

Abstract
Tumor necrosis factor α (TNF-α) plays a central role in the pathophysiology of Alzheimer’s disease (AD). Food and Drug Administration–approved biologic TNF-α inhibitors are thus a potential treatment for AD, but they do not cross the blood-brain barrier. In this short review, we discuss the involvement of TNF-α in AD, challenges associated with the development of existing biologic TNF-α inhibitors for AD, and potential therapeutic strategies for targeting TNF-α for AD therapy.
Keywords: Alzheimer’s disease, TNF-α, blood-brain barrier, biologic TNF-α inhibitors
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia worldwide and represents one of the most serious health issues for the elderly. An estimated 5.4 million Americans have AD, and this number is expected to triple in 2050 due to lack of medical breakthroughs to stop, prevent, or slow the disease.1 Over the years, research efforts have focused extensively on delineating the mechanisms and identifying targets involved in AD pathogenesis; however, to date, no disease-modifying therapy has been clinically approved for AD.
The 2 major neuropathological hallmarks of AD are extracellular deposition of amyloid beta (Aβ)–containing senile plaques and intracellular tau-containing neurofibrillary tangles (NFTs) in the brain.2 Aβ plaque formation is considered relatively specific to AD pathology; however, NFTs are found associated with other disorders as well.3 Aβ plaque formation involves a cascade of events beginning with the sequential cleavage of the amyloid precursor protein (APP), a large transmembrane protein, by proteolytic enzymes β-secretase and γ-secretase. Cleavage by γ-secretase releases Aβ, peptides which are usually composed of 40 to 42 amino acid residues, into the extracellular space where they aggregate into oligomers, β-sheet–structured fibrils, followed by clumping into Aβ plaques.4 Some studies suggest that Aβ pathology more closely reflects the preclinical (asymptomatic) stage of AD, whereas accumulation of tau tangles more closely reflects the onset of clinical symptoms in AD.5 Among the numerous targets explored to treat AD, anti-Aβ approaches directed toward removal of Aβ and/or decreasing its production (eg, inhibition of γ-secretase, active immunotherapy, and anti-Aβ antibodies) have shown promising results in animal models of AD6–8 and have been at the forefront of AD research. Most of the studies investigating the effects of anti-Aβ approaches have had limited success in mid- to late-stage clinical development and have failed to reach primary clinical end points.9 A recent study investigated the effect of a selective tau aggregation inhibitor in patients with mild-to-moderate AD. This anti-tau approach, however, failed to reach primary outcome measures.10
More recent research has pivoted focus to cytokine-mediated neuroinflammation as a major contributor to the development of AD, and evidence suggests that inflammation promotes pathological processes that lead to AD.11–13 Among the cytokines involved in neuroinflammation, tumor necrosis factor α (TNF-α) is the most studied and plays an essential role in the cytokine cascade during an inflammatory response. Although the levels of TNF-α in the periphery and central nervous system (CNS) of healthy adults are maintained at very low levels, the levels of this cytokine are significantly elevated in blood14 and CNS12 of patients with AD, and many clinical and animal studies have demonstrated a link between excess TNF-α levels in the brain and AD.15 Here, we focus on the involvement of TNF-α in AD, challenges associated with the development of existing biologic TNF-α inhibitors (TNFIs) for AD, and potential therapeutic strategies for targeting TNF-α for AD therapy.
