

Abstract
The selective estrogen receptor (ER) modulator tamoxifen inhibits ER signaling in breast cancer cells, and it is used for the treatment of ER-positive breast cancer. However, this type of cancer often acquires resistance to tamoxifen, and a better understanding of the molecular mechanisms underlying tamoxifen resistance is required. In this study, we established tamoxifen-resistant (TAM-R) breast cancer cells by long-term tamoxifen treatment of ER-positive breast cancer MCF7 cells. In TAM-R cells, expression of not only ERα, a major form of ER in breast cancer, but also its transcriptional partner forkhead box protein A1 (FOXA1) was found to be reduced. In contrast, activation of the transcription factor nuclear factor-κB (NF-κB) and expression of its target IL6 were increased in these cells. Stable expression of FOXA1, but not ERα, reduced the expression of IL6 in the FOXA1- and ERα-negative breast cancer MDA-MB-231 cells and TAM-R cells, without affecting the activation of the NF-κB signaling pathways. Conversely, FOXA1 knockdown induced IL6 expression in MCF7 cells. Chromatin immunoprecipitation assays revealed that FOXA1 bound to the promoter region of IL6 and repressed recruitment of the NF-κB complex to this region. TAM-R cells were found to have high mammosphere-forming activity, characteristics of cancer stem cells, and this activity was suppressed by NF-κB and IL6 signaling inhibitors. Taken together, these results suggest that FOXA1 suppresses expression of IL6 through inhibition of NF-κB recruitment to the IL6 promoter in an ERα-independent manner and that reduction in FOXA1 expression induces IL6 expression and contributes to cancer stem cell-like properties in TAM-R cells.
Keywords: breast cancer, cancer stem cells, estrogen receptor, interleukin 6 (IL-6), NF-kappa B (NF-KB)
Introduction
Breast cancer is the most common type of cancer among women. The majority of breast cancer has an expression of estrogen receptors (ERs).2 Between the two members of the ER family, ERα and ERβ, ERα is the dominant form found in breast cancer (1). Because progression of ER-positive breast cancer depends on estrogen signaling, breast cancer can be treated with targeting estrogen signaling with ER inhibitors (2). For example, the selective ER modulator tamoxifen has been used for treatment of breast cancer patients. However, ER-positive breast cancer frequently acquires resistance to tamoxifen after long-term treatment, and therefore, overcoming tamoxifen resistance with a novel therapeutic strategy is needed to treat breast cancer patients (3, 4).
Although the molecular mechanisms underlying tamoxifen resistance are not yet fully understood, previous studies have shown that the transcription factor nuclear factor-κB (NF-κB) is involved in tamoxifen resistance (5–8). We previously found that NF-κB is constitutively activated and promotes estrogen-independent proliferation in ER-negative breast cancer cells (9). NF-κB is reported to induce breast cancer progression through various target genes, such as the inflammatory cytokines IL6 and IL8 (10, 11). Moreover, NF-κB is thought to be involved in the maintenance and progression of breast cancer stem cells via expression of IL6 and the NOTCH ligand JAG1 (10, 12) or stimulation of nuclear export of the cell cycle inhibitor p27 (13).
NF-κB is an important regulator of the genes necessary for proliferation and differentiation of various types of cells (14). The NF-κB family is composed of five different proteins, including RelA, RelB, c-Rel, p105/p50, and p100/p52. Two distinct NF-κB-signaling pathways have been proposed: the canonical pathway, which activates the RelA-p50 complex; and the non-canonical pathway, which activates the RelB-p52 complex. Activation of the NF-κB pathways occurs in response to various cytokine and growth stimuli, leading to phosphorylation of inhibitors of NF-κB (IκB) family proteins and p100 proteins by the IκB kinase (IKK) complex, followed by IκB degradation and p100 processing into p52, and subsequent nuclear translocation of NF-κB (14, 15).
Most ER-positive breast cancers express the transcriptional regulator FOXA1, which is responsible for opening up chromatin to allow for recruitment of ERα to the promoter regions of its target genes (1). Although ERα and FOXA1 are shown to suppress malignancy of breast cancer cells (16, 17), their involvement in NF-κB activation and tamoxifen resistance is not fully understood. In this study, we established tamoxifen-resistant breast cancer cells (TAM-R) by long-term tamoxifen treatment of MCF7 cells, an ERα-positive human breast cancer cell line, and we analyzed the involvement of ERα and FOXA1 in NF-κB activation and tamoxifen resistance in breast cancer.
Results
Establishment of TAM-R and long-term estrogen-deprived breast cancer cells
We established the TAM-R breast cancer cell line by treating ERα-positive breast cancer MCF7 cells with tamoxifen (4-OHT, 5 μm) for more than 1 year (Fig. 1A). Treatment with 4-OHT at 5 μm for 6 days reduced the viability of MCF7 cells by ∼50%, and more than 5 μm 4-OHT, such as 10 and 20 μm, killed MCF7 completely within 3 days probably because of non-specific toxic effects (Fig. 1B). To establish TAM-R cells having strong resistance to tamoxifen, we proceeded to use the highest concentration of 4-OHT in the range of the concentrations that show no apparent side effects. Therefore, we decided to treat MCF7 with 4-OHT at 5 μm to establish TAM-R cells. Treatment with 4-OHT at 5 μm reduced cell viability of MCF7 by 60–70% within 1 week and then arrested growth of the residual cells for ∼2 months. After 2 months of treatment, the cell number was gradually increased, and cell culture was continued in the presence of 5 μm 4-OHT. After 1 year of culture, the cell mixture grown under treatment with 4-OHT was used as TAM-R cells. To compare with estrogen deprivation-resistant breast cancer, we also established the long-term estrogen-deprived (LTED) MCF7 cells by culturing with estrogen-deprived media for more than 1 year. TAM-R cells showed a flat morphology, but LTED cells formed more membrane protrusions than parental cells (Fig. 1A). Although tamoxifen treatment or hormone-depleted culture conditions significantly suppressed proliferation of parental MCF7 cells, TAM-R and LTED cells were resistant to tamoxifen treatment and hormone-depleted culture conditions, respectively (Fig. 1C). Interestingly, mRNA and protein expression of ERα and its transcriptional partner FOXA1 (1) was significantly reduced in TAM-R cells but not in LTED cells (Fig. 1, D and E). Expression of ERα targets, such as X-box-binding protein 1 (XBP1), trefoil factor 1 (TFF1), and cyclin D1 (CCND1), was also significantly reduced in TAM-R cells (Fig. 1F). Estrogen (estradiol, E2)-stimulated induction of these genes was also abolished in TAM-R cells (Fig. 1G). These results suggest that acquired tamoxifen resistance significantly reduces the expression levels of ERα, FOXA1, and ERα targets.
Figure 1.

