

Abstract
Metabolic products and environmental factors constantly damage DNA. To protect against these insults and maintain genome integrity, cells have evolved mechanisms to repair DNA lesions. One such mechanism involves Rad3, a master kinase coordinating the DNA damage response. Rad26 is a functional subunit of the Rad3-Rad26 complex and is responsible for bringing the kinase to sites of DNA damage. Here, I present the crystal structure of Rad26 and identify the elements important for recruiting Rad3. The structure suggests that Rad26 is a dimer with a conserved interface in the N-terminal part of the protein. Biochemical data showed that Rad26 uses its C-terminal domain and the flanking kinase-docking motif to bind specific HEAT repeats in Rad3. Analysis of the reconstituted Rad3-Rad26 heterotetrameric complex with electron microscopy enabled me to propose a structural model for its quaternary structure. In conclusion, these results suggest that Rad26 exists as a dimer and provide crucial insight into how Rad3 is recruited and incorporated into the Rad3-Rad26 DNA repair complex.
Keywords: cell signaling, crystal structure, DNA damage response, structure-function, tertiary structure, Rad26 (ATRIP), Rad3 (ATR), kinase signaling
Introduction
Maintaining genome integrity is a constant challenge for all living cells. Metabolic products and environmental factors are continuously damaging the DNA, and it is important for cells to respond and repair these errors to maintain viability and prevent diseases. Eukaryotic cells are dependent on the DNA-damage response signaling pathway to safeguard the DNA. Rad3 (ATR in mammals) and Tel1 (ATM in mammals) are the master kinases that transduce signals of DNA damage to downstream effectors. Rad3 and Tel1 are members of the PI3K-related kinase family (1, 2) that are characterized by their giant size and similar sequence and domain architecture (3). All PI3K-related kinases are structurally alike with an expanded helical HEAT repeat domain in the N terminus, followed by FAT (FRAP-ATM-TRRAP) and FAT C-terminal (FATC) domains encapsulating the conserved kinase domain (3, 4). Rad3 binds Rad26 (ATRIP in mammals), and this complex is considered the functional unit (5). Upon DNA damage, Rad3 and Tel1 are recruited to the DNA, and once activated, they phosphorylate hundreds of downstream effector proteins that collectively promote cell-cycle arrest and DNA repair (6). Both Rad3 and Tel1 show preference for serine and threonine residues followed by glutamine or hydrophobic residues, and they phosphorylate an overlapping set of effector proteins. The best understood downstream targets of both Rad3 and Tel1 are the protein kinases Chk1 and Cds1 (Chk2 in mammals), that serve to reduce cyclin-dependent kinase activity leading to cell-cycle arrest and DNA repair before replication or mitosis continues.
An essential part of the DNA-damage response is a surveillance system that directly monitors errors in the DNA. Double-strand breaks (DSBs)2 activate Tel1, and here the MRN complex acts as the DNA damage sensor that recruit Tel1 to DSBs. Rad3 is activated by single-stranded DNA (ssDNA) and is therefore also activated during every S phase where it regulates the firing of replication origins and repair of damaged replication forks to prevent premature onset of mitosis (7). Replication protein A (RPA) is a heterotrimeric complex that recognizes and coats exposed ssDNA and hereby serves to mark these vulnerable sites in the DNA. Rad26 binds the oligonucleotide/oligosaccharide-binding (OB) fold of replication protein A via its most N-terminal helix and thus localizes Rad3 to ssDNA (8, 9). Recruitment to ssDNA is not itself enough to activate Rad3 because binding of both the 9-1-1 (Rad9-Rad1-Hus1) complex and Rad4 (TopBP1) is necessary for full Rad3 activation (10, 11).