TNF-α and TNF-α Receptors
TNF-α, first recognized for its antitumor activity,16 is one of the main inflammatory cytokines involved in initiating and propagating an inflammatory response. TNF-α is a monomeric (17 kDa) nonglycosylated type 2 transmembrane protein that belongs to a superfamily of ligand/receptor proteins called the TNF/TNF receptor (TNF/TNFR) superfamily proteins. TNF-α is inserted into the membrane as a homotrimer which is then cleaved into a 51-kDa soluble trimer via proteolytic cleavage by the TNF-α–converting enzyme.17 In the central nervous system, TNF-α can be synthesized in the brain by microglia,18 neurons,19 and astrocytes.20
TNF-α binds to 2 receptor subtypes, TNFR1 (p55-R) and TNFR2 (p75-R), each having distinct signaling pathways differentiated by the presence of an intracellular death domain (DD).21 TNFR1, which contains the DD, is ubiquitously expressed in various cell types and is activated through either soluble TNF-α (sTNF-α) or membrane TNF-α (mTNF-α).22 TNFR1, once activated, is involved in divergent effects including cell proliferation, activation, antiviral activity, and primarily in TNF-α–mediated apoptosis and cytotoxicity. TNFR2 has a higher affinity for TNF-α, is activated preferentially by mTNF-α, and is primarily expressed at low levels by cells of the immune system and endothelial cells.22 TNFR2 lacks the DD and can trigger signaling cascades that activate pro-inflammatory and pro-survival pathways through the activation of the cellular inhibitor of apoptosis proteins 1 and 2, nuclear factor κB, and phosphatidylinositol 3-kinase–dependent signaling pathways.23 Some studies also report an indirect role of TNFR2 in TNF-α–mediated cytotoxicity via the enhancement of TNFR1-mediated cytotoxicity, but no induction of cytotoxicity by TNFR2 alone.24 Overall, the general consensus is that TNFR1 exerts pro-apoptotic functions, whereas TNFR2 typically promotes cell survival and proliferation.23
TNF-α Involvement in AD
The role of TNF-α in the pathophysiology of AD has been examined both in clinical and animal studies. One of the earliest evidence suggesting the involvement of TNF-α in AD pathophysiology was its presence around the Aβ plaque in postmortem human AD brains.25 Following studies in human AD brains showed that TNFR1 signaling is required for Aβ-induced neuronal death, and although TNFR1 protein levels are significantly greater in AD brains compared with nondemented brains, TNFR2 protein levels are lower.26 Furthermore, an increase in the binding affinity of TNF-α to TNFR1, but a decrease in binding affinity to TNFR2, was observed.26 Genetic association studies showed that chromosome 12p region encoding for the TNFR1 gene and the chromosome 1p region encoding for the TNFR2 gene are associated with late-onset AD.27 The role of TNF-α in AD pathology was further suggested by studies in which significant elevation of TNF-α levels in the cerebrospinal fluid (CSF)28 and serum29,30 of patients with AD correlated with disease progression.30
The reported clinical findings regarding the role of TNF-α in AD pathology are consistent with observations made in mouse models of AD. Elevated TNF-α levels were observed in the brain tissues of AD transgenic mice.31 Furthermore, elevated TNF-α levels were associated with intraneuronal Aβ immunoreactivity in the entorhinal cortex, and these elevated TNF-α levels correlated with cognitive deficits in AD mice.32 The role of TNF-α signaling in abnormal APP processing, Aβ plaque accumulation, tau-related pathology, and cell death has also been reported. Deletion of TNFR1 in AD transgenic mice lowered Aβ formation, Aβ plaque burden, β-secretase 1 (BACE1) expression, and cognitive deficits.33 However, ablation of both TNFR1 and TNFR2 exacerbated Aβ and tau pathology due to aggravation of TNFR1-mediated AD pathology resulting from silencing of TNFR2.34 Furthermore, genetic inactivation of TNFR1 signaling in AD transgenic mice prevented intraneuronal accumulation of Aβ,35 whereas genetic deletion of TNFR2 exacerbated AD pathology in a transgenic mouse model of AD.36 In AD transgenic mice, TNF-α increased Aβ production through upregulation of both β-secretase expression37 and γ-secretase activity,38 and chronic neuronal TNF-α expression resulted in extensive neuronal cell death.19 Excess TNF-α levels in the brain can disrupt clearance of Aβ by inhibiting microglial clearance of Aβ,39 cause synaptic dysfunction,40 and accelerate disease progression and cognitive decline.41 Overall, TNF-α–driven processes are involved at multiple stages of AD pathophysiology and disease progression (Figure 1).
Figure 1.
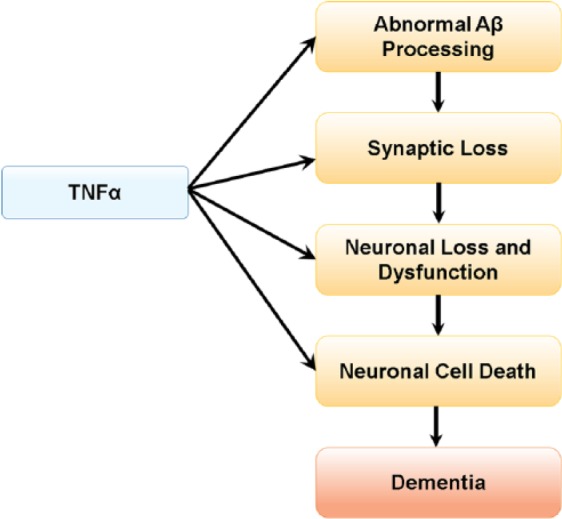
Central role of TNF-α in AD pathophysiology. Elevated TNF-α levels in AD may enhance Aβ production, decrease Aβ clearance, increase neuronal loss and cell death, and are implicated in cognitive decline in AD. Aβ indicates amyloid beta; AD, Alzheimer’s disease; TNF-α, tumor necrosis factor α.