Reduction in FOXA1 expression in TAM-R cells. A, normal MCF7, TAM-R, and LTED cells were visualized by phase-contrast microscopy. B, viabilities of normal MCF7 cells treated with the indicated concentrations of 4-OHT for 6 days were detected by MTT assay. The viability of cells cultured in 4-OHT-free media was set to 1. The results represent the mean ± S.D. (n = 3).***, p < 0.001 as calculated by Student's t test. C, viabilities of normal MCF7, TAM-R, and LTED cells cultured under the indicated conditions for 3 days were detected by MTT assay, as in B. The viability of cells cultured in normal media was set to 1. D, normal MCF7, TAM-R, and LTED cells were analyzed for ERα (ESR1) and FOXA1 mRNA expression by qPCR analyses. The level of GAPDH mRNA expression was used to normalize the data. The expression level of ERα or FOXA1 mRNA in normal MCF7 cells was set to 1. The results represent the mean ± S.D. (n = 3). ***, p < 0.001; **, p < 0.01 as calculated by Student's t test. E, normal MCF7, TAM-R, and LTED cells were analyzed for ERα and FOXA1 expression by Western blotting (WB). Actin was used as a loading control. F, mRNA expression of XBP1, TFF1, and CCND1 was analyzed by qPCR analyses, as in D. G, mRNA expression of XBP1, TFF1, and CCND1 in normal MCF7 and TAM-R cells stimulated or unstimulated with E2 (100 nm, 3 h) was analyzed by qPCR analyses, as in D.
Activation of the canonical and non-canonical NF-κB pathways in TAM-R cells
Because previous studies found that tamoxifen-resistant phenotypes are associated with NF-κB signaling (5–8), we analyzed NF-κB activation in TAM-R and LTED cells. Phosphorylation of RelA and processing of p100 were significantly increased in TAM-R cells (Fig. 2A). Expression of RelB, whose expression is induced by the canonical NF-κB pathway (18), was up-regulated in these cells (Fig. 2, A and B). mRNA expression of NIK (MAP3K14), a crucial activator of the non-canonical NF-κB pathway (19), was also up-regulated (Fig. 2B). We found an increase in tumor necrosis factor-α (TNF-α)-induced phosphorylation of IKK, IκBα, and RelA in TAM-R cells (Fig. 2C), which is characteristic of activation of the canonical NF-κB pathway. TNF-like weak inducer of apoptosis (TWEAK)-stimulated and receptor activator of NF-κB ligand (RANKL)-stimulated induction of NIK and RelB proteins were also increased in these cells (Fig. 2, D and E). TAM-R cells showed higher activity to induce the NF-κB-EGFP (enhanced green fluorescent protein) reporter, which expresses EGFP driven by three NF-κB-binding sites, than parental cells (Fig. 2F). These results suggest that activation of the canonical and non-canonical NF-κB pathways is enhanced in TAM-R cells under both stimulated and unstimulated conditions.
Figure 2.

Sustained activation of NF-κB in TAM-R cells. A, normal MCF7, TAM-R, and LTED cells were analyzed by Western blotting (WB) with the indicated antibodies. B, mRNA expression of NIK (MAP3K14) and RELB was analyzed by qPCR analyses, as in Fig. 1D. C–E, normal MCF7, TAM-R, and LTED cells were stimulated with TNF-α (C), TWEAK (D), or RANKL (E) for the indicated times and analyzed by Western blotting with the indicated antibodies. F, normal MCF7 and TAM-R cells were transfected with pcDNA/3κB-EGFP reporter vector together with pCAG/TR vector. At 24 h after transfection, cells were lysed and subjected to Western blotting with the indicated antibodies. G, mRNA expression of IL6 and IL8 (CXCL8) was analyzed by qPCR analyses, as in Fig. 1D. H, normal MCF7, TAM-R, and LTED cells were treated with brefeldin A (5 μg/ml) for 2 h and then analyzed by Western blotting with the indicated antibodies. I, TAM-R cells were treated with SC-514 (100 μm) or DMSO (dimethyl sulfoxide, 0.1%) for 8 h and then analyzed for NIK, RELB, IL6, and IL8 mRNA expression by qPCR analyses. The level of GAPDH mRNA expression was used to normalize the data. The expression level of DMSO-treated cells was set to 1. The results represent the mean ± S.D. (n = 3). **, p < 0.01 as calculated by Student's t test. J, viabilities of normal MCF7 and TAM-R cells cultured under the indicated conditions (100 μm SC-514, 0.1% DMSO) for 3 days were detected by MTT assay, as in Fig. 1C. **, p < 0.01 as calculated by Student's t test.
Previous studies have shown that IL6 and IL8 (CXCL8) are commonly targeted by NF-κB to promote breast cancer progression (10, 11). Therefore, we analyzed expression of these genes in TAM-R cells. Expression of both IL6 and IL8 was significantly increased in these cells (Fig. 2G). However, induction of IL8 expression was more enhanced in LTED cells than TAM-R cells, and the IKK inhibitor SC-514 suppressed expression of IL6, but not IL8, in TAM-R cells (Fig. 2I). Expression of IL6 was up-regulated in TAM-R cells at the protein level (Fig. 2H). Notably, SC-514 significantly reduced viability of TAM-R cells, but not MCF7 cells (Fig. 2J). These results suggest that IL6 is a primary target of NF-κB and is involved in progression of TAM-R cells.
FOXA1-mediated suppression of IL6 expression in breast cancer cells
ERα and FOXA1 have been shown to be involved in suppression of malignant breast cancer phenotypes (16, 17). Because TAM-R cells have reduced expression of these genes (Fig. 1, C and D), we investigated whether these genes are involved in the suppression of NF-κB and IL6 expression in breast cancer. We stably expressed ERα or FOXA1 in the breast cancer cell line MDA-MB-231, which lacks endogenous expression of ERα and FOXA1 and has sustained activation of NF-κB (9, 16, 20). It is interesting to note that FOXA1 expression strongly reduced IL6 expression under the unstimulated condition (Fig. 3A) and strongly suppressed TNF-α-induced IL6 expression without apparent effects on activation of the canonical NF-κB pathway (Fig. 3, B and D). Although TWEAK-stimulated induction of NIK and RelB proteins was not suppressed by FOXA1 expression, mRNA expression of these proteins was slightly reduced in MDA-MB-231 cells (Fig. 3, A and C). In contrast to FOXA1, ERα expression had no apparent effects on the mRNA expression of IL6, IL8, NIK, and RelB, and on TNF-α- or TWEAK-induced NF-κB activation.
Figure 3.

Reduction in IL6 expression in FOXA1-expressing MDA-MB-231 cells. A, MDA-MB-231 cells stably expressing FOXA1, ERα, or empty vector were analyzed for IL6, IL8 (CXCL8), NIK (MAP3K14), and RELB mRNA expression by qPCR analyses, as in Fig. 1D. B and C, MDA-MB-231 cells stably expressing FOXA1, ERα, or empty vector were stimulated with TNF-α (B) or TWEAK (C) for the indicated times and then analyzed by Western blotting (WB) with the indicated antibodies. D, MDA-MB-231 cells stably expressing FOXA1, ERα, or empty vector were stimulated with TNF-α and then analyzed for IL6 and IL8 mRNA expression by qPCR analyses. The level of GAPDH mRNA expression was used to normalize the data. The expression level of unstimulated control cells was set to 1. The results represent the mean ± S.D. (n = 3).