To gain mechanistic insight into Rad26 and how it is able to recruit Rad3, I determined its crystal structure. This shows that Rad26 forms a compact unit with an unexpected fold of non-classical helical repeats. Biochemical analysis demonstrated that a conserved C-terminal domain together with a conserved kinase-docking motif is required for Rad3 kinase recruitment and further that the binding surface on Rad3 is located within a few HEAT repeats. The structure of Rad26 also provides insight into the oligomeric state of the protein, and the data support that Rad26 uses a large and highly conserved N-terminal domain for dimerization. Small-angle X-ray scattering (SAXS) experiments confirm Rad26 dimerization in solution, and analytic gel filtrations show that Rad3 and Rad26 likely exist as a heterotetrameric complex that can be directly observed using single-particle electron microscopy.
Results
Structure of Rad26
To understand Rad26 structurally, I crystallized the protein from Myceliophthora thermophila devoid of the flexible N terminus (residues 373–841, hereafter referred to as Rad26). M. thermophila is a thermophile fungus evolutionary closer to fission yeast compared with budding yeast or human, and the Schizosaccharomyces pombe nomenclature is therefore used throughout. The structure was solved by single-wavelength anomalous dispersion method using selenomethionine-substituted Rad26 (supplemental Fig. S1, A and B). Eight of twelve possible selenium sites were found, and in the initial experimental electron density maps Rad26 was clearly seen (supplemental Fig. S1C). The Rad26 structure was built, and the selenium sites served as good makers to assign the correct amino acid registry. The Rad26 structure was refined using data extending to 3.15 Å resolution with a final free R factor of 23.5% and excellent geometry (Table 1), and most side chains were well defined in the final electron density map even in the high b-factor regions (supplemental Fig. S1D). Rad26 shows an unexpected fold and is built of helical repeat with unique arrangements distinct from the more common HEAT or ARM units (Fig. 1). The N-terminal domain (NTD) of Rad26 (α1-α2) is not forming repeats, but instead the two helices are arranged perpendicular to each other with an extended loop region in between. The central part of the structure is formed by five helical repeats (α3-α4, α5-α7, α8-α10, α11-α12, and α13-α15) forming an α-solenoid arrangement similar to HEAT and ARM structures. The C-terminal domain (CTD) comprises the last three helices in the structure where the helical repeats diverges and shows a large positive twist of ∼45° between the helical axes compared with the preceding helical repeats. The last 10 C-terminal residues (hereafter referred to as the kinase-docking motif) are too flexible to be clearly defined in the electron density maps. The closest structural homologs to Rad26 as found by a Dali search is HspBP1 (Protein Data Bank entry 1xqr, root mean square deviation of 3.0 Å over 244 residues), which acts as a nucleotide-exchange factor for Hsp70 (12). HspBP1 is built of regular ARM repeats and superposes surprisingly well over the central part of Rad26, exemplifying the low sequence requirement (9% identity between Rad26 and HspBP1) for this basic helical fold.
Table 1.
Data collection and refinement statistics
| Protein | M. thermophila Rad26 (373–841, SeMet) |
|---|---|
| Protein Data Bank code | 5LOI |
| Data collection | |
| Wavelength (Å) | 0.979 |
| Space group | I 4 2 2 |
| a, b, c (Å) | 210.8, 210.8, 104.8 |
| α, β, γ (°) | 90, 90, 90 |
| Total reflections | 540,554 |
| Unique reflections | 20,640 |
| Resolution range (Å) | 105.4–3.15 (3.27–3.15)a |
| Completeness (%) | 99.4 (100.0) |
| Redundancy | 26.2 (26.8) |
| Rmerge (%) | 12.3 (478.1) |
| Rpim (%) | 2.5 (93.7) |
| CC½ (%) | 99.9 (61.2) |
| I/σ(I) | 23.0 (0.8) |
| Wilson B-factor (Å2) | 140.6 |
| Refinement | |
| Rwork (%)/Rfree (%) | 20.7/23.5 |
| Number of atoms | 3013 |
| B-factors (Å2) | 147.1 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angels (°) | 1.02 |
| Estimated coordinate error (Å) | 0.51 |
| Estimated phase error (°) | 28.5 |
| Ramachandran (%) (favored/allowed/outliers) | 97.4/2.6/0.0 |
| Rotamer outliers (%) | 0.0 |
| MolProbity clashscore | 6.97 |
a The highest resolution shell is in parentheses.