Targeting TNF-α in AD
The most potent TNFIs are biologic drugs that are Food and Drug Administration (FDA) approved for the treatment of peripheral inflammatory conditions including Crohn disease, rheumatoid arthritis, and psoriatic arthritis. These biologic TNFIs include TNF-α–specific monoclonal antibodies (MAbs) (infliximab, adalimumab, golimumab, and certolizumab), and recombinant fusion proteins (etanercept).15 TNF-α–specific MAbs bind to either mTNF-α or sTNF-α thus preventing TNF-α signaling mediated by both TNFRs. However, recombinant fusion protein TNFI (etanercept) is comprised of the extracellular domain (ECD) of the TNFR2 fused to the c-terminal of the Fc domain of human IgG1. The fusion protein also binds to both mTNF-α and sTNF-α with high affinity preventing TNF-α signaling mediated by both TNFRs.23
The protective effects of TNF-α inhibition using these biologic TNFIs have been demonstrated both in clinical and experimental AD (Table 1). Inhibition of sTNF-α signaling by intracerebral infusion of a dominant negative TNFI prevented preplaque Aβ-associated pathology in a transgenic mouse model of AD.35 Intracerebroventricular injection of the anti-TNF-α MAb, infliximab, reduced TNF-α levels, Aβ plaques and tau-phosphorylation,42 and cognitive deficits43,44 in mouse models of AD. In a small open-label 6-month study in humans, perispinal etanercept administration (dose range: 25–50 mg per week) resulted in significant improvement in 3 standard measures of cognition: the AD Assessment Scale-Cognitive Subscale, the Severe Impairment Battery, and the Mini-Mental State Examination in patients with AD.45 These observed clinical improvements are hypothesized to be due to rapid penetration of etanercept into the CSF following perispinal administration and mediated by nonsynaptic signal transduction mechanisms of etanercept. However, in a recent study, perispinal injection of etanercept resulted in perispinal drug accumulation, but consistent intracranial delivery of the drug was not observed.46 In a recent case report, intrathecal administration of infliximab resulted in rapid cognitive improvement in a female patient with AD,47 and larger controlled trials are warranted. Collectively, both clinical and preclinical evidence underscore the therapeutic potential of biologic TNFIs for AD therapy.
Table 1.
Summary of studies focusing on neutralization of brain TNF-α with biologic TNFIs in AD.

One hindrance to the further development of the FDA-approved biologic TNFIs for AD is their limited blood-brain barrier (BBB) penetration,48 and invasive delivery approaches may not be practical or safe for chronic dosing for AD. Recently, modulation of peripheral TNF-α activity by biologic TNFIs has shown promise in transgenic mouse models of AD49; however, in a randomized double-blind phase 2 clinical trial, weekly subcutaneous injections of etanercept did not improve cognition, global function, or behavior in a small group of subjects with mild-to-moderate AD dementia.50 Further studies will be needed to determine whether peripheral modulation of TNF-α offers clinical improvement in AD.