We next analyzed the effects of reconstitution of FOXA1 or ERα in TAM-R cells. Reconstitution of each gene did not affect basal activation of the NF-κB pathways in TAM-R cells (Fig. 4A). Although the mRNA expression of IL8 and RelB was not changed by reconstitution of each gene, IL6 expression was significantly reduced at mRNA and protein levels by reconstitution of FOXA1 but not ERα (Fig. 4, B and C). Reconstitution of FOXA1 slightly increased the proliferation rate of TAM-R cells, whereas its reconstitution recovered susceptibility to 4-OHT in TAM-R cells (Fig. 4, D and E).
Figure 4.

Reduction in IL6 expression in FOXA1-reconstituted TAM-R cells. A, TAM-R cells stably expressing FOXA1, ERα, or empty vector were analyzed by Western blotting (WB) with the indicated antibodies. B, TAM-R cells stably expressing FOXA1, ERα, or empty vector were analyzed for IL6, IL8 (CXCL8), and RELB mRNA expression by qPCR analyses, as in Fig. 1D. C, TAM-R cells stably expressing FOXA1, ERα, or empty vector were treated with brefeldin A (5 μg/ml) for 2 h and then analyzed by Western blotting with the indicated antibodies. D, TAM-R cells stably expressing FOXA1 or empty vector were seeded at 5,000 cells/well in 96-well plates. At 1, 3, or 6 days after incubation, cell growth was analyzed by MTT assay. Cell growth levels are shown as relative to that of the empty vector-expressing cells at day 1. The results represent the mean ± S.D. (n = 3). *, p < 0.05 as calculated by Student's t test. E, viabilities of TAM-R cells stably expressing FOXA1 or empty vector cultured under the indicated conditions for 3 days were detected by MTT assay, as in Fig. 1C. The viability of cells cultured in 4-OHT-free media was set to 1. *, p < 0.05 as calculated by Student's t test.
The role of endogenous FOXA1 in MCF7 cells was analyzed with siRNA-mediated knockdown. FOXA1 knockdown significantly induced mRNA expression of IL6, NIK, and RelB (Fig. 5A). Although TNF-α-induced activation of the canonical NF-κB pathway was not affected by FOXA1 knockdown, TWEAK-induced expression of NIK and RelB proteins was slightly increased (Fig. 5, B and C). We also performed FOXA1 knockdown and FOXA1/ERα double knockdown and analyzed their effects on NF-κB-EGFP reporter activity and IL6 protein expression in MCF7 cells. Although FOXA1 knockdown or FOXA1/ERα double knockdown did not affect the reporter activity, IL6 protein expression was significantly increased by FOXA1 knockdown and FOXA1/ERα double knockdown (Fig. 5, D and E). Notably, ERα knockdown had no apparent effect on IL6 protein expression, and FOXA1 knockdown was sufficient for IL6 induction. Taken together, these results suggest that expression of FOXA1 represses mRNA expression of IL6 independently of ERα expression without affecting activation of the canonical NF-κB pathway in breast cancer cells. In addition, FOXA1, to a lesser extent, inhibits activation of the non-canonical NF-κB pathway through suppression of NIK mRNA expression.
Figure 5.

Change in IL6 expression in FOXA1-depleted MCF7 cells. A, MCF7 cells transfected with siRNA against FOXA1 (siFOXA1) or luciferase (siLuc) for 48 h were analyzed for IL6, IL8 (CXCL8), NIK (MAP3K14), and RELB mRNA expression by qPCR analyses. The level of GAPDH mRNA expression was used to normalize the data. The expression level of siLuc-transfected cells was set to 1. The results represent the mean ± S.D. (n = 3). ***, p < 0.001; **, p < 0.01; *, p < 0.05 as calculated by Student's t test. B and C, MCF7 cells transfected with siFOXA1 or siLuc for 48 h were stimulated with TNF-α (B) or TWEAK (C) for the indicated times and then analyzed by Western blotting with the indicated antibodies. D, MCF7 cells transfected with siLuc, siFOXA1, or siFOXA1/siERα for 48 h. After incubation, cells were transfected again with pcDNA/3κB-EGFP reporter vector together with pCAG/TR vector. At 24 h after transfection, cells were lysed and subjected to Western blotting with the indicated antibodies. E, MCF7 cells transfected with siLuc, siFOXA1, or siFOXA1/siERα for 48 h. After incubation, cells were treated with brefeldin A (5 μg/ml) for 2 h and then analyzed by Western blotting with the indicated antibodies.
Binding of FOXA1 to the IL6 and NIK promoter regions
FOXA1 was previously shown to function as a transcriptional repressor (16, 21), and therefore, we examined its binding to the promoter regions of IL6 and NIK genes. DNA sequences of the promoter regions of these genes (5,000 nucleotides upstream from the transcription start site of each gene) were analyzed by JASPAR, a transcription factor binding profile database (22), and three putative FOXA1-binding sites were found in each promoter region (Fig. 6, A and F). Chromatin immunoprecipitation (ChIP) assays revealed that FOXA1 bound to two of three putative binding sites (binding site 2 (BS2) and BS3) in the promoter region of each gene in MCF7 cells (Fig. 6, B and G). ChIP assays also revealed that FOXA1 knockdown promotes the binding of RelA to the IL6 promoter region in MCF7 cells and that stable expression of FOXA1 conversely suppressed this binding in MDA-MB-231 cells and TAM-R cells (Fig. 6, C–E). These results suggest that FOXA1 functions as a transcriptional repressor of both IL6 and NIK by binding to their promoter regions and that the binding of FOXA1 to the IL6 promoter region represses the recruitment of the canonical NF-κB complex to this region.
Figure 6.

Binding of FOXA1 to the IL6 and NIK promoter regions. A, schematic diagram of IL6 promoter region. Putative FOXA1-binding sites predicted by the JASPAR database are indicated by the open boxes, and corresponding sequences are shown below. TSS, transcription start site; SS, sense strand; AS, antisense strand. B, MCF7 cells were analyzed by ChIP assays with anti-FOXA1 or control antibody to evaluate the binding activity of FOXA1 to the IL6 promoter region. The percentage input values are shown as relative to that of the control immunoprecipitates. The results represent the mean ± S.D. (n = 3). ***, p < 0.001 as calculated by Student's t test. C, MCF7 cells transfected with siFOXA1 or siLuc for 48 h were analyzed by ChIP assays with anti-RelA or control antibody to evaluate the binding activity of RelA to the IL6 promoter region, as in B, except that the percentage input values are shown as relative to that of the control immunoprecipitates from siLuc-transfected cells. *, p < 0.05 as calculated by Student's t test. D and E, MDA-MB-231 (D) or TAM-R cells (E) stably expressing FOXA1 or empty vector were analyzed by ChIP assays with anti-RelA or control antibody to evaluate the binding activity of RelA to the IL6 promoter region, as in B, except that the percentage input values are shown as relative to that of the control immunoprecipitates from empty vector-expressing cells. ***, p < 0.001; **, p < 0.01 as calculated by Student's t test. F, schematic diagram of NIK (MAP3K14) promoter region. Putative FOXA1-binding sites are indicated by the open boxes, as in A. G, MCF7 cells were analyzed by ChIP assays with anti-FOXA1 or control antibody to evaluate the binding activity of FOXA1 to the NIK promoter region, as in B. ***, p < 0.001; *, p < 0.05 as calculated by Student's t test.
Increased sphere-forming activity in TAM-R cells
IL6 induces cancer stem cell (CSC) formation in breast cancer cells (10), and increased sphere-forming activity is major characteristic of CSCs (23). Interestingly, we found that TAM-R cells had higher sphere-forming activity than control or LTED cells (Fig. 7, A and B). ALDEFLUOR assay, another method to detect CSC (24), was also performed, and TAM-R cells were found to have a higher ALDEFLUOR-positive population than control MCF7 cells (Fig. 7, C and D). Notably, the sphere-forming activity was significantly suppressed by treatment with the IKK inhibitor SC-514, which suppresses the NF-κB pathways, or the JAK1/2 inhibitor ruxolitinib, which suppresses the IL6 pathway (Fig. 7, A and B) (25). Moreover, knockdown of IL6 significantly reduced the sphere-forming activity in TAM-R cells (Fig. 7, E–G). These results suggest that the increased activity of NF-κB and IL6 expression contributes to CSC-like properties in TAM-R cells (Fig. 8, A and B).
Figure 7.

Increased mammosphere formation in TAM-R cells. A, mammospheres from normal MCF7, TAM-R, and LTED cells cultured with mammosphere media in the presence of DMSO, SC-514, or ruxolitinib and visualized by phase-contrast microscopy. B, colony-forming units were calculated and are presented as the mean ± S.D. (n = 3). **, p < 0.01 as calculated by Student's t test. C, ALUDEFLUOR assays of normal MCF7 and TAM-R cells. Percentages of ALDEFLUOR-positive cells are shown in the gates. D, percentages of ALDEFLUOR-positive cells of normal MCF7 and TAM-R cells treated with or without DEAB. The results represent the mean ± S.D. (n = 3). ***, p < 0.001 as calculated by Student's t test. E, TAM-R cells transfected with siLuc or siIL6 for 48 h. After incubation, cells were treated with brefeldin A (5 μg/ml) for 2 h and then analyzed by Western blotting (WB) with the indicated antibodies. F, TAM-R cells transfected with siLuc or siIL6 for 24 h were subjected to mammosphere formation assays. Mammospheres were visualized by phase-contrast microscopy. G, colony-forming units were calculated and are presented as the mean ± S.D. (n = 3). *, p < 0.05 as calculated by Student's t test. H, gene expression profiles of tamoxifen-resistant or control ERα-positive breast tumors from mouse xenograft models. Each tumor has five independent samples. Dashed lines show average expression levels in control tumors.
Figure 8.
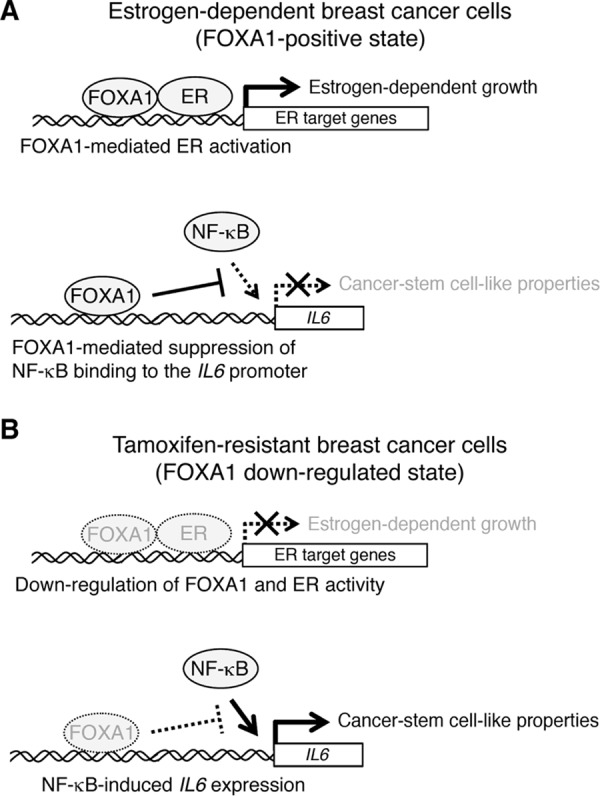
Model of the roles of FOXA1 in breast cancer cells. A and B, FOXA1 induces expression of ER target genes by promoting recruitment of ER to their promoter regions. In contrast, FOXA1 suppresses IL6 expression through inhibiting the recruitment of the canonical NF-κB complex to the IL6 promoter region. In estrogen-dependent breast cancer cells, FOXA1-mediated ER activation promotes estrogen-dependent cell growth (A). In TAM-R cells, expression of FOXA1 is significantly reduced, and this reduction causes expression of IL6 and leads to cancer stem cell-like properties (B).
Finally, we analyzed the gene expression profiles of tamoxifen-resistant breast tumors established from primary ERα-positive breast tumors in an in vivo tamoxifen treatment model (26). Among five replicates, the three tamoxifen-resistant breast tumors (m27, m58, and m69) showed slightly increased expression of IL6 and decreased expression of FOXA1 and ERα as compared with control ERα-positive tumors (Fig. 7H). These results suggest that about half of breast tumors with acquired resistance to tamoxifen may have increased IL6 expression caused by reduction in expression of FOXA1 and ERα as seen in TAM-R cells established in this study.
Discussion
FOXA1 is a member of the forkhead box transcription factor family and binds to the consensus element A(A/T)TRTT(G/T)RYTY through the forkhead DNA-binding domain (27). The major role of FOXA1 is to open up the tightly coiled chromatins and induce activation of other transcription factors, including ERα. Therefore, FOXA1 is crucial for expression of ERα target genes and development of estrogen-dependent tissues (1). FOXA1 is also known to act as a transcriptional repressor. In mouse liver cells, Foxa1 suppresses Il6 gene expression by recruiting the glucocorticoid receptor, which represses Il6 expression, to the Il6 promoter region (21). In breast cancer cells, FOXA1 represses expression of reprimo (RPRM) by forming a complex with ER and histone deacetylase 7 (HDAC7) (28). HDACs remove acetyl groups from histones, increase chromatin compaction, and repress transcription, whereas the FOXA1-ER-HDAC7 complex represses gene expression in an HDAC7 activity-independent manner. Recent studies reported that FOXA1 represses expression of genes involved in tumor malignancy independently of ER (16).
In this study, we found that FOXA1 suppresses gene expression of IL6 and, to a lesser extent, NIK in an ER-independent manner. Although the precise molecular mechanism of FOXA1-mediated inhibition of expression of these genes is still unknown, our present data suggest that FOXA1 functions as a transcriptional repressor by binding to the promoter regions of these genes. At the IL6 promoter, the canonical NF-κB complex binds to the region close to the transcription start site (TSS) and promotes IL6 gene expression (29). In contrast, the FOXA1-binding sites are located at the regions that are several thousands (BS2) or hundreds (BS3) of bases far away from the TSS. Therefore, we hypothesize that the FOXA1-binding sites can spatially interact with a region close to the TSS of the IL6 gene in the folded chromosomes and that FOXA1 can affect the binding of NF-κB to the IL6 promoter region.
Previous studies revealed that NF-κB is constitutively activated in ER- or FOXA1-negative breast cancer cells and that elevated expression of NIK is involved in this constitutive NF-κB activation (9). Because NIK mainly activates the non-canonical NF-κB, sustained activation of the canonical NF-κB pathway in TAM-R cells is likely to be driven by a NIK-independent mechanism. We hypothesize that reduction in TFF1 expression is involved in this sustained activation of the canonical NF-κB pathway. TFF1-deficient mice show overactivation of the canonical NF-κB pathway, NF-κB-dependent inflammation, and tumor formation in gastric tissues, indicating that TFF1 suppresses activation of the canonical NF-κB pathway (30). Because TFF1 expression is substantially reduced in TAM-R cells (Fig. 1, F and G), loss of TFF1-mediated repression may be involved in the elevated activation of the canonical NF-κB pathway in these cells. Notably, our present data show that FOXA1 expression is also reduced in TAM-R cells and that FOXA1 suppresses IL6 expression by inhibiting the binding of NF-κB to the IL6 promoter region. This suggests that reduced expression of TFF1 and FOXA1 induces IL6 expression and contributes to CSC-like properties in TAM-R cells.
Although FOXA1 expression was reduced in TAM-R cells, the molecular mechanism underlying this phenomenon is unclear. Previously, estrogen-dependent ER activity was suggested to induce FOXA1 expression (27). In contrast, we found that FOXA1 expression was not affected in LTED cells (Fig. 1, D and E), where estrogen-dependent ER activity was almost completely suppressed. Recent studies have shown that RNAi-mediated ERα silencing reduces FOXA1 expression and that unliganded ERα is likely to be involved in its expression (31). Because long-term treatment of tamoxifen causes a reduction in ERα expression, this treatment would inhibit estrogen-dependent and -independent ERα roles and lead to down-regulation of FOXA1 expression.
Our previous studies showed that FOXA1 is required for proliferation of estrogen-dependent breast cancer cells (32). Furthermore, our present data show that depletion of FOXA1 expression is involved in phenotypic conversion from estrogen-dependent breast cancer cells to estrogen-independent TAM-R cells. We hypothesize that this dual function of FOXA1 is derived from the difference in the period of FOXA1 depletion. Knockdown-mediated short-term (within ∼1 week) FOXA1 silencing causes an acute reduction in ER target gene expression, which is required for proliferation of estrogen-dependent breast cancer cells. In contrast, long-term (more than 1 year) FOXA1 reduction by tamoxifen treatment releases FOXA1-mediated inhibition of gene expression involved in malignancy and estrogen-independent growth of tumors (Fig. 8, A and B). For better understanding of the roles of FOXA1 in breast cancer cells, whole genome chromatin immunoprecipitation and deep sequencing analyses of FOXA1 and identification of its direct target genes will be required.
In conclusion, our results suggest that reduction in FOXA1 expression contributes to the CSC-like phenotype in TAM-R cells by inducing recruitment of the canonical NF-κB complex to the IL6 promoter to promote its gene expression. Because tamoxifen resistance is a serious problem in treatment of breast cancer patients, gene therapy designated to introduce FOXA1 in TAM-R breast cancer tissues may be an effective strategy to overcome the resistance.
Experimental procedures
Cells and transfection
MCF7 and MDA-MB-231 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Nissui, Tokyo, Japan) containing 5% fetal bovine serum (FBS). Tamoxifen-resistant MCF7 cells (TAM-R) were generated by treatment with 5 μm 4-OHT (Enzo Life Sciences, Farmingdale, NY) for more than 1 year. Estrogen deprivation-resistant MCF7 cells (LTED) were generated by treatment with phenol red-free DMEM (Wako, Tokyo, Japan) consisting of 5% charcoal-stripped FBS (Thermo Fisher Scientific, Pittsburgh, PA) for more than 1 year. To establish stable cell pools, we first improved transcription efficiency by replacing the promoter sequences of the pIRESpuro3 vectors (TaKaRa, Shiga, Japan) from the cytomegalovirus (CMV) promoter to the hybrid CMV early enhancer/chicken β-actin (CAG) promoter, which was derived from the pEBMultiNeo vector (Wako) by digestion with SpeI and EcoRV (pIRESpuro3-CAG). Next, cDNAs that encode FOXA1 and ERα (ESR1) were generated by PCR from the reverse-transcribed product of MCF7 total RNA and then subcloned into the pIRESpuro3-CAG vector. An N-terminal Myc tag was fused to the ERα cDNA. MDA-MB-231 and TAM-R cells were transfected with these vectors by Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer's instruction, and transfected cell pools were selected for resistance to puromycin. siRNAs for FOXA1 (sense, 5′-GAACAGGCACUGCAAUACUCGCCUU-3′, and antisense, 5′-AAGGCGAGUAUUGCAGUGCCUGUUC-3′), ESR1 (sense, 5′-GCUACUGUGCAGUGUGCAAUGACUA-3′, and antisense, 5′-UAGUCAUUGCACACUGCACAGUAGC-3′), and IL6 (sense, 5′-GAACGAAUUGACAAACAAATT-3′, and antisense, 5′-UUUGUUUGUCAAUUCGUUCTG-3′) were purchased from Thermo Fisher Scientific. siRNA for luciferase (sense, 5′-GCGCUGCUGGUGCCAACCCTT-3′, and antisense, 5′-GGGUUGGCACCAGCAGCGCTT-3′) was purchased from Hokkaido System Sciences (Hokkaido, Japan). siRNAs were transfected into cells (33 nm) with Lipofectamine 2000 following the manufacturer's instruction. E2 was purchased from Sigma. E2 stimulation was performed as described previously (33).
Antibodies
The following antibodies were used: ERα (catalog no. 8644), IκBα (catalog no. 4818), phospho-IκBα (catalog no. 2859), IKKβ (catalog no. 8943), phospho-IKKα/β (catalog no. 2697), NIK (catalog no. 4994), phospho-RelA (catalog no. 4922), and RelB (catalog no. 4922) from Cell Signaling Technology (Beverly, MA); FOXA1 (sc-6553) and RelA (sc-8008) from Santa Cruz Biotechnologies (Santa Cruz, CA); actin (MAB1501R) and p100/p52 (05–361) from Merck Millipore (Darmstadt, Germany); GFP (mFX75) from Wako; tetracycline repressor (TR, TET01) from MoBiTech (Goettingen, Germany); and IL6 (ab6672) from Abcam (Cambridge, MA). Horseradish peroxidase (HRP)-F(ab′)2-bound secondary antibodies were purchased from GE Healthcare.
Western blotting
Western blotting was performed with enhanced chemiluminescence (Merck Millipore) as described previously (34). TNF-α (10 ng/ml, PeproTech, Rocky Hill, NJ), TWEAK (100 ng/ml, R&D Systems, Minneapolis, MN), and RANKL (100 ng/ml, Wako) were used separately for cell treatments. Whole cell lysates prepared in SDS-sample buffer were subjected to SDS-PAGE and electrotransfered onto polyvinylidene difluoride membranes. Protein bands were detected with their respective antibodies and analyzed using a ChemiDoc XRS+ image analyzer (Bio-Rad). The intensity of chemiluminescence was measured using Quantity One software (Bio-Rad).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cells were plated 5,000 cells/well in a 96-well plate and incubated for 3 days at 37 °C with DMEM in the presence or absence of ethanol (0.2%) or 4-OHT (5 μm), or with the phenol red-free DMEM containing 5% charcoal-stripped FBS. After the incubation period, the solution in each well was replaced with phenol red-free DMEM containing 0.5 mg/ml MTT (Dojindo Kumamoto, Japan) and incubated for an additional 1 h. The resulting crystallized product was dissolved in 100 μl of 100% DMSO (dimethyl sulfoxide) and measured at 595 nm by Filter Max F5 microplate reader (Molecular Devices, Sunnyvale, CA).
Quantitative real time PCR (qPCR) analysis
Total RNAs were isolated from cells with the RNAiso Plus reagent (TaKaRa), and cDNAs were synthesized from 0.5 μg of each RNA preparation using the ReverTra Ace qPCR RT kit (Toyobo, Osaka, Japan) following the manufacturer's instruction. We used the following primers for PCR: GAPDH, 5′-GGA GCG AGA TCC CTC CAA AAT-3′ (sense) and 5′-GGC TGT TGT CAT ACT TCT CAT GG-3′ (antisense); ESR1, 5′-CCC ACT CAA CAG CGT GTC TC-3′ (sense) and 5′-CGT CGA TTA TCT GAA TTT GGC CT-3′ (antisense); FOXA1, 5′-GCA ATA CTC GCC TTA CGG CT-3′ (sense) and 5′-TAC ACA CCT TGG TAG TAC GCC-3′ (antisense); MAP3K14, 5′-CGG AAA GTG GGA GAT CCT GAA-3′ (sense) and 5′-GGG CGA TGA TAG AGA TGG CAG-3′ (antisense); RELB, 5′-CAG CCT CGT GGG GAA AGA C-3′ (sense) and 5′-GCC CAG GTT GTT AAA ACT GTG C-3′ (antisense); IL6, 5′-ACT CAC CTC TTC AGA ACG AAT TG-3′ (sense) and 5′-CCA TCT TTG GAA GGT TCA GGT TG-3′ (antisense); CXCL8, 5′-ATG ACT TCC AAG CTG GCC GT-3′ (sense) and 5′-TTA CAT AAT TTC TGT GTT GGC-3′ (antisense); XBP1, 5′-GCG CCT CAC GCA CCT G-3′ (sense) and 5′-GCT GCT ACT CTG TTT TTC AGT TTC C-3′ (antisense); TFF1, 5′-GTG TCA CGC CCT CCC AGT-3′ (sense) and 5′-GGA CCC CAC GAA CGG TG-3′ (antisense); and CCND1, 5′-TGG AGG TCT GCG AGG AAC AGA A-3′ (sense) and 5′-TGC AGG CGG CTC TTT TTC A-3′ (antisense). After initial denaturation at 95 °C for 1 min, PCR was performed for 40 cycles (15 s at 95 °C and 45 s at 60 °C) using the Thunderbird SYBR Green polymerase kit (Toyobo) and Eco real time PCR system (Illumina, San Diego).
Reporter assay
The reporter vector (pcDNA/3κB-EGFP) that expresses EGFP driven by three NF-κB-binding sites was constructed as follows. The cDNA encoding EGFP was isolated from pEGFP-C1 vector by PCR with the primers 5′-AAA GGA TCC ACC ATG GTG AGC AAG GGC GAG GAG-3′ (sense) (BamHI site is underlined) and 5′-TTT GAA TTC TCA CTT GTA CAG CTC GTC CAT G-3′ (antisense) (EcoRI site is underlined). The isolated cDNA was cloned into the BamHI and EcoRI sites in the pcDNA3 vector (pcDNA/EGFP). A DNA fragment with three NF-κB-binding sites and the thymidine kinase minimal promoter was synthesized, digested with BglII, and cloned into the BglII and BamHI sites of pcDNA/EGFP vector whose CMV promoter was pre-removed by digestion with the same enzymes. The sequence of the synthesized DNA fragment: 5′-AAAAGATCTGGCCTCGGCGGCCAAGCTTGCATGCCAGGGGACTTTCCCAATGGGGAATTCCCCAATGGGGAATTTCCCTCTCTAGAGGATCCGGCCCCGCCCAGCGTCTTGTCATTGGCGAATTCGAACACGCAGATGCAGTCGGGGCGGCGCGGTCCGAGGTCCACTTCGCATATTAAGGTGACGCGTGTGGCCTCGAACACCGAGCGACCCTGCAGCGACCCGCTTAACAGCGTCAACAGCGTGCCGCAGATAGATCTAAA-3′ (NF-κB-binding sites are shown in bold, the minimal promoter is underlined, and BglII sites are shown in italic). Cells were transfected with the constructed reporter vector together with pCAG/TR vector by Lipofectamine 2000. pCAG/TR vector (35), which express TR driven by the strong constitutive CAG promoter, was used to assess transfection efficiency. At 24 h after transfection, cells were lysed with the SDS-sample buffer and subjected to Western blotting.
ChIP assay
The ChIP assay was performed as described previously (34, 36). Chromatin solution (2 ml) was incubated overnight with 2 μg of appropriate antibodies. For the FOXA1 ChIP analysis, FOXA1 antibody (sc-6553) or control goat antibody (sc-3887; Santa Cruz Biotechnologies) were used. For the RelA ChIP analysis, RelA antibody (sc-8008) and control mouse antibody (MOPC21; Sigma) were used. DNA was purified and subjected to qPCR analysis as described above. We used the following PCR primers: IL6 promoter site 1 (between −4,818 and −4,596 from the transcription start site of the IL6 gene predicted from RefSeq NM_000600.4), 5′-TGG CCT CTC TTC ATG GAT TC-3′ (sense) and 5′-GCA TTG GCT GGT TTC CTC TA-3′ (antisense); IL6 promoter site 2 (between −2,135 and −1,910), 5′-CGA TAT AGC CGA GCT GGA AG-3′ (sense) and 5′-AGC TGT TTG ATC CTG GCT GT-3′ (antisense); IL6 promoter site 3 (between −1,334 and −1,087), 5′-AAA TGC CCA ACA GAG GTC AC-3′ (sense) and 5′-TTC CCT CAG GAT GGT GTC TC-3′ (antisense); MAP3K14 promoter site 1 (between −3,999 and −3,750 from the transcription start site of the MAP3K14 gene predicted from RefSeq NM_003954.4), 5′-ATG GCT CAG CAC CAT AAA CC-3′ (sense) and 5′-GAG ACC CAG GAT TCA AAC CA-3′ (antisense); MAP3K14 promoter site 2 (between −2,309 and −2,079), 5′-AAC GGA GAG GCC TTG CTT AT-3′ (sense) and 5′-TTC CCT TGA CAC CTC TGG TC-3′ (antisense); and MAP3K14 promoter site 3 (between −1,055 and −846), 5′-CCT TGC TAT ATT GCC CAG GA-3′ (sense) and 5′-GGT GGG GAG TAG GGA AGG TA-3′ (antisense). Percent input values were calculated by comparing Ct values of input and immunoprecipitated fractions and were shown as ratios relative to those of control samples.
Mammosphere formation assays
Mammosphere formation assays were performed as described previously, with some modifications (23). Briefly, cells were detached from culture dishes by trypsinization. The cell suspensions were centrifuged at 210 × g for 3 min; supernatants were removed, and cells were washed with 5 ml of phosphate-buffered saline (PBS). The cell suspensions were centrifuged at 210 × g for 3 min; supernatants were removed, and cells were resuspended with mammosphere media (DMEM/F-12 (Wako) supplemented with B27 (Thermo Fisher Scientific) and recombinant epidermal growth factor (20 ng/ml; PeproTech)). The cell suspensions were passed through a syringe three times with 26-gauge needles to ensure formation of single-cell suspensions, and cell concentration was adjusted to 600 cells/ml. These cell suspensions (1 ml) were plated out in triplicate onto a 24-well ultra-low attachment plate (Corning). Cells were supplemented with SC-514 (100 μm, FOCUS Biomolecules, Plymouth Meeting, PA), ruxolitinib (300 nm, Selleck Chemicals, Houston, TX), or DMSO (0.3%) and then incubated at 37 °C for 5 days without moving the plates and without replenishing the media. After 5 days, the number of mammospheres with a diameter greater than 50 μm was counted using a microscope fitted with a graticule, and colony-forming units were calculated by dividing the number of mammospheres by the number of cells plated.
ALUDEFLUOR assay
Cells were detached from culture dishes by trypsinization. The detached cells were passed through a syringe three times with 26-gauge needles to ensure formation of single-cell suspensions. The obtained cell suspensions (1 × 106 cells) were subjected to ALUDEFLUOR assay as per the manufacturer's instruction (StemCell Technologies, Vancouver, Canada). The aldehyde dehydrogenase inhibitor diethylaminobenzaldehyde (DEAB) was used to detect aldehyde dehydrogenase activity. ALDEFLUOR-positive cells were detected by Guava EazyCyte flow cytometer (Merck Millipore) per the manufacturer's instruction.
Author contributions
Noritaka Yamaguchi designed the study, performed the experiments, and wrote the paper. Y. N. and Naoto Yamaguchi analyzed the data.
This work was supported in part by Grants-in-aid for Scientific Research C 16K08227 (to Noritaka Yamaguchi) and 15K07922 (to Naoto Yamaguchi), Scientific Research from the Promotion and Mutual Aid Corporation for Private Schools of Japan (Kyoto Pharmaceutical University and Chiba University), and grants from The Hamaguchi Foundation for the Advancement of Biochemistry (to Noritaka Yamaguchi) and the Japan Foundation for Applied Enzymology (to Naoto Yamaguchi). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
- ER
- estrogen receptor
- TAM-R
- tamoxifen-resistant
- EGFP
- enhanced GFP
- qPCR
- quantitative PCR
- 4-OHT
- tamoxifen
- TWEAK
- TNF-like weak inducer of apoptosis
- RANKL
- receptor activator of NF-κB ligand
- CSC
- cancer stem cell
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- LTED
- long-term estrogen-deprived
- DEAB
- diethylaminobenzaldehyde
- IKK
- IκB kinase
- TSS
- transcription start site
- E2
- 17β-estradiol
- HDAC
- histone deacetylase
- TR
- tetracycline repressor.
References
- 1. Carroll J. S., Liu X. S., Brodsky A. S., Li W., Meyer C. A., Szary A. J., Eeckhoute J., Shao W., Hestermann E. V., Geistlinger T. R., Fox E. A., Silver P. A., and Brown M. (2005) Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43 [DOI] [PubMed] [Google Scholar]
- 2. Ali S., and Coombes R. C. (2002) Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2, 101–112 [DOI] [PubMed] [Google Scholar]
- 3. Ellis A. J., Hendrick V. M., Williams R., and Komm B. S. (2015) Selective estrogen receptor modulators in clinical practice: a safety overview. Expert Opin. Drug Saf. 14, 921–934 [DOI] [PubMed] [Google Scholar]
- 4. Rondón-Lagos M., Villegas V. E., Rangel N., Sánchez M. C., and Zaphiropoulos P. G. (2016) Tamoxifen resistance: emerging molecular targets. Int. J. Mol. Sci. 17, E1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yde C. W., Emdal K. B., Guerra B., and Lykkesfeldt A. E. (2012) NFκB signaling is important for growth of antiestrogen resistant breast cancer cells. Breast Cancer Res. Treat. 135, 67–78 [DOI] [PubMed] [Google Scholar]
- 6. Gu Z., Lee R. Y., Skaar T. C., Bouker K. B., Welch J. N., Lu J., Liu A., Zhu Y., Davis N., Leonessa F., Brünner N., Wang Y., and Clarke R. (2002) Association of interferon regulatory factor-1, nucleophosmin, nuclear factor-κB, and cyclic AMP response element binding with acquired resistance to Faslodex (ICI 182,780). Cancer Res. 62, 3428–3437 [PubMed] [Google Scholar]
- 7. Nehra R., Riggins R. B., Shajahan A. N., Zwart A., Crawford A. C., and Clarke R. (2010) BCL2 and CASP8 regulation by NF-κB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. FASEB J. 24, 2040–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. deGraffenried L. A., Chandrasekar B., Friedrichs W. E., Donzis E., Silva J., Hidalgo M., Freeman J. W., and Weiss G. R. (2004) NF-κB inhibition markedly enhances sensitivity of resistant breast cancer tumor cells to tamoxifen. Ann. Oncol. 15, 885–890 [DOI] [PubMed] [Google Scholar]
- 9. Yamaguchi N., Ito T., Azuma S., Ito E., Honma R., Yanagisawa Y., Nishikawa A., Kawamura M., Imai J., Watanabe S., Semba K., and Inoue J. (2009) Constitutive activation of nuclear factor-κB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 100, 1668–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iliopoulos D., Hirsch H. A., and Struhl K. (2009) An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ginestier C., Liu S., Diebel M. E., Korkaya H., Luo M., Brown M., Wicinski J., Cabaud O., Charafe-Jauffret E., Birnbaum D., Guan J. L., Dontu G., and Wicha M. S. (2010) CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Invest. 120, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamamoto M., Taguchi Y., Ito-Kureha T., Semba K., Yamaguchi N., and Inoue J. (2013) NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat. Commun. 4, 2299. [DOI] [PubMed] [Google Scholar]
- 13. Zhang W., Tan W., Wu X., Poustovoitov M., Strasner A., Li W., Borcherding N., Ghassemian M., and Karin M. (2013) A NIK-IKKα module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 23, 647–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghosh S., and Karin M. (2002) Missing pieces in the NF-κB puzzle. Cell 109, S81–S96 [DOI] [PubMed] [Google Scholar]
- 15. Hayden M. S., and Ghosh S. (2004) Signaling to NF-κB. Genes Dev. 18, 2195–2224 [DOI] [PubMed] [Google Scholar]
- 16. Bernardo G. M., Bebek G., Ginther C. L., Sizemore S. T., Lozada K. L., Miedler J. D., Anderson L. A., Godwin A. K., Abdul-Karim F. W., Slamon D. J., and Keri R. A. (2013) FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene 32, 554–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Belguise K., Kersual N., Kirsch K. H., Mineva N. D., Galtier F., Chalbos D., and Sonenshein G. E. (2007) Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 9, 470–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bren G. D., Solan N. J., Miyoshi H., Pennington K. N., Pobst L. J., and Paya C. V. (2001) Transcription of the RelB gene is regulated by NF-κB. Oncogene 20, 7722–7733 [DOI] [PubMed] [Google Scholar]
- 19. Sun S. C. (2011) Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neve R. M., Chin K., Fridlyand J., Yeh J., Baehner F. L., Fevr T., Clark L., Bayani N., Coppe J. P., Tong F., Speed T., Spellman P. T., DeVries S., Lapuk A., Wang N. J., et al. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z., White P., Tuteja G., Rubins N., Sackett S., and Kaestner K. H. (2009) Foxa1 and Foxa2 regulate bile duct development in mice. J. Clin. Invest. 119, 1537–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mathelier A., Fornes O., Arenillas D. J., Chen C. Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R., Zhang A. W., Parcy F., Lenhard B., Sandelin A., and Wasserman W. W. (2016) JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44, D110–D115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shaw F. L., Harrison H., Spence K., Ablett M. P., Simões B. M., Farnie G., and Clarke R. B. (2012) A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 17, 111–117 [DOI] [PubMed] [Google Scholar]
- 24. Charafe-Jauffret E., Ginestier C., Iovino F., Wicinski J., Cervera N., Finetti P., Hur M. H., Diebel M. E., Monville F., Dutcher J., Brown M., Viens P., Xerri L., Bertucci F., Stassi G., et al. (2009) Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 69, 1302–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodriguez-Barrueco R., Yu J., Saucedo-Cuevas L. P., Olivan M., Llobet-Navas D., Putcha P., Castro V., Murga-Penas E. M., Collazo-Lorduy A., Castillo-Martin M., Alvarez M., Cordon-Cardo C., Kalinsky K., Maurer M., Califano A., and Silva J. M. (2015) Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR−/HER2+ breast cancers. Genes Dev. 29, 1631–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cottu P., Bièche I., Assayag F., El Botty R., Chateau-Joubert S., Thuleau A., Bagarre T., Albaud B., Rapinat A., Gentien D., de la Grange P., Sibut V., Vacher S., Hatem R., Servely J. L., et al. (2014) Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clin. Cancer Res. 20, 4314–4325 [DOI] [PubMed] [Google Scholar]
- 27. Bernardo G. M., and Keri R. A. (2012) FOXA1: a transcription factor with parallel functions in development and cancer. Biosci. Rep. 32, 113–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malik S., Jiang S., Garee J. P., Verdin E., Lee A. V., O'Malley B. W., Zhang M., Belaguli N. S., and Oesterreich S. (2010) Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol. Cell. Biol. 30, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McFarland B. C., Hong S. W., Rajbhandari R., Twitty G. B. Jr, Gray G. K., Yu H., Benveniste E. N., and Nozell S. E. (2013) NF-κB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS ONE 8, e78728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soutto M., Belkhiri A., Piazuelo M. B., Schneider B. G., Peng D., Jiang A., Washington M. K., Kokoye Y., Crowe S. E., Zaika A., Correa P., Peek R. M. Jr., and El-Rifai W. (2011) Loss of TFF1 is associated with activation of NF-κB-mediated inflammation and gastric neoplasia in mice and humans. J. Clin. Invest. 121, 1753–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caizzi L., Ferrero G., Cutrupi S., Cordero F., Ballaré C., Miano V., Reineri S., Ricci L., Friard O., Testori A., Corà D., Caselle M., Di Croce L., and De Bortoli M. (2014) Genome-wide activity of unliganded estrogen receptor-alpha in breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 111, 4892–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamaguchi N., Ito E., Azuma S., Honma R., Yanagisawa Y., Nishikawa A., Kawamura M., Imai J., Tatsuta K., Inoue J., Semba K., and Watanabe S. (2008) FoxA1 as a lineage-specific oncogene in luminal type breast cancer. Biochem. Biophys. Res. Commun. 365, 711–717 [DOI] [PubMed] [Google Scholar]
- 33. Yamaguchi N., Shibazaki M., Yamada C., Anzai E., Morii M., Nakayama Y., Kuga T., Hashimoto Y., Tomonaga T., and Yamaguchi N. (2016) Tyrosine phosphorylation of the pioneer transcription factor FoxA1 promotes activation of estrogen signaling. J. Cell. Biochem. in press [DOI] [PubMed] [Google Scholar]
- 34. Yamaguchi N., Yuki R., Kubota S., Aoyama K., Kuga T., Hashimoto Y., Tomonaga T., and Yamaguchi N. (2015) c-Abl-mediated tyrosine phosphorylation of JunB is required for adriamycin-induced expression of p21. Biochem. J. 471, 67–77 [DOI] [PubMed] [Google Scholar]
- 35. Kasahara K., Nakayama Y., Sato I., Ikeda K., Hoshino M., Endo T., and Yamaguchi N. (2007) Role of Src-family kinases in formation and trafficking of macropinosomes. J. Cell. Physiol. 211, 220–232 [DOI] [PubMed] [Google Scholar]
- 36. Agata Y., Katakai T., Ye S. K., Sugai M., Gonda H., Honjo T., Ikuta K., and Shimizu A. (2001) Histone acetylation determines the developmentally regulated accessibility for T cell receptor γ gene recombination. J. Exp. Med. 193, 873–880 [DOI] [PMC free article] [PubMed] [Google Scholar]