

Abstract
The regulatory control of cardiac endoplasmic reticulum (ER) stress is incompletely characterized. As ER stress signaling upregulates the E3-ubiquitin ligase Parkin, we investigated the role of Parkin in cardiac ER stress. Parkin knockout mice exposed to aortic constriction-induced cardiac pressure-overload or in response to systemic tunicamycin (TM) developed adverse ventricular remodeling with excessive levels of the ER regulatory C/EBP homologous protein CHOP. CHOP was identified as a Parkin substrate and its turnover was Parkin-dose and proteasome-dependent. Parkin depletion in cardiac HL-1 cells increased CHOP levels and enhanced susceptibility to TM-induced cell death. Parkin reconstitution rescued this phenotype and the contribution of excess CHOP to this ER stress injury was confirmed by reduction in TM-induced cell death when CHOP was depleted in Parkin knockdown cardiomyocytes. Isogenic Parkin mutant iPSC-derived cardiomyocytes showed exaggerated ER stress induced CHOP and apoptotic signatures and myocardium from subjects with dilated cardiomyopathy showed excessive Parkin and CHOP induction. This study identifies that Parkin functions to blunt excessive CHOP to prevent maladaptive ER stress-induced cell death and adverse cardiac ventricular remodeling. Additionally, Parkin is identified as a novel post-translational regulatory moderator of CHOP stability and uncovers an additional stress-modifying function of this E3-ubiquitin ligase.
Introduction
The E3-ubiquitin ligase Parkin is enriched in multiple organs1, 2, resides in various subcellular locations3, 4 and its cognate ubiquitylation substrates modulate diverse cellular functions spanning from the regulation of gene transcription, protein stability, endoplasmic reticulum (ER) stress and mitochondrial quality control5–8. Given that mutations in the gene encoding for Parkin predispose to early onset Parkinson Disease9, our understanding of its regulatory functions in cellular homeostasis may shed insight into potential targets for the modulation of disease.
The most well characterized role of Parkin has been defined in the orchestration of adaptive mitochondrial autophagy (mitophagy) in the context of ‘de-energized’ mitochondria10. This adaptive Parkin-mediated mitophagy effect is evident in the heart following exposure to direct ischemia-reperfusion injury and following triggering of cardiac mitochondrial adaptive programing in response to ischemic preconditioning11, 12. Recently, Parkin has also been found to be essential in the turnover of mitochondria during cardiac development13. These studies highlight that: (i) the role of Parkin in protecting against mitochondrial stressors is operational in-vivo; (ii) Parkin plays an important role in mitochondrial maturation during cardiac development; and (iii) the heart may be a useful organ to explore additional putative regulatory roles of Parkin in response to biological stress.
We reasoned that cardiac pressure-overload biomechanical stress would be an additional pathology, to enable characterization of chronic Parkin-linked adaptive and/or maladaptive programming. One such chronically engaged program evident during cardiac hypertrophy is ER stress14. Interestingly, ER stress initially evokes adaptive programming, but when chronically engaged facilitate apoptosis15, 16. ER stress has been found to upregulate Parkin17, 18, and this E3-ligase is implicated in the modulation of ER stress protein folding6, 19, 20. To further characterize ER stress and to investigate the role of Parkin in this integrated stress response program we explored cardiac remodeling in response to thoracic aortic constriction (TAC) and in response to tunicamycin-activated ER stress in Parkin knockout (KO) mice.
We found that Parkin levels were robustly increased following TAC in wild type (WT) mice and that Parkin deficient mice exhibit exaggerated cardiac hypertrophy and contractile dysfunction in response to TAC. In parallel to this adverse remodeling, the C/EBP homologous protein (CHOP), a known regulator of ER stress initiated apoptosis, was disproportionally increased in Parkin KO mice. Furthermore, we identified CHOP as a Parkin ubiquitylation substrate and showed that CHOP turnover is regulated by Parkin. Additionally excess ER stress was evident in human primary fibroblasts from subjects with genetic mutations in PARK2 and in Parkin deficient iPSC-derived cardiomyocytes. Together these data show that Parkin modulates ER stress via the post-translational control of CHOP levels to blunt adverse remodeling in response to pressure-overload and in response to systemic ER stress.
Results
Parkin depletion enhances cardiac hypertrophy and contractile dysfunction in response to pressure-overload
To explore the role of Parkin in adaptation to pressure-overload Parkin WT and KO mice were subjected to TAC followed by sequential imaging of cardiac structure and function over a 14-week period. By 10 weeks it was noted that the KO mice had evidence of exaggerated left ventricular dilatation (Fig. 1A) and worsening ejection fraction (45.14 ± 4.07% in KO-TAC vs. 49.24 ± 3.9% in WT-TAC) (Fig. 1B). By 14 weeks of TAC, the KO mice showed evidence of excessive hypertrophy and concomitant reduction in ejection fraction (39.32 ± 2.57% in KO-TAC vs. 46.91 ± 6.14% in WT-TAC) as measured by cardiac MRI (Fig. 1B–D). This measurement was confirmed by the direct assessment of cardiac mass indexed to tibial length (Supplemental Fig. S1A) and by histologic assessment of left-ventricular wall dimensions (Supplemental Fig. S1B). Quantifications of these changes are shown in supplemental Table S1. To assess whether the fetal gene program was differentially regulated in KO mice, transcript levels of genes encoding contractile proteins and cardiac natriuretic peptides were assessed. The fetal reprogramming of the contractile genes was activated to a similar extent in both strains with the development of cardiac hypertrophy (Supplemental Fig. S1C–F). However, in keeping with the increased ventricular dilatation, the transcripts encoding atrial natriuretic and brain natriuretic proteins (ANP and BNP), which are secreted in response to excessive chamber wall stress and fluid overload, were induced to a significantly greater extent in KO mice (Supplemental Fig. S1G,H).
Figure 1.
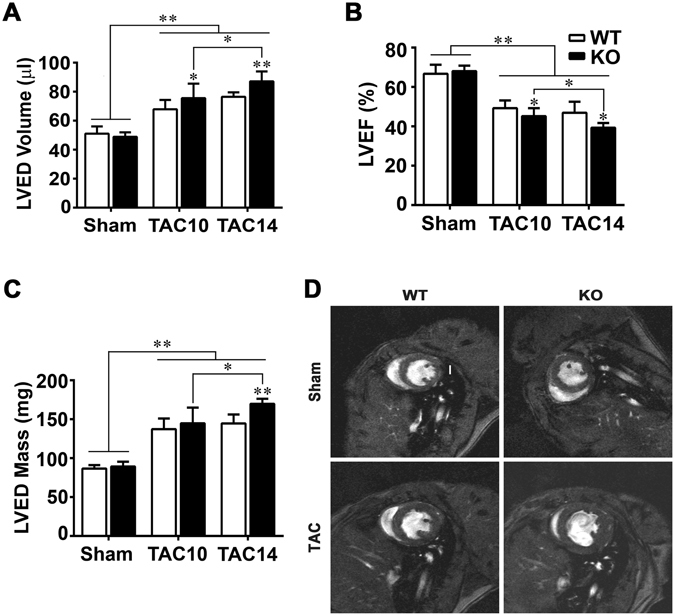
Adverse cardiac remodeling is exaggerated in the absence of Parkin. MRI quantification of (A) left ventricular end diastolic (LVED) volume, (B) LV ejection fraction (EF), and (C) LVED myocardial mass at 10 and 14 weeks following TAC. Data are shown as means ± SD (n = 6–12 per group). *p < 0.05 and **p < 0.01, compared to the corresponding controls. (D) Representative ventricular short-axis MRI images at end-diastole in mice at 14 weeks post-procedure. MRI Image was prepared 480 × 500 pixels resolution is 117 × 117 sq. micron in plane. The vertical white scale bar in the WT sham image represents 1-mm.
