Ther. 2001;296:426–433. [PubMed] [Google Scholar]
- 34.Ma YC, Huang J, Ali S, Lowry W, Huang XY. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102:635–646. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- 35.Mori M, Tsushima H. Vanadate activates RhoA translocation in association with contracting effects in ileal longitudinal smooth muscle of guinea pig. J Pharmacol Sci. 2004;95:443–451. doi: 10.1254/jphs.fp0030576. [DOI] [PubMed] [Google Scholar]
- 36.Derewenda U, Oleksy A, Stevenson AS, Korczynska J, Dauter Z, Somlyo AP, et al. The crystal structure of RhoA in complex with the DH/PH fragment of PDZRhoGEF, an activator of the Ca2+ sensitization pathway in smooth muscle. Structure. 2004;12:1955–1965. doi: 10.1016/j.str.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki N, Nakamura S, Mano H, Kozasa T. Gα12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc Natl Acad Sci USA. 2003;100:733–738. doi: 10.1073/pnas.0234057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.