Biologic drugs such as etanercept, ECD of the TNFR2, can be reengineered to enable BBB penetration by fusion of the biologic drug to a BBB molecular Trojan horse (MTH).51 A BBB MTH is an endogenous peptide or peptidomimetic monoclonal antibody that undergoes receptor-mediated transport across the BBB via endogenous peptide receptors, such as the transferrin receptor (TfR) or the insulin receptor. This binding triggers receptor-mediated transport of the MAb across the BBB, which ferries the biologic drug into the brain.52 One such BBB-penetrating TNFR, specific for the mouse, has been engineered and tested in mouse models of neurological diseases (Figure 2). The amino terminus of the human TNFR2 ECD is fused to the carboxyl terminal of the heavy chain of the chimeric TfRMAb to engineer a fusion protein designated as cTfRMAb-TNFR.53 This orientation frees the amino terminus of the cTfRMAb that is involved in binding to the BBB TfR and places the TNFR ECD in a dimeric configuration. Because both human and mouse monomeric TNF-α share high amino acid identity, and the human TNFR binds mouse TNF-α with high affinity,54 the cTfRMAb-TNFR fusion protein involves the human TNFR and not the mouse TNFR. The cTfRMAb-TNFR fusion protein binds to both the mouse TfR and TNF-α with high affinity evident from the low binding constants (Kd = 2.6 ± 0.3 nM for mouse TfR and 96 ± 34 pM for TNF-α). The cTfRMAb-TNFR is thus a bifunctional molecule that allows (a) rapid noninvasive transvascular delivery of the TNFR across the BBB using a receptor-mediated transcytosis approach and (b) once in the brain, the TNFR can sequester excess TNF-α. A complete pharmacokinetic analysis of this BBB-penetrating biologic TNFI, the cTfRMAb-TNFR fusion protein, showed that systemic routes (intraperitoneal, subcutaneous, or intravenous) of administration result in brain concentrations of the cTfRMAb-TNFR fusion protein that are 20-fold to 50-fold greater than the concentration of TNF-α in pathologic conditions of the brain.55 The intravenous route of administration is preferred for acute neurological conditions, such as ischemic stroke, whereas the intraperitoneal or subcutaneous routes of administration are more practical for dosing during chronic conditions including AD. The cTfRMAb-TNFR fusion protein was found to be protective in both acute (ischemic stroke) and chronic (Parkinson disease) mouse models of neurological diseases56–58 and is currently being investigated in a mouse model of AD.
Figure 2.

Schematic of the molecular Trojan horse (MTH) technology to ferry biologic TNF-α inhibitors (TNFIs) into the brain. The TNFI of interest in the figure is a tumor necrosis factor α receptor (TNFR). A fusion protein of TNFR and a blood-brain barrier (BBB) MTH, such as the chimeric monoclonal antibody against the mouse transferrin receptor (cTfRMAb), can be engineered and this is designated as cTfRMAb-TNFR. Following systemic injection, the cTfRMAb-TNFR fusion protein binds to the BBB transferrin receptor (TfR)1 and is transported across the BBB from blood to brain via the BBB TfR that undergoes receptor-mediated transcytosis.2 Once in the brain, the TNFR domain of the fusion protein can sequester excess TNF-α in the brain.3
For use in humans, a fusion protein of the ECD of the TNFR2 and a chimeric or humanized antibody against the human insulin receptor (HIRMAb), designated as the HIRMAb-TNFR fusion protein, has been engineered.48 The HIRMAb is the most potent MTH engineered for the human brain and cross-reacts with the insulin receptor of the Old World primates. In rhesus monkeys, the brain uptake of a 0.2 mg/kg dose of the HIRMAb-TNFR fusion protein after intravenous injection was 3 ± 0.1% ID/100 g brain compared with the brain uptake of the TNFR:Fc fusion protein which was 0.23 ± 0.06% ID/100 g brain. Furthermore, chronic administration of a high dose (20 mg/kg) of a HIRMAb-based fusion protein was found to be safe in rhesus monkeys.59
Conclusions
A plethora of clinical and animal studies strongly suggest an involvement of TNF-α in the pathophysiology of AD. The FDA-approved biologic TNFIs are thus a potential treatment for AD; however, these large molecules have limited BBB penetration. Clinical studies using the perispinal route of administration for biologic TNFIs have shown encouraging results in small open-label trials; however, larger controlled trials are required to confirm these results. Another approach currently under investigation is to target peripheral TNF-α rather than brain TNF-α. However, this approach has not shown any cognitive improvement in a clinical setting so far, and further studies are required to determine the effect of peripheral TNF-α modulation on AD pathology. Novel drug delivery strategies, such as the MTH technology, enable noninvasive delivery of biologic TNFIs to the brain and target both peripheral and brain TNF-α. Considering the multifactorial role of brain TNF-α in AD pathophysiology, such noninvasive drug delivery strategies may be a reasonable approach to deliver biologic TNFIs to the brain for AD treatment.
Footnotes
PEER REVIEW: Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 858 words, excluding any confidential comments to the academic editor.
FUNDING: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is funded by the Alzheimer’s Association grant RG-15-361188 (R.K.S.).
DECLARATION OF CONFLICTING INTERESTS: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RC wrote the first draft of the manuscript. RC, K-LY, and RKS contributed to the writing of the manuscript and agree with manuscript results and conclusions. RKS developed the structure and arguments for the paper and made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
As a requirement of publication, author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. The external blind peer reviewers report no conflicts of interest.
REFERENCES
- 1.Alzheimer’s Association Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 3.Joachim CL, Selkoe DJ. The seminal role of beta-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis Assoc Disord. 1992;6:7–34. doi: 10.1097/00002093-199205000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Citron M. Alzheimer’s disease: treatments in discovery and development. Nat Neurosci. 2002;5:1055–1057. doi: 10.1038/nn940. [DOI] [PubMed] [Google Scholar]
- 5.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79:110–119. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 7.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 8.Dovey HF, John V, Anderson JP, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 9.Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rafii MS. Targeting tau protein in Alzheimer’s disease. Lancet. 2016;388:2842–2844. doi: 10.1016/S0140-6736(16)32107-9. [DOI] [PubMed] [Google Scholar]
- 11.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarkowski E, Andreasen N, Tarkowski A, Blennow K. Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:1200–1205. doi: 10.1136/jnnp.74.9.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCaulley ME, Grush KA. Alzheimer’s disease: exploring the role of inflammation and implications for treatment. Int J Alzheimers Dis. 2015;2015:515248. doi: 10.1155/2015/515248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fillit H, Ding WH, Buee L, et al. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- 15.Cheng X, Shen Y, Li R. Targeting TNF: a therapeutic strategy for Alzheimer’s disease. Drug Discov Today. 2014;19:1822–1827. doi: 10.1016/j.drudis.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 16.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotox-in-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang P, Hung MC, Klostergaard J. Human pro-tumor necrosis factor is a homotrimer. Biochemistry. 1996;35:8216–8225. doi: 10.1021/bi952182t. [DOI] [PubMed] [Google Scholar]
- 18.Janelsins MC, Mastrangelo MA, Oddo S, LaFerla FM, Federoff HJ, Bowers WJ. Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease mice. J Neuroinflammation. 2005;2:23. doi: 10.1186/1742-2094-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janelsins MC, Mastrangelo MA, Park KM, et al. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol. 2008;173:1768–1782. doi: 10.2353/ajpath.2008.080528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci U S A. 1989;86:6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 22.Sedger LM, McDermott MF. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants—past, present and future. Cytokine Growth Factor Rev. 2014;25:453–472. doi: 10.1016/j.cytogfr.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 23.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss T, Grell M, Hessabi B, et al. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor 80: requirement of the TNF receptor-associated factor-2 binding site. J Immunol. 1997;158:2398–2404. [PubMed] [Google Scholar]
- 25.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Cheng X, Yang L, He P, Li R, Shen Y. Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer’s disease and non-demented patients. J Alzheimers Dis. 2010;19:621–630. doi: 10.3233/JAD-2010-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perry RT, Collins JS, Harrell LE, Acton RT, Go RC. Investigation of association of 13 polymorphisms in eight genes in southeastern African American Alzheimer disease patients as compared to age-matched controls. Am J Med Genet. 2001;105:332–342. doi: 10.1002/ajmg.1371. [DOI] [PubMed] [Google Scholar]
- 28.Tarkowski E, Liljeroth AM, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61:255–260. doi: 10.1016/s0361-9230(03)00088-1. [DOI] [PubMed] [Google Scholar]
- 29.Alvarez A, Cacabelos R, Sanpedro C, Garcia-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. 2007;28:533–536. doi: 10.1016/j.neurobiolaging.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 30.Paganelli R, Di Iorio A, Patricelli L, et al. Proinflammatory cytokines in sera of elderly patients with dementia: levels in vascular injury are higher than those of mild-moderate Alzheimer’s disease patients. Exp Gerontol. 2002;37:257–263. doi: 10.1016/s0531-5565(01)00191-7. [DOI] [PubMed] [Google Scholar]
- 31.Sly LM, Krzesicki RF, Brashler JR, et al. Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res Bull. 2001;56:581–588. doi: 10.1016/s0361-9230(01)00730-4. [DOI] [PubMed] [Google Scholar]
- 32.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 33.He P, Zhong Z, Lindholm K, et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montgomery SL, Mastrangelo MA, Habib D, et al. Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology: implications for chronic pan-TNF-alpha suppressive therapeutic strategies in the brain. Am J Pathol. 2011;179:2053–2070. doi: 10.1016/j.ajpath.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McAlpine FE, Lee JK, Harms AS, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34:163–177. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang H, He P, Xie J, Staufenbiel M, Li R, Shen Y. Genetic deletion of TNFRII gene enhances the Alzheimer-like pathology in an APP transgenic mouse model via reduction of phosphorylated IκBα. Hum Mol Genet. 2014;23:4906–4918. doi: 10.1093/hmg/ddu206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto M, Kiyota T, Horiba M, et al. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- 39.Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lourenco MV, Clarke JR, Frozza RL, et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18:831–843. doi: 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 41.McGeer EG, McGeer PL. Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- 42.Shi JQ, Shen W, Chen J, et al. Anti-TNF-alpha reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011;1368:239–247. doi: 10.1016/j.brainres.2010.10.053. [DOI] [PubMed] [Google Scholar]
- 43.Medeiros R, Prediger RD, Passos GF, et al. Connecting TNF-alpha signaling pathways to iNOS expression in a mouse model of Alzheimer’s disease: relevance for the behavioral and synaptic deficits induced by amyloid beta protein. J Neurosci. 2007;27:5394–5404. doi: 10.1523/JNEUROSCI.5047-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim DH, Choi SM, Jho J, et al. Infliximab ameliorates AD-associated object recognition memory impairment. Behav Brain Res. 2016;311:384–391. doi: 10.1016/j.bbr.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed. 2006;8:25. [PMC free article] [PubMed] [Google Scholar]
- 46.Roerink ME, Groen RJ, Franssen G, Lemmers-van de Weem B, Boerman OC, van der Meer JW. Central delivery of iodine-125-labeled cetuximab, etanercept and anakinra after perispinal injection in rats: possible implications for treating Alzheimer’s disease. Alzheimers Res Ther. 2015;7:70. doi: 10.1186/s13195-015-0149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi JQ, Wang BR, Jiang WW, et al. Cognitive improvement with intrathecal administration of infliximab in a woman with Alzheimer’s disease. J Am Geriatr Soc. 2011;59:1142–1144. doi: 10.1111/j.1532-5415.2011.03445.x. [DOI] [PubMed] [Google Scholar]
- 48.Boado RJ, Hui EK, Lu JZ, Zhou QH, Pardridge WM. Selective targeting of a TNFR decoy receptor pharmaceutical to the primate brain as a receptor-specific IgG fusion protein. J Biotechnol. 2010;146:84–91. doi: 10.1016/j.jbiotec.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Detrait ER, Danis B, Lamberty Y, Foerch P. Peripheral administration of an anti-TNF-α receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-α levels and memory deficits in mice. Neurochem Int. 2014;72:10–13. doi: 10.1016/j.neuint.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Butchart J, Brook L, Hopkins V, et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84:2161–2168. doi: 10.1212/WNL.0000000000001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardridge WM. Biologic TNFα-inhibitors that cross the human blood-brain barrier. Bioeng Bugs. 2010;1:231–234. doi: 10.4161/bbug.1.4.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pardridge WM. Biopharmaceutical drug targeting to the brain. J Drug Target. 2010;18:157–167. doi: 10.3109/10611860903548354. [DOI] [PubMed] [Google Scholar]
- 53.Zhou QH, Boado RJ, Hui EK, Lu JZ, Pardridge WM. Brain-penetrating tumor necrosis factor decoy receptor in the mouse. Drug Metab Dispos. 2011;39:71–76. doi: 10.1124/dmd.110.036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scallon B, Cai A, Solowski N, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 55.Sumbria RK, Zhou QH, Hui EK, Lu JZ, Boado RJ, Pardridge WM. Pharmacokinetics and brain uptake of an IgG-TNF decoy receptor fusion protein following intravenous, intraperitoneal, and subcutaneous administration in mice. Mol Pharm. 2013;10:1425–1431. doi: 10.1021/mp400004a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sumbria RK, Boado RJ, Pardridge WM. Brain protection from stroke with intravenous TNFα decoy receptor-Trojan horse fusion protein. J Cereb Blood Flow Metab. 2012;32:1933–1938. doi: 10.1038/jcbfm.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou QH, Sumbria R, Hui EK, Lu JZ, Boado RJ, Pardridge WM. Neuroprotection with a brain-penetrating biologic tumor necrosis factor inhibitor. J Pharmacol Exp Ther. 2011;339:618–623. doi: 10.1124/jpet.111.185876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sumbria RK, Boado RJ, Pardridge WM. Combination stroke therapy in the mouse with blood-brain barrier penetrating IgG-GDNF and IgG-TNF decoy receptor fusion proteins. Brain Res. 2013;1507:91–96. doi: 10.1016/j.brainres.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 59.Boado RJ, Hui EK, Lu JZ, Pardridge WM. AGT-181: expression in CHO cells and pharmacokinetics, safety, and plasma iduronidase enzyme activity in Rhesus monkeys. J Biotechnol. 2009;144:135–141. doi: 10.1016/j.jbiotec.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]