Diverse stress signatures are exaggerated following pressure-overload in Parkin KO mice
Oxidative stress and lipid peroxidation end products contribute to cardiac remodeling and play a role in ER stress signaling and apoptosis21–23. We therefore evaluated whether signatures of these modifications were modified in the absence of Parkin. We found that the extent of nitrotyrosine (Supplemental Fig. S2A,B) and 4-hydroxynonenal (Supplemental Fig. S2C,D) modifications of myocardial proteins were significantly higher in KO mice (2.6-fold and 2.0-fold vs. WT-TAC, respectively).
In light of the role of Parkin in stress-induced mitophagy, we explored mitophagy signatures in isolated mitochondria from sham and TAC mice. Here, the mitochondrial accumulation of an autophagy mediator (the lipidated form of the microtubule-associated protein 1A/1B-light chain 3 (LC3-II)) and the autophagy receptor (e.g. p62)24 were increased in mitochondria in response to TAC irrespective of the genotype. However, and consistent with the role of Parkin in mitophagy control the relative levels of accumulation of LC3-II and p62 were modestly reduced in the TAC KO mice (Supplemental Fig. S2E–G). Despite this, the mitochondrial content was not appreciably different, given that the mitochondrial DNA copy number (Supplemental Fig. S2H), and the levels of the mitochondrial proteins including medium chain acetyl-CoA dehydrogenase and cytochrome c were not differentially expressed in response to TAC comparing the two genotypes (Supplemental Fig. S2I,J). Additionally there was also no discernable phenotype difference in mitochondrial structural integrity in the sham or TAC mice by electron microscopy (Supplemental Fig. S2K).
Pressure-overload induced ER stress is increased in Parkin KO mice hearts
To begin to explore ER stress directly we then assayed Parkin and ER stress levels in WT mice and found that Parkin and ER stress regulatory proteins were highly upregulated at the transcript and protein levels in response to TAC (Supplemental Fig. S3A,B). Comparing these data to the expression in Parkin KO hearts, it was interesting to note that the transcript levels of the genes encoding CHOP, Activating Transcription Factor 4 (ATF4), Bip/Grp78, and Activating Transcription Factor 6 (ATF6) were all significantly higher in WT compared to KO mice hearts in response to TAC (Fig. 2A). The steady-state protein levels of Bip/Grp78, ATF4 and cleaved ATF6 were similarly induced by pressure-overload in the two genotypes (Fig. 2B), as were the transcript level of spliced XBP1 (Supplemental Fig. 3C). In contrast, steady-state protein levels of ER stress regulatory protein CHOP was disproportionately more robustly increased in the TAC KO mice hearts (Fig. 2B and Supplemental Fig. S3D). As excess CHOP orchestrates maladaptive ER stress pathways15 and the genetic depletion of CHOP ameliorates maladaptive pressure-overload remodeling in the heart25, we evaluated whether cognate downstream transcriptional CHOP targets were differentially regulated in Parkin KO mice. Here we found the CHOP targets linked to apoptosis including GADD34, DR5, TRB3 and Bim15 were excessively induced at 14 weeks following TAC in the KO mice (Fig. 2C). Furthermore, as CHOP activation can result in adverse effects via PARP cleavage26, we assessed the extent of this modification in response to TAC. Consistent with the canonical upstream events, the extent of PARP cleavage was markedly higher in TAC Parkin KO mice (Fig. 2D and Supplemental Fig. S3E).
Figure 2.
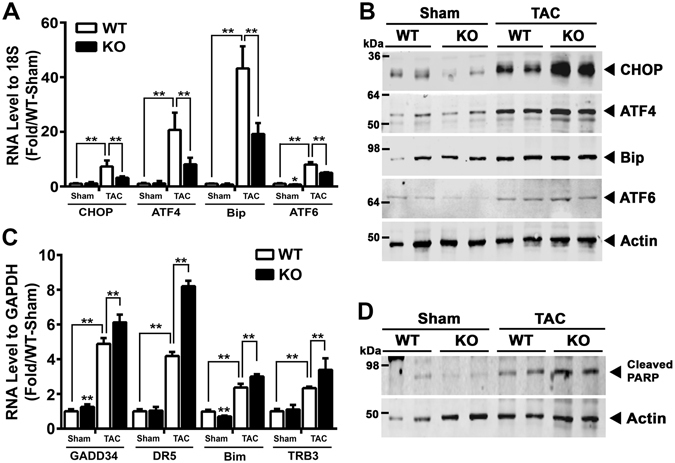
Parkin modulates ER stress during adverse remodeling in response to pressure-overload. (A) Quantification of ER stress responsive transcript (CHOP, ATF4, Bip and ATF6) levels in Parkin WT and KO mouse hearts in response to TAC. (B) Representative immunoblot showing expression of ER stress responsive proteins (CHOP, ATF4, Bip, and ATF6) in response to TAC. (C) RNA expression levels of CHOP target genes (GADD34, DR5, Bim and TRB3) following 14 weeks of TAC. Mean ± SD. **P < 0.01, vs. controls (n = 6 per group). (D) Representative immunoblot of cleaved PARP levels in Parkin WT and KO mouse hearts in response to TAC. The immunoblots in Fig. 2B and D are cropped images and the full membranes are depicted in a supplemental file.
CHOP is a Parkin substrate and its degradation is Parkin and ubiquitin proteasome dependent
We next investigated the putative regulatory effects of Parkin on CHOP biology. We firstly assessed the protein interaction between Parkin and CHOP. Cotransfection and bidirectional co-immunoprecipitation studies showed robust interactions between CHOP and Parkin (Fig. 3A,B). Interestingly, the N-terminal alpha-helical domain of CHOP, which is known to bind to other regulatory proteins27, 28, was found to be the required domain for its interaction with Parkin (Fig. 3C). Morover, in high Parkin-expressing SH-SY5Y cells, the endogenous interaction of Parkin with CHOP was found in response to ER stress induction (Fig. 3D).
Figure 3.