Figure 1.
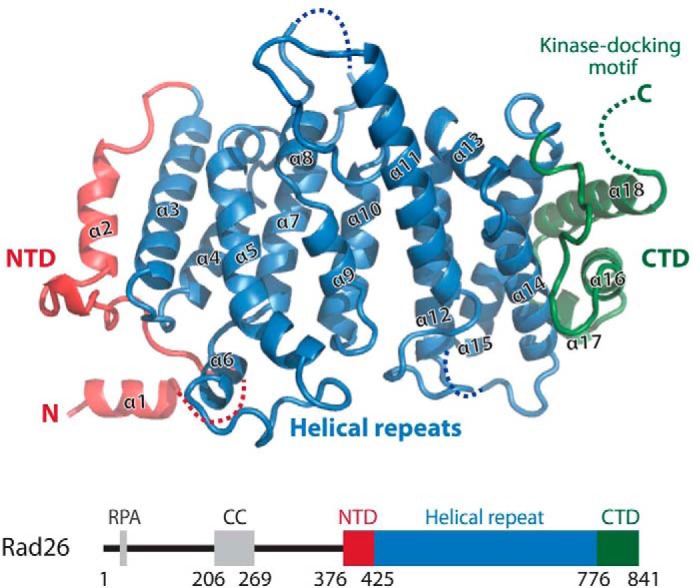
Crystal structure of Rad26. Shown is a cartoon representation of Rad26 colored as indicated schematically below with residue numbers showing the domain boundaries. The NTD, the central helical repeat, and the CTD are shown with helix numbers. Missing loops in the structure are indicated together with the flexible C-terminal kinase-docking motif. RPA, replication protein A.
Rad26 interacts with Rad3 through specific HEAT repeats
Next I wanted to experimentally validate that Rad26 is able to bind the Rad3 kinase, which is vital for correct function. Pulldown experiments using recombinant purified Rad3 fragments of its N-terminal HEAT domain and histidine-tagged Rad26 (residues 373–841) confirmed that the structural domain of Rad26 binds Rad3 (residues 36–899) (Fig. 2A, lanes 1–3). The slightly shorter Rad3 (residues 1–719) fragment failed to bind Rad26, showing that residues 719–899 in Rad3 are vital for Rad26 binding (Fig. 2A, lanes 4 and 5). Additionally, pulldown with the N-terminal truncated form of Rad3 including residues 495–899 likewise showed binding of Rad26 to this fragment (Fig. 2A, lanes 6 and 7), showing that the first 494 residues in Rad3 are not required for Rad26 binding, which contradicts previous yeast two-hybrid reports (13). To validate these pulldown results, I measured the physical interaction between Rad26 and Rad3 using bio-layer interferometry. Histidine-tagged Rad26 (residues 373–841) were immobilized on anti-His biosensors, and association and dissociation to different concentrations of Rad3 were measured. From the sensorgrams obtained, the equilibrium dissociation constant (KD) values were calculated for Rad3 (residues 36–899) to 9.4 ± 5.9 nm and for Rad3 (residues 495–899) to 7.9 ± 2.2 nm with fits of 0.999 and 0.997, respectively. In agreement with the pulldown experiments, no detectable binding was observed with Rad3 (residues 1–719), showing that residues 719–899 in Rad3 are needed for Rad26 binding.
Figure 2.

Rad26 binds to specific HEAT repeats in Rad3. A, pulldown experiments using tagged Rad26 and different fragments of untagged Rad3 covering most of its N-terminal HEAT domain. The top gel is the bound fraction, and the bottom gel is the corresponding unbound fraction from each pulldown experiment. B, sensorgrams show surface binding in nm as a result of Rad26 binding to increasing concentrations of Rad3. C, results are summarized schematically.
Conserved structural elements in Rad26 recruit Rad3
The PIKK kinases family members Rad3(ATR), Tel1(ATM), and DNA-PKcs bind a conserved kinase-docking motif located in the C termini of their respective activating proteins Rad26 (ATRIP), Nbs1, and Ku80 (14). To understand in more detail how Rad26 recruits Rad3, I conducted structure guided mutations in Rad26 and examined their effects on Rad3 binding. Rad3 from M. thermophila contains a conserved C terminus, which has similarity to the kinase-docking motif in human ATRIP, Nbs1, and Ku80 (Fig. 3A). Wild-type Rad26 (residues 373–841) associates with Rad3 (Fig. 3B, lanes 1–3), but a truncated version of Rad26 lacking the last 12 residues lost most of its binding to Rad3 (Fig. 3B, lane 4). Mutating conserved residues in the motif (Asp-832/Glu-833 or Glu-839) into alanines did not have any observable effect on Rad3 binding (Fig. 3B, lanes 5 and 6). Interestingly, a GST-fused version containing only the last 12 C-terminal residues failed to recruit Rad3, showing that additional elements in Rad26 are important for Rad3 binding (Fig. 3B, lane 7). The notion that other elements in Rad26 are required for Rad3 recruitment is supported by the observation that Rad26 lacking the kinase-docking motif still associates with measurable amounts of Rad3 (Fig. 3B, lane 4). Examination of the Rad26 structure showed that the CTD has a distinct acidic patch neighboring the kinase-docking motif (Fig. 3C). Furthermore, this part of the structure is highly conserved between Rad26 orthologs (Fig. 3C), arguing that the CTD might be the structural element responsible for full Rad3 recruitment. To test this, I mutated conserved glutamates in this region and measured the relative effect these mutants had on binding Rad3 in pulldown experiments (Fig. 3D and supplemental Fig. S2A). Mutating Glu-804/Glu-805 had a marginal effect, and mutation of Glu-816/Glu-819 or Glu-823/Glu-826 had no observable effect, whereas mutating Glu-810 into an alanine resulted in a more dramatic decrease in Rad3 binding (Fig. 3D). Together, these experiments show that Rad26 uses both its C-terminal kinase-docking motif and its conserved acidic CTD to bind and recruit Rad3.
Figure 3.

Conserved structural elements in Rad26 recruits Rad3. A, alignment of the C-terminal kinase-docking motif of M. thermophila (Mt) Rad26 with human (Hs) ATRIP, KU80, and NBS1. B, pulldown experiments of Rad3 using tagged Rad26 mutated in the kinase-docking motif as indicated. The top gel is the bound fraction, and the bottom gel is the corresponding unbound fraction from each pulldown experiment. C, surface representation of Rad26 colored according to electrostatic potential with a distinct acidic C-terminal patch marked. Below is Rad26 colored by surface conservation with highly conserved areas marked at both the CTD and the NTD. D, close-up of the CTD of Rad26 showing the acidic patch of conserved glutamine residues that was mutated in the pulldown experiments. The relative effect of Rad26 mutants in their ability to pull down Rad3 is shown in a scatter plot (n = 3, see supplemental Fig. S2A).
Support for a dimeric Rad26
Rad26 contains an N-terminal coiled-coil domain believed to be essential for dimerization (13). However, I find that Rad26 can form a dimer independently of the coiled-coil domain. Analytic gel filtration chromatography analysis of the Rad26 fragment comprising residues 373–841 without the coiled-coil domain shows that the protein elutes in two distinct peaks, indicating different oligomeric states (supplemental Fig. S3A). Inspection of the crystal lattice shows that one molecule of Rad26 makes extensive contact to a neighboring Rad26 molecule (Fig. 4, A and B). Additionally, the most N-terminal helix (α1) from one molecule extends and interacts with the concave helical repeats of the neighboring molecule, and this helix-swap stabilizes a Rad26 dimer. Because this putative dimer interface is formed across a special position crystallographic 2-fold axis, I reprocessed the data in lower symmetry (I4) with one dimer in the asymmetric unit instead of the I422 symmetry, which has only the monomer in the asymmetric unit, thereby allowing the two Rad26 molecules to be different. The loop region (residues 385–390) connecting α1 to the rest of the molecule is flexible, but even in the lower I4 symmetry space group, this connection is not visible in the electron density, arguing that I422 symmetry is correct. Sterically, it would be possible for α1 to fold back on itself, and this could occur when Rad26 is in a monomeric form. This crystal-packing dimer has a PISA (15) score of 1.0, which implies that the interface is important for complex formation. The N-terminal part of two Rad26 molecules comes together in a “handshake” fashion and forms an intimate interface involving a total of eight helices. The interface footprint is more than 1907 Å2 and involves 51 residues that make hydrogen bonds and numerous hydrophobic interactions to the neighboring Rad26 molecule (Fig. 4C). Furthermore, the large dimerization surface between two Rad26 molecules very nicely explains the high level of sequence conservation seen in this area (Figs. 3C and 4B). To validate that Rad26 makes a dimer in solution I performed SAXS experiment on the protein (residues 185–841) containing the coiled-coil part (supplemental Table S1). Comparing the experimental scattering curve to the theoretical scattering curves revealed a good fit of the Rad26 dimer described above (χ2 = 2.5) but a very poor fit to the Rad26 monomer (χ2 = 32.0) (Fig. 4D). Furthermore, low resolution ab initio reconstruction in DAMMIN produced 20 similar models that could be convincingly averaged and superimposes well with the crystal dimer of Rad26 (Fig. 4E). Thus, the SAXS data are in good agreement with the observed Rad26 dimer in the crystal.
Figure 4.

Support for a dimeric Rad26. A, top view of the Rad26 dimer with one molecule colored in light orange, and the other molecule colored as in Fig. 1. The 2-fold symmetry axis is indicated at the dimer interface. B, side view of the Rad26 dimer showing the highly conserved NTD is the interface site between the two molecules. C, the footprint of the Rad26 dimer interface is in blue marked with a dotted line, and residues that contribute hydrogen bonds and strong hydrophobic interaction (above 0.44 kcal/mol) are indicated. D, CRYSOL fit of the theoretical scattering for the Rad26 crystal dimer (red) and monomer (blue) to the experimental scattering with the Guinier plot and the pair distance distribution P(r) below. E, low resolution ab initio DAMMIN model of Rad26 (spheres) with the Rad26 crystal dimer superimposed. F, analytic gel filtration of the Rad3-Rad26 complex absorbance (abs.). G, proposed model for the organization of the heterotetrameric Rad3-Rad26 complex.
Proposed model for a heterotetrameric Rad3-Rad26 complex
The orientation of the Rad26 dimer and the finding that the CTD together with the kinase-docking motif are the interaction site with Rad3 opens the possibility that two molecules of Rad3 can bind a Rad26 dimer simultaneously. To investigate this further, I did analytic gel filtration chromatography on isolated Rad3 and Rad26 and the complex between them. Because I and others (13) have observed that the Rad3-Rad26 complex is more stable if the coiled coil of Rad26 is present, I used a Rad26 construct encompassing residues 185–841. Rad3 (residues 36–899) and Rad3 (residues 185–841) elute as single monodisperse peaks (Fig. 4F). Masses estimated by comparing their elution volumes with those of known molecular mass standards corresponded well with the actual molecular mass of a dimer Rad26 (145.1 kDa) and a monomer Rad3 (95.8 kDa). When Rad26 was preincubated together with Rad3, the complex elutes as a higher molecular mass species close to position of the 440-kDa standard, indicating a potential heterotetrameric 2:2 complex (336.7 kDa) (Fig. 4F).
To gain structural information of how Rad3-Rad26 is organized, I used EM to investigate negative-stained samples of the purified complex (supplemental Fig. S4A). Particles were clearly resolved, and the best micrographs were further processed. Reference-free 2D class averaging of more than 5000 particles shows that the Rad3-Rad26 complex has a dimeric nature, and several classes contain protrusions from the center of the particles (supplemental Fig. S4A). The longest axis measures ∼150 Å, which corresponds nicely with the length of the Rad26 dimer (∼110 Å) plus the diameter of two HEAT repeats from Rad3 (∼20 Å each). The internal symmetry observed is most likely created by the Rad26 dimer that positions two opposing binding sites for Rad3 that are rotated 180° relative to each other. A very similar dimeric architecture was recently observed in a 3D reconstruction of the orthologs complex in yeast Mec1-Ddc2 (16). Based on the combined data, it is possible to suggest a model of how Rad3-Rad26 assembles into a heterotetrameric complex, in which the Rad26 dimer is the central scaffold binding two molecules of Rad3 through their HEAT domains (Fig. 4G).
Discussion
The crystal structure of Rad26 reveals a compact helical protein with a highly conserved CTD that together with a flexible kinase-docking motif recruits Rad3. The data support that Rad26 forms a dimer that is probably further strengthened by its N-terminal coiled-coil domain. The coiled-coil domains in Rad26 orthologs are very well conserved, and it has been demonstrated very elegantly that the coiled-coil domain in human ATRIP is important for oligomerization and increased the affinity for ATR (13, 17). However, Xenopus ATRIP was shown to form oligomers and could bind ATR independently of the coiled-coil domain, showing differences between species (18).
The organization of the suggested Rad26 dimer exposes two Rad3 binding sites orientated in opposite directions, and electron microscopy of purified Rad3-Rad26 suggests how this complex is arranged at low resolution. The dimensions and dimeric architecture of the Rad3-Rad26 complex is in good agreement with a recent single-particle electron microscopy reconstruction of yeast Mec1-Ddc2 (16). Interestingly, the oligomeric states of other PIKKs are known to be important for their function. Tel1 forms inactive dimers or higher-order multimers that dissociates after autophosphorylation into active monomers in response to DSB (19). DNA-PKcs important for non-homologous end joining after DSB forms monomers and dimers and associates with the Ku70/Ku80 heterodimers (20–22). The mammalian target of rapamycin regulator of cell metabolism is a functional dimer in humans and yeast (23–26). It has previously been shown that Rad3-Rad26 can form oligomers, but the exact nature and stoichiometry of this complex was not known (13, 27–29). The data now support that Rad26 is a dimer and most likely forms a 2:2 complex with Rad3; however, the dynamics and biological role of this oligomeric state remain elusive. Recently, it was shown that the Nek1 kinase associates and stabilizes the Rad3-Rad26 complex and primes it for an effective DNA-damage response, suggesting that Nek1-mediated phosphorylation is important for Rad3-Rad26 stability (30).
Rad3 binds Rad26 through its N-terminal HEAT repeats, and here I show that the interaction site is within a few HEAT repeats in the middle and located within residues 719–899 (∼881–1085 in human ATR). Yeast two-hybrid experiments have previously shown that human ATRIP interact with the very N terminus of ATR (residues 30–346) (31), but I find that the first 495 residues (approximately residues 1–472 in human) are not required for effective binding. The mapped Rad26 interaction site in Rad3 near the middle of the HEAT domain overlaps with the HEAT region in Tel1 that binds Nbs1 (32). Furthermore, Ku70/Ku80 interacts with DNA-PKcs through at least two binding sites: one near the kinase domain and one within the HEAT repeats (33, 34), but future studies will show whether Rad26, Nbs1, and Ku70/Ku80 share more properties in the way they interact with their respective kinases. It is, however, clear that not only do Rad3, Nbs1, and Ku70/K80 recruit their kinases to sites of DNA damage, but they also play an important role for kinase activity and regulation.
Another interesting similarity between the PIKKs is that they share the same chaperone system important for maturation of functional kinase complexes. The Tel2-Tti1-Tti2 (TTT) complex interacts with newly synthesized PIKKs and promotes proper folding of these large HEAT containing proteins in conjunction with the heat shock chaperone systems (35–37). The subunit Tel2 of the TTT complex binds the HEAT repeat of Rad3 and other PIKKS and promotes their stability and association with their binding partners (35, 36). Tel2 binds to a similar stretch of HEAT repeats in the middle of the domain, as does Rad26, suggesting that Tel2 occupies the binding sites until Rad26 is recruited and properly associated, hence the stabilizing function of the TTT complex for newly synthesized PIKKS. The crystal structure of Rad26 presented here shows strong similarity to the chaperone-binding protein HspBP1, which is part of the Hsp70 system, and it is tempting to speculate that the structural similarity reveals some common features in how the chaperone system and the interacting proteins bind their kinases. At least it has been shown that Hsp70 co-purifies with ATR-ATRIP in human cells and is part of the TTT complex (5, 36), suggesting further general principles in how the PIKKs are maturated and engaged in complex formation. In summary, this study reports the crystal structure of Rad26 and suggests it exists as a dimer and further gives insight into how Rad3 is recruited and arranged in a Rad3-Rad26 heterotetrameric complex.
Experimental procedures
Protein preparation
Rad3 and Rad26 from the thermophilic fungus M. thermophila was cloned into a modified pETDuet-1 vector, and proteins were expressed in Escherichia coli LOBSTR cells (Kerafast, Boston, MA) (38). Rad3 constructs were N-terminally fused to a SUMO tag, and both Rad3 and Rad26 constructs had a C-terminal His tag. Point mutations and truncations were generated by inverse PCR. Bacterial cultures were grown at 37 °C to an A600 of ∼0.6 and shifted to 18 °C for induction overnight with 0.2 mm isopropyl β-d-thiogalactopyranoside. The cells were lysed, cleared by centrifugation, loaded onto nickel-Sepharose 6 FF affinity resins (GE Healthcare), and extensively washed with (50 mm potassium phosphate, pH 7.6, 500 mm NaCl, 40 mm imidazole, and 5 mm β-mercaptoethanol) before being eluted with 300 mm imidazole. The proteins were further purified by cation-exchange chromatography on a HiTrapS FF (GE Healthcare) and eluted with a linear gradient from 0.05 to 1.2 m NaCl. For crystallization and for usage in pulldown experiments, the tags were removed with 3C protease, followed by a second cation-exchange chromatography step. The proteins were finally purified via gel filtration on either a Superdex 200 10/300 or Superose I 6 3.2/300 in GF buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm DTT).
Crystallization and structure determination
Purified selenomethionine-derivatized Rad26 protein (residues 373–841) was concentrated to 4–8 mg/ml and mixed with reservoir solution in a 1:1 ratio and crystallized by vapor diffusion against a reservoir of 0.1 m bis-Tris, pH 6.5, 10% (w/v) PEG 10K, and 0.2 m potassium sodium tartrate. Crystals were cryoprotected by transferring them stepwise into reservoir solution supplemented with 30% (w/v) ethylene glycol prior to flash-freezing in liquid nitrogen. Anomalous data were collected at the K absorption edge of selenium (Se) (λ = 0.979 Å) at the Beamline 24ID-C at Argonne National Laboratory equipped with a Pilatus detector. Data reduction was done with XDS (39), and heavy-atom substructure was determined using SHELXC/D/E (40), and interpretable electron density maps were obtained after density modification. Iterative model building was done in Coot (41), whereas coordinates and temperature factors were refined in phenix.refine (42) and iMDFF (43). The final Rad26 model contains residues 374–831 except for three loops (residues 385–390, 539–546, and 694–746) that were poorly defined in the electron density maps. The electrostatic potential was calculated using APBS plugin in PyMOL, and surface conservation was calculated using CONSURF (44). The data and refinement statistics are summarized in Table 1.
Pulldowns
All pulldown experiments were performed in PB buffer (20 mm potassium phosphate, pH 7.6, 200 mm NaCl, 40 mm imidazole, and 5 mm β-mercaptoethanol). Histidine-tagged Rad26 (5 μg) was incubated with untagged Rad3 (12 μg) on ice for 1 h to allow complex formation followed by the addition of 10 μl of equilibrated nickel-affinity resin. The mixtures were