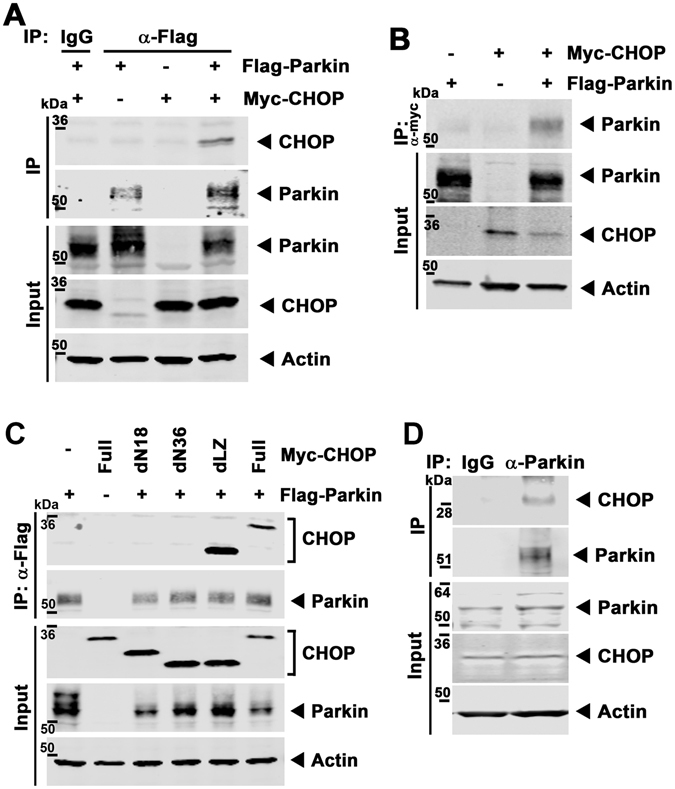
Parkin interacts with CHOP. (A) Representative gel by immunoprecipition with anti-Flag M2 agarose and immunoblotting with an anti-CHOP antibody in HL-1 cells transfected with pcDNA-Myc-CHOP constructs and p3 × Flag-Parkin. (B) Representative immunoblot with the reciprocal pull-down with anti-Myc agarose and immunoblot analysis with Parkin antibody. (C) Representative immunoblot for physical interaction in 293 cells transfected with the various deletion pcDNA-Myc-CHOP constructs and p3 × Flag-Parkin following immunoprecipitation with anti-Flag M2 agarose and immunoblot analysis with an anti-CHOP and Parkin antibody. dN18, 18-amino acid deletion of N-terminal CHOP; dN36, 36-amino acid deletion of N-terminal CHOP; dLZ, Leucine-Zipper domain deletion of CHOP. (D) Representative immunoblot with the endogenous protein interaction in thapsigargin-treated SH-SY5Y cells following pull-down with Parkin antibody and immunoblot analysis with CHOP and Parkin antibody. The immunoblots in this figure are cropped images and the full membranes are depicted in a supplemental file.
As TAC elicits excess CHOP in the absence of Parkin we then explored the effect of Parkin overexpression on endogenous CHOP levels in HL-1 cardiomyocytes. Here we found that Parkin, in a dose-dependent manner, progressively diminished steady-state levels of endogenous CHOP (Fig. 4A) without the modulation of the corresponding transcript levels (Supplemental Fig. S4A). To explore whether Parkin modulated CHOP protein levels in direct response to ER stress, Parkin shRNA HL-1 cells were exposed to TM. Here CHOP levels were more robustly induced in Parkin depleted cells (Fig. 4B and Supplemental Fig. S4B) without any difference in the induction of CHOP transcript levels (Supplemental Fig. S4C). To evaluate whether increased Parkin levels increased CHOP turnover, Parkin was overexpressed in TM treated cells in the presence or absence of the translational inhibitor, cycloheximide (CHX). In the presence of the CHX, the turnover of endogenous CHOP was significantly increased in the presence of Parkin (Fig. 4C and Supplemental Fig. S4D).
Figure 4.
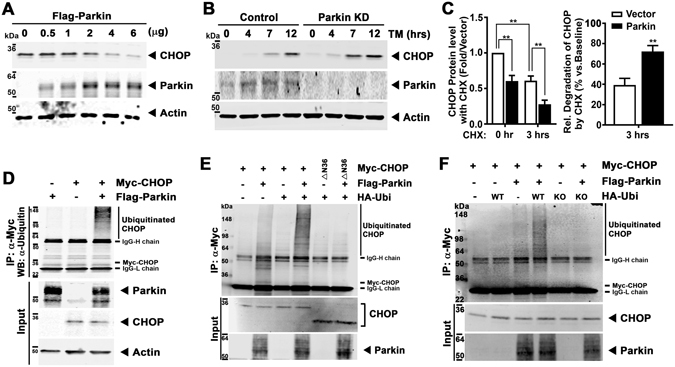
Parkin poly-ubiquitylates CHOP protein and modulates CHOP steady-state levels. (A) Degradation of endogenous CHOP with increasing doses of Parkin overexpression after TM administration for 12 hrs. (B) Immunoblot of temporal levels of endogenous CHOP in response to TM administration in control shRNA and Parkin shRNA HL-1 cells. (C) Quantification of endogenous CHOP protein level at 3 hrs of cycloheximide (CHX - 5 μg/ml) incubation in control and Parkin overexpressing cells compared to no CHX incubation. The cells are exposed to TM for 12 hrs followed by CHX. Mean ± SD. **P < 0.01, vs. controls. (D) Ubiquitylation of CHOP in HL-1 cells transfected with constructs of Myc-CHOP and Flag-Parkin following immunoprecipitation with anti-Myc agarose and immunoblot analysis with an ubiquitin antibody. (E) Ubiquitylation of CHOP in HL-1 cells transfected with constructs of Myc-CHOP, Flag-Parkin, and HA-Ubi (WT) following immunoprecipitation with anti-Myc agarose and immunoblot analysis with an ubiquitin antibody. △N36, 36-amino acid deletion of N-terminal CHOP. (F) Ubiquitylation of CHOP in HL-1 cells transfected with constructs of Myc-CHOP, Flag-Parkin, and HA-Ubi (WT and KO) following immunoprecipitation with anti-Myc agarose and immunoblot analysis with an ubiquitin antibody. HA-Ubi-KO, Lysine residues were substituted for arginine. The immunoblots in this Figure are cropped images and the full membranes are depicted in a supplemental file.
As Parkin is an E3-ubiquitin ligase we then explored whether the ubiquitylation of CHOP was modulated by Parkin. Immunoprecipitation of Myc-tagged CHOP with the subsequent immunoblot analysis with an ubiquitin antibody showed that excess Parkin increased ubiquitylation of CHOP with a pattern consistent of polyubiquitylation (Fig. 4D). Similarly, the co-expression of Parkin with ubiquitin, polyubiquitylated wildtype CHOP but not not the ΔN36 CHOP construct which we previously showed negated CHOP interaction with Parkin (Fig. 4E). In addition, the substitution of ubiquitin lysine residues with arginine similarly abolished Parkin mediated polyubiquitylation of CHOP (Fig. 4F). In parallel, and consistent with CHOP degradation by the ubiquitin-proteasome pathway, the exposure to the proteasome inhibitor MG132 increased CHOP levels in WT and Parkin KD cells. This being in contrast to the lack of increased CHOP levels by inhibition of the endosome/lysosomal degradation pathway by bafilomycin and/or chloroquine (Supplemental Fig. S4E,F).
Parkin levels modulate cell death susceptibility in HL-1 cells
Given the effect of Parkin on CHOP levels and following our observations of increased PARP cleavage in TAC Parkin KO hearts we then exposed HL-1 cardiomyocytes to TM to evaluate their susceptibility to apoptosis. Parkin knockdown HL-1 cells showed increased PARP and caspase 3 cleavage (Fig. 5A and Supplemental Fig. S5A) and increased annexin V staining vs. control shRNA HL-1 cells (Fig. 5B). The reconstitution of Parkin in the stable KD HL-1 cells diminished cell death as measured by the reduction in PARP and caspase 3 cleavage (Fig. 5C and Supplemental Fig. S5B,C), Annexin V staining, and reactive oxygen species levels compared to Parkin KD cells (Fig. 5D and Supplemental Fig. S5D,E). In parallel, the concurrent siRNA knockdown of CHOP in Parkin deplete HL-1 cells reversed the detrimental effects of tunicamycin on reactive oxygen species levels and cell death (Fig. 5E and Supplemental Fig. S5F,G).
Figure 5.
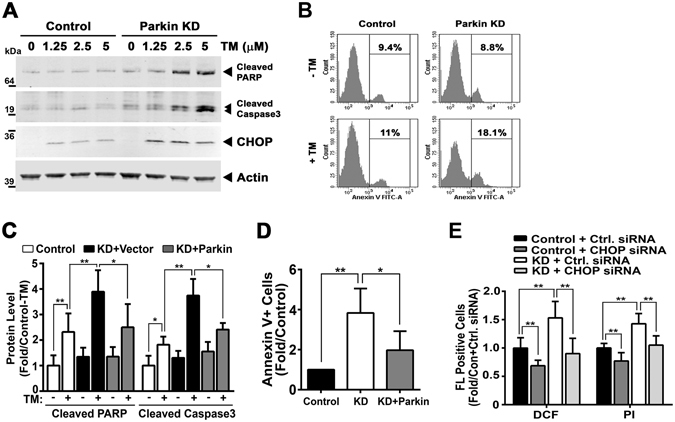
Parkin knockdown HL-1 cells to Tunicamycin show increased their susceptibility to apoptosis. (A) Immunoblot of the Cleaved PARP and Caspase3 protein after increasing doses of TM (0, 1.25, 2.5, and 5 μg/ml) for 24 hrs in control and Parkin KD HL-1 cells. (B) Representative flow cytometry image showing the increased Annexin V staining after TM administration (50 μg/ml) in Parkin KD cells vs. control shRNA HL-1 cells. (C) Quantification of PARP and caspase 3 cleavage level after reintroducing wildtype Parkin following the transfection of 2 µg of the Parkin expression plasmid. (D) Histogram showing the reduced Annexin V staining after TM in Parkin KD cells following the reconstitution of Parkin. (E) Histogram showing the relative reduction in DCF and PI staining in HL-1 cells in response to TM comparing Parkin to Parkin and CHOP knockdown. **P < 0.01, compared to the corresponding controls. The immunoblots in Fig. 5A are cropped images and the full membranes are depicted in a supplemental file.
Cardiac dysfunction is exaggerated by ER stress in Parkin KO mice
To directly evaluate whether ER stress itself differentially exacerbated cardiac dysfunction WT and Parkin KO mice were exposed to intraperitoneal TM. In concordance with the TAC studies, the KO mice showed greater left ventricular dilatation and a reduction in ejection fraction (Fig. 6A,B and Supplemental Table S2). This adverse remodeling was accompanied by higher levels of CHOP and Bip/Grp78 in the KO compared to WT murine hearts in response to TM administration (Fig. 6C,D). This effect also appeared to be due to the preferential induction of apoptosis, as measured by caspase 3 cleavage (Fig. 6E,F).
Figure 6.
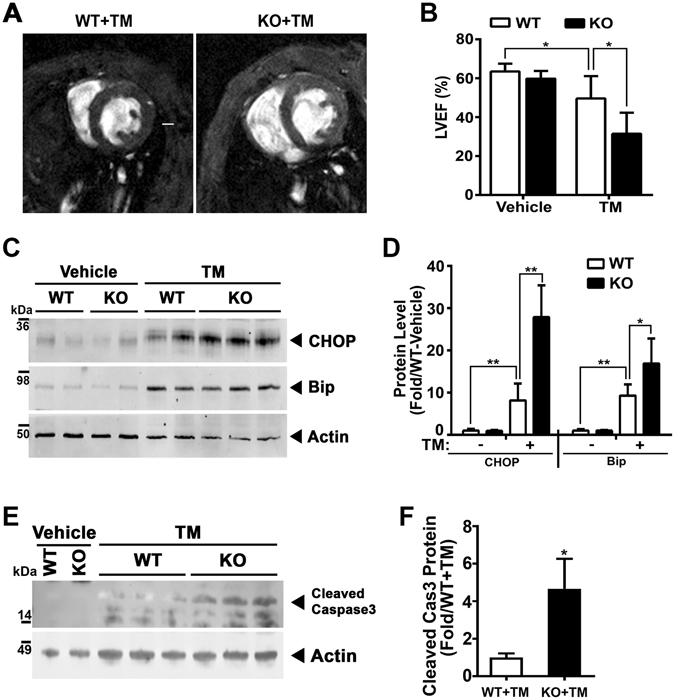
ER stress perturbations exacerbate cardiac dysfunction of Parkin KO mice. (A) Representative MRI image shows the cross section of heart of Parkin WT and KO mice 48 hrs. after systemic Tunicamycin (TM) administration. The white scale bar in the WT + TM image represents 1-mm. (B) MRI analysis of LVEF in WT and Parkin KO mice following the IP administration of Tunicamycin (2 mg/kg body weight). Mean ± SD. *P < 0.05, vs. controls. (n = 4–6 per group). Representative immunoblot (C) and histogram (D) showing the quantitation of CHOP and Bip/Grp78 in LV tissue extracted from the TM treated mice. (E) Representative immunoblot of the cleaved Caspase3 in LV tissue extracted from the TM treated mice. (F) Histogram showing the quantitation of cleaved Caspase 3. Mean ± SD. *p < 0.05 and **p < 0.01, compared to the WT-Vehicle controls (n = 4 per group). The immunoblots in Fig. 6C and E are cropped images and the full membranes are depicted in a supplemental file.
Evidence of Parkin-dependent CHOP levels in human samples and the induction of this ER stress program in non-ischemic dilated cardiomyopathy
We then determined whether this role of Parkin is operational in skin fibroblasts extracted from individuals with early onset Parkinson disease with associated PARK2 mutations compared to control subjects. Basal CHOP levels were low and induced by TM with a significantly exaggerated CHOP induction in PARK2 mutant subject samples (Fig. 7A and Supplemental Fig. S6). Since Parkin levels are low in human fibroblasts, we then went on to assay whether this Parkin-CHOP regulatory interaction was operational in control and isogenic Parkin mutant inducible pluripotential stem cell (iPSC) derived cardiomyocytes. Here too, the disruption of Parkin resulted in elevated CHOP levels following exposure to TM in parallel with evidence of elevated PARP and Caspase-3 cleavage (Fig. 7B,C). The differentiation of iPSC’s into cardiomyocytes was evident by increased levels of the myocardial contractile protein including troponins I and T (Supplemental Fig. S7A). The CRISPR targeted disruption of Parkin was confirmed at both the sequence and steady-state protein levels (Supplemental Fig. S7B).
Figure 7.
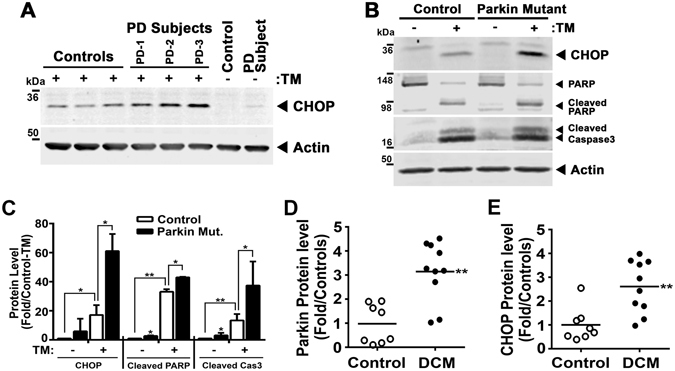
ER stress is evident in human Parkin mutant tissues. (A) Representative immunoblot of steady-state CHOP levels in controls and PARK2 mutant primary human fibroblasts exposed to TM (5 μg/ml) for 7 hrs. PD-1, deletion of Exon 3–4 and deletion of 255A; PD-2, mutation of GLN34ARG; PD-3, deletion of Exon 3 and mutation of ARG42PRO. Representative immunoblot (B) and histogram (C) showing the quantitation of CHOP Cleaved PARP, and Cleaved Caspase3 in the iPS cell-derived cardiomyocytes exposed to TM (5 μg/ml) for 48 hrs. Cardiomyocytes are differentiated from control and Parkin mutant induced pluripotent stem cells (iPSC’s). Data are shown as mean ± SD. *P < 0.05 and **P < 0.01, vs. control cells. Relative protein expression of Parkin (D) and CHOP (E) in 10 human dilated cardiomyopathy (DCM) heart tissues and 8 control subjects by immunoblot analysis. **P < 0.01, vs. control subjects. The immunoblots in Fig. 7A and B are cropped images and the full membranes are depicted in a supplemental file.
Finally, to evaluate whether this regulatory program may be operational in the human heart, we explored the protein levels of Parkin and CHOP in explanted hearts from unused control donor and from dilated cardiomyopathy patients. In parallel to the findings in WT mice in response to TAC, Parkin and CHOP levels were induced in the dilated cardiomyopathy heart samples, further supporting the activation of this program in human cardiomyopathy (Fig. 7D,E).
Discussion
Cardiac remodeling in response to pressure-overload activates a wide array of efficiency and quality control programs including: alterations in contractile protein biology; the modulation of metabolic pathways to sustain bioenergetics and to control/ameliorate redox stressors; and the capacity for distinct organelles within the myocardium to orchestrate self-preservation and repair. Endoplasmic reticulum is one such organelle implicated in biomechanical stress adaptation, although mediators regulating the ER response are not as well characterized as other remodeling programs29. In concert with prior studies that implicate a role for Parkin in the control of ER biology, we showed that the absence of Parkin exacerbated adverse pressure-overload induced cardiac remodeling and evoked excess myocardial ER stress and apoptotic signatures. Additionally, Parkin was found to directly interact with, and modulate the protein stability of the pivotal ER stress regulatory protein CHOP. These findings identify a new Parkin substrate in the modulation of cardiac adaptation, and identify this E3-ubiquitin ligase in the post-translational regulatory control of ER stress.
The ER is responsible for protein folding, maturation, and trafficking, as well as functioning as a critical organelle in the regulation of cellular calcium homeostasis30. In response to cellular stressors, the ER can exhibit short-term adaptive effects, mediated in part by the ER stress response and/or, when stressful triggers are excessive or chronically engaged, perpetuate damage via ER stress signaling and apoptosis15. This biphasic role of ER stress is operational in the heart as evident by protection against acute ischemia-reperfusion injury following modest pharmacologic ER stress induction31 and where the genetic depletion of CHOP attenuates adverse remodeling following chronic pressure-overload25. The role of Parkin in ER stress biology has not been extensively characterized, although Parkin has previously been shown to be induced by ER stress17, and its upregulation is controlled, in part, by the ER stress activating transcription factor ATF418. Additionally Parkin has been found to interact with CHIP and Hsp70 proteins to regulate ER protein folding19. In this study we found that the ER stress-induced transcription factor CHOP is a Parkin binding partner/substrate and that, in an ubiquitin/proteasome-dependent manner, Parkin modulates CHOP protein stability. Interestingly, adverse effects of CHOP can be alleviated when the adaptive pathways of ER stress are coordinately engaged32. In contrast to this parallel regulation, we found that in response to pressure-overload, the Parkin KO mice had diminished induction of the ER stress transcript levels with a disproportionate elevation in CHOP protein levels. In contrast, the induction of ATF4, ATF6 and Bip were similar between the WT and KO mice following TAC. These data, coupled with the in-vitro data showing that Parkin enhanced CHOP degradation and turnover in a dose-dependent manner, identify a new regulatory pathway in the control of chronic pressure-overload ER stress. The summation of these data support that the induction of Parkin in pressure-overload functions as an adaptive response to diminish CHOP levels to prevent/attenuate, in part, adverse ER-stress remodeling. Interestingly, additional regulatory proteins have been identified that control ubiquitin-proteosomal degradation of CHOP28, 33, suggesting potential context specific redundancy in the control of CHOP proteasomal degradation.
Parkin has previously been shown to mediate ameliorative effects via the attenuation of redox stress34, 35 and via the augmentation of mitochondrial autophagy11, 12. Although these adaptive programs were not the major focus of this study we did find evidence that these pathways were adversely affected in the absence of Parkin in response to chronic (14 weeks) cardiac pressure-overload. The apparent less robust contribution of mitophagy to this phenotype may reflect our temporal assessment of mitophagy signatures in this model. This is in contrast to where mitophagy had been found to be most active and important in the first week of cardiac adaptation to pressure overload36. At the same time it is conceivable that additional Parkin effects may be operational in cardiac remodeling in response to pressure-overload, including roles for Parkin in PARIS degradation as a mediator of mitochondrial biogenesis8, and/or in the maintenance of additional mitochondrial quality control programs7, 37–39. These pathways have not been investigated in this study and their role in this integrated adaptation would be useful to explore. In addition, and as opposed to the relatively extensive understanding of the regulatory control of Parkin activity in the mitochondria by the mitochondrial targeted PINK140, 41, activation of Parkin’s extramitochondrial functioning has been less well characterized. Emerging data shows that the phosphorylation and nitrosylation of Parkin may modulate these extramitochondrial activity42–44. However, in this manuscript, we have not identified the regulatory control of Parkin activation in response to pressure-overload or ER stress.
The findings from this study expand our understanding of the regulatory role of ER stress in pressure-overload induced cardiac remodeling. We focused on the regulation of CHOP by Parkin, given that excessive CHOP is linked to maladaptive ER stress. Our data suggests that ER stress induction of Parkin in response to cardiac pressure-overload is an adaptive regulatory effect and that the role of Parkin in modulation of the steady-state levels of CHOP may function to maintain adaptive rather than progression to maladaptive ER stress signaling. This concept is further supported by the induction of Parkin and CHOP in cardiac samples from patients with dilated cardiomyopathy and evidence of the Parkin-dependent CHOP expression and evidence of increased apoptotic signatures in iPSC derived cardiomyocytes exposed to ER stress. However, the most compelling data supporting the role of Parkin in the blunting of ER stress is evident when WT and KO mice were directly exposed to systemic tunicamycin. Here, the reduction in ejection fraction and dilatation of the left ventricle was profoundly more evident in the absence of Parkin and these KO mice showed evidence of increased apoptosis. The model depicting the role of Parkin in this regulatory pathway is shown as a schematic (Fig. 8). At the same time we found that mediators of ER stress control of protein folding are also excessively induced in response to pressure overload (data not shown). Although Parkin had previously been shown to modulate protein folding6, 19, 20 it’s role of this ER-regulated program in cardiac hypertrophic pathophysiology remains to be explored.
Figure 8.
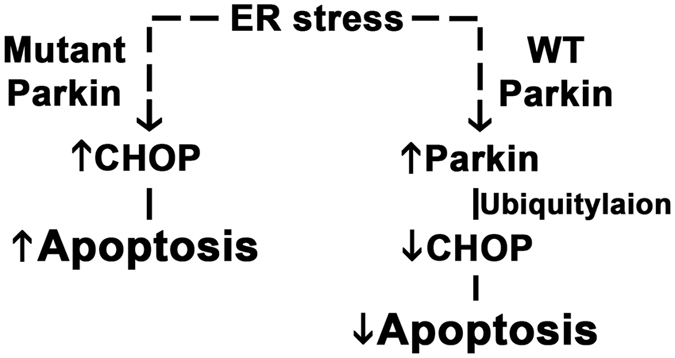
Working model of Parkin-mediated regulation during cardiac adaptation to pressure-overload.
We conclude that this study identifies a novel post-translational regulatory control node in the regulation of CHOP that, we postulate, may be operational to alleviate adverse effects of chronic ER stress activation. This program has been found to be active in response to cardiac pressure-overload and in human subjects with end-stage heart failure. Furthermore, these data identify an additional adaptive function of Parkin to attenuate adverse effects of chronic ER stress. Interestingly, ER stress is linked to Parkinson disease19, 45 and whether the loss of this ER stress regulatory control may contribute to increased susceptibility to early onset Parkinson disease in patients with mutations in PARK2 is an intriguing concept that warrants investigation.
Methods
Animal Models and Human Tissues
Parkin knockout mice (129/Sv × C57BL/6 background) were obtained from the Jackson Laboratory and backcrossed (>10 generations) into the C57BL/6J strain. Tunicamycin (2 mg/kg body weight) was administrated by intraperitoneal injection and the cardiac contractile dysfunction was measured 48 hrs later. All animal procedures were conducted using an animal protocol that was approved by the National Heart Lung, and Blood Institute (NHLBI) Animal Care and Use Committee and all experiments were performed in accordance with NIH adopted National Research Council 2011 Guide for the Care and Use of Laboratory Animals. The diseased human heart samples were obtained from the Duke University Heart Transplant Program and the control samples from the National Disease Research Interchange (NDRI) tissue collection facility in Philadelphia, PA, USA. The National Institutes of Health (NIH) Office of Human Research Protections (OHRP) granted an exemption from IRB oversight for the scientific analysis of these deidentified human tissue samples.
Transverse Aortic Constriction (TAC)
Parkin male mice at 14–15 weeks of age were subjected to TAC or sham surgeries. Briefly, the left chest of each mouse was opened to identify the thoracic aorta by blunt dissection at the second intercostal space after anesthetization with 1–3% Isoflurane and the absence of any toe pinch reflex. We then performed TAC using 7–0 silk sutures to band the thoracic aorta against 26-gauge needle for Parkin mice. The needle was removed before closing the thoracic cavity. A similar procedure, without constricting the aorta, was performed in the sham-operated group.
Cell Culture Studies and Flow Cytometry
HL-1 cardiomyocytes (a gift from Dr. Claycomb) were cultured in 0.02% gelatin/12.5 mg/ml fibronectin-coated culture dishes with Claycomb medium (Sigma) containing 10% fetal bovine serum, 0.1 mM norepinephrine, and 2 mM L-glutamine with exchange of fresh media every three days46. 293 (human embryonic kidney cells) and SH-SY5Y (human neuroblastoma) cells were obtained from ATCC and the primary human fibroblasts from the Parkinson’s disease (PD) patients with PARK2 mutation were purchased from Coriell Institute47. For flow cytometry analysis, HL-1 cells were incubated with Annexin V-FITC (Fluorescein isothiocyanate, eBioscience) and Propidium Iodide (PI, eBioscience) for 15 mins and 10 µM DCF (2′,7′-dichlorofluorescein, Invitrogen) for 1 hr, respectively and then were analyzed using the FACSCanto (BD).
Isogenic iPSC-derived Cardiomyocytes
A gRNA sequence (TGCTAAGCGACAGG GGGTTC, followed by “CGG” PAM sequence) was selected from Exon 2 of human PARK2 gene by using online CRISPR design software at crispr.mit.edu with high ranking/specificity scores48. To enhance CRISPR/Cas9 editing efficiency, the gRNA sequence was cloned into a modified pX458 all-in-one CRISPR/Cas9 vector49, where CBh promoter was replaced with CAG promoter and the first nucleotide of gRNA was replaced with G50. Surveyor NHEJ assay was used to confirm that resultant PARK2-targeting CRISPR/Cas9 vector (pCAG458-PARK2-gRNA1) edited human 293T cells at the efficiency of ~39.5% (data not shown). Then 1 million ND1.4 human iPSCs were transfected with 5 ug pCAG458-PARK2-gRNA1 vector using lipofectamine 3000 (ThermoFisher) and following the manufacturer’s manual. Two days later, the GFP+ transfected iPSCs were sorted and plated on 10-cm Matrigel coated tissue culture plate at 10,000 cells/plate with 10 µM Rock inhibitor and E8 medium (ThermoFisher). Two weeks later, small single colonies were picked, expanded, and screened for PARK2 editing using PCR primers (F: 5′-gtttgcaggtcactgacgaa and R: 5′-ggaatccccaggcactattt). The gene-edited clones with homozygous or compound heterozygous indels were further confirmed by TOPO cloning and sequencing to select those with frame-shift indels that generate premature stop codon.
To induce cardiomyocyte differentiation from human iPSCs, 80% confluent iPSCs grown on Matrigel were cultured with basal differentiation medium (BDM) which contains E8 medium minus bFGF, TGF-β, and insulin, but plus 1X chemically defined lipid concentrate (Life Technologies) and 1x Pen Strep (Life Technologies). For the first 24 hrs of induction, 5 µM GSK3-beta inhibitor CHIR99021 (Tocris) was added to BDM. From day 2 to day 5, 3 µM Wnt inhibitor IWP2 (Tocris) and 3 µg/ml heparin (Sigma) were added to BDM. From day 7, >90% cells differentiated into beating cardiomyocytes expressing cardiac Troponin T and 20 µg/ml insulin (Sigma) was added to BDM to maintain cardiomyocytes with medium change every 2–3 days.
Plasmids Transfection and Transduction
pcDNA plasmid encoding WT-Parkin was obtained as gifts from Dr. Olga Corti (Pitié-Salpêtrière Hospital, Paris, France) and then subcloned into p3xFlag vectors (Sigma). pcDNA-Myc-CHOP plasmids were purchased from Addgene and we then generated three deletion constructs of CHOP with 18-amino acid deletion of N-terminal (dN18), 36-amino acid deletion of N-terminal (dN36), and Leucine-Zipper domain deletion (dLZ). Transfection was performed using 1–2 µg of these plasmids with X-tremeGene HP (Roche) and Amaxa nucleofector (Lonza) according to manufacturer’s instructions. For stable knockdown of Parkin in HL-1 cells, cells were transducted with Lentivirus carrying mouse Parkin shRNA (SHCLNV-NM_016694, Sigma) and were selected with medium containing 1 µg/ml puromycin for 4 weeks. For knockdown of CHOP in HL-1 cells, cells were transfected with 400 pmol Control and Ddit3 SMARTpool siRNA (Dharmacon) with X-tremeGene siRNA (Roche).
MRI Imaging
Cardiac MRI was used to quantify RV and LV function and morphology in the mice as previously described51. In brief, experiments were carried out in a 7.0T, 16 cm horizontal Bruker MR imaging system (Bruker) with Bruker ParaVision 5.1 software. Mice were anesthetized with 1.5–2.5% isoflurane and imaged with ECG and respiratory detection using a 35 mm m2m birdcage volume coil (m2m Imaging). Magnevist (Bayer Healthcare) diluted 1:10 with sterile 0.9% saline, was administered IV at a dose of 0.3 mmol Gd/kg body weight. Short axis gradient echo cine images of the heart were acquired (TR/TE = 10–11/3.4 ms, 4 averages, 1.0 mm slice thickness, 2.8–3.0 cm field of view, 256 × 256 matrix, 8–10 slices, respiratory and ECG-gated). Images were processed with CAAS MRV FARM software (Pie Medical Imaging) to determine left and right ventricle (LV and RV) ejection fraction, related functional parameters, LV and RV volumes and myocardial mass.
Immunoblot and Immunoprecipitation analyses
Immunoprecipitation (IP) and immunoblot analysis were performed as described2. Total proteins from cells and tissues were extracted using RIPA buffer (50 mM Tris-HCl, pH 8.0, 0.5% deoxycholic acid, 1% NP-40, 0.1% sodium dodecyl sulfate and 0.5 M NaCl) supplemented with protease inhibitor cocktails and phosphatase inhibitors (Sigma). The lysates were separated by 4–20% Tris-Glycine or 4–12% Bis-Tris gel (Invitrogen) and transferred to nitrocellulose or PVDF membranes. Antibodies were purchased from Cell signaling (Parkin, Bip/Grp78, Ubiquitin, Nitrotyrosine, PARP, Caspase 3, VDAC, Cytochrome C, and GAPDH), Sigma (LC3), Progen (p62), Oxis international (4-hydroxynonenal, 4-HNE), Santa Cruz Biotechnology (CHOP, ATF4, Myc, and Troponin I), Novus (MCAD), Proteintech (ATF6), and Millipore (Actin). For immunoprecipitation (IP), cells were extracted using the lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 0.5% NP-40 and 300 mM NaCl) containing protease inhibitor cocktails and phosphatase inhibitors. Protein extract was incubated with antibodies against Myc or anti-Flag M2 agarose (Sigma)/anti-myc agarose (Pierce) overnight at 4 °C. For antibody conjugates, protein G agarose (Sigma) was added and incubated for 4 hrs. The agarose beads were washed with the lysis buffer and the supernatant with loading buffer was used for immunoblot analysis. For crosslinking protein interaction analysis, SH-SY5Y cells were treated with 25 mM thapsigargin for 5 hrs and crosslinked with 0.4% PFA for 20 mins on ice. The crosslink reaction was stopped with 125 mM glycine solution and the cells were lysed in Co-IP buffer (20 mM Tris-HCl pH 8, 137 mM NaCl, 1% NP-40, 2 mM EDTA, and 10% Glycerol). Parkin monoclonal antibody (Cell Signaling) and IgG antibody as control was added and incubated overnight at 4 °C. For antibody conjugates, sera-mag protein A/G magnetic beads (GE Healthcare Life Sciences) were incubated for 4 hrs and then washed with wash buffer (10 mM Tris-HCl pH 7.4, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, and 0.5% Triton X-100).
Quantitative Real-time PCR
Quantitative real-time PCR was performed as described47. Total RNA was isolated from heart using Tripure isolation reagent (Roche). Reverse transcription of total RNA was performed using Superscrip III first-strand synthesis system (Invitrogen) according to the manufacturer’s protocol. Quantitative real-time PCR was performed using SYBR green PCR master mix (Applied Biosystems and Roche) and run on Opticon2 DNA engine (Bio-Rad) and Light cycler 96 systems (Roche). All reactions were normalized using GAPDH, TBP, or 18S. Primers were purchased from predesigned primers of QuantiTect primer assays (Qiagen)52.
Statistical Analyses
Data are expressed as means ± SD for the indicated number of observations. Statistical significance between groups was determined using two-tailed Student’s test when analyzing the response between groups. Multiple comparison analysis was performed using ANOVA. P value < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
We thank Dr. Olga Corti from Pitié-Salpêtrière Hospital (France) for the pcDNA-Parkin/∆Ubi constructs, Dr. William C. Claycomb from Louisiana State University Medical Center (New Orleans, LA) for the HL-1 cells and assistance from the NHLBI Electron Microscopy (Camron Keshavarz and Erin Stempinski), the murine phenotyping core (Audrey Noguchi) and Flow Cytometry (J. Phil McCoy Jr.) Core facilities. The ND1.4 iPSC line was obtained from the laboratory of Dr. James Thomson from the University of Wisconsin. The Troponin T monoclonal antibody was developed by the Developmental Studies Hybridoma Bank, and created for the NICHD of the NIH and maintained at The Department of Biology of The University of Iowa, Iowa City. This research was supported by the Division of Intramural Research program of the NHLBI and by funding from the Michael J. Fox Foundation (MNS).
Author Contributions
K.H., S.H. and M.N.S. designed experiments; K.H., S.H., K.S., S.M., T.T.N., M.V.S., A.N., H.S., and S.A.A. performed experiments; Y.L., J.Z. and E.M. provided samples and reagents; S.A.A. and E.M. made conceptual and editorial contributions and K.H. and M.N.S. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-02339-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Huynh DP, Dy M, Nguyen D, Kiehl TR, Pulst SM. Differential expression and tissue distribution of parkin isoforms during mouse development. Brain research. Developmental brain research. 2001;130:173–181. doi: 10.1016/S0165-3806(01)00234-6. [DOI] [PubMed] [Google Scholar]
- 2.Cesari R, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5956–5961. doi: 10.1073/pnas.0931262100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimura H, et al. Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Annals of neurology. 1999;45:668–672. doi: 10.1002/1531-8249(199905)45:5<668::AID-ANA19>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson BN, Berger AK, Cortese GP, Lavoie MJ. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6283–6288. doi: 10.1073/pnas.1113248109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. The Journal of biological chemistry. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 7.Muller-Rischart AK, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Molecular cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 8.Shin JH, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 10.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature reviews. Molecular cell biology. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubli DA, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. The Journal of biological chemistry. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang C, et al. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PloS one. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong G, et al. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459–aad2459. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maron BJ, Ferrans VJ, Roberts WC. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. The American journal of pathology. 1975;79:387–434. [PMC free article] [PubMed] [Google Scholar]
- 15.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nature cell biology. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okada K, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 17.Wang HQ, Imai Y, Kataoka A, Takahashi R. Cell type-specific upregulation of Parkin in response to ER stress. Antioxidants & redox signaling. 2007;9:533–542. doi: 10.1089/ars.2006.1522. [DOI] [PubMed] [Google Scholar]
- 18.Bouman L, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell death and differentiation. 2011;18:769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai Y, et al. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Molecular cell. 2002;10:55–67. doi: 10.1016/S1097-2765(02)00583-X. [DOI] [PubMed] [Google Scholar]
- 20.Imai Y, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/S0092-8674(01)00407-X. [DOI] [PubMed] [Google Scholar]
- 21.Ago T, et al. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation research. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sciarretta S, et al. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circulation research. 2013;113:1253–1264. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin MH, et al. Lipid peroxidation end product 4-hydroxy-trans-2-nonenal triggers unfolded protein response and heme oxygenase-1 expression in PC12 cells: Roles of ROS and MAPK pathways. Toxicology. 2014;315:24–37. doi: 10.1016/j.tox.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Webster BR, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. Journal of cell science. 2013;126:4843–4849. doi: 10.1242/jcs.131300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu HY, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–369. doi: 10.1161/CIRCULATIONAHA.109.917914. [DOI] [PubMed] [Google Scholar]
- 26.Lim MP, Devi LA, Rozenfeld R. Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell death & disease. 2011;2:e170. doi: 10.1038/cddis.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohoka N, Hattori T, Kitagawa M, Onozaki K, Hayashi H. Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. The Journal of biological chemistry. 2007;282:35687–35694. doi: 10.1074/jbc.M703735200. [DOI] [PubMed] [Google Scholar]
- 28.Jeong K, et al. Cyclophilin B is involved in p300-mediated degradation of CHOP in tumor cell adaptation to hypoxia. Cell death and differentiation. 2014;21:438–450. doi: 10.1038/cdd.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annual review of physiology. 2013;75:49–67. doi: 10.1146/annurev-physiol-030212-183707. [DOI] [PubMed] [Google Scholar]
- 30.Minamino T, Kitakaze M. ER stress in cardiovascular disease. Journal of molecular and cellular cardiology. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 31.Petrovski G, et al. Cardioprotection by endoplasmic reticulum stress-induced autophagy. Antioxidants & redox signaling. 2011;14:2191–2200. doi: 10.1089/ars.2010.3486. [DOI] [PubMed] [Google Scholar]
- 32.Woo CW, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nature cell biology. 2009;11:1473–1480. doi: 10.1038/ncb1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi Y, Xia P. Cellular inhibitor of apoptosis protein-1 (cIAP1) plays a critical role in beta-cell survival under endoplasmic reticulum stress: promoting ubiquitination and degradation of C/EBP homologous protein (CHOP) The Journal of biological chemistry. 2012;287:32236–32245. doi: 10.1074/jbc.M112.362160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuroda Y, Mitsui T, Kunishige M, Matsumoto T. Parkin affects mitochondrial function and apoptosis in neuronal and myogenic cells. Biochemical and biophysical research communications. 2006;348:787–793. doi: 10.1016/j.bbrc.2006.06.201. [DOI] [PubMed] [Google Scholar]
- 35.Palacino JJ, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. The Journal of biological chemistry. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 36.Shirakabe A, et al. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lutz AK, et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. The Journal of biological chemistry. 2009;284:22938–22951. doi: 10.1074/jbc.M109.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka A, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. The Journal of cell biology. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caulfield TR, Fiesel FC, Springer W. Activation of the E3 ubiquitin ligase Parkin. Biochem Soc Trans. 2015;43:269–274. doi: 10.1042/BST20140321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koyano F, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 42.Avraham E, Rott R, Liani E, Szargel R, Engelender S. Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. The Journal of biological chemistry. 2007;282:12842–12850. doi: 10.1074/jbc.M608243200. [DOI] [PubMed] [Google Scholar]
- 43.Ko HS, et al. Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao D, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Omura T, Kaneko M, Okuma Y, Matsubara K, Nomura Y. Endoplasmic reticulum stress and Parkinson’s disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxidative medicine and cellular longevity. 2013;2013:239854–7. doi: 10.1155/2013/239854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. American journal of physiology. Heart and circulatory physiology. 2004;286:H823–829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 47.Kim KY, et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. The Journal of clinical investigation. 2011;121:3701–3712. doi: 10.1172/JCI44736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ran FA, et al. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nature biotechnology. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 51.Holmstrom KM, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. Journal of molecular and cellular cardiology. 2015;85:178–182. doi: 10.1016/j.yjmcc.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen G, et al. Chemically defined conditions for human iPSC derivation and culture. Nature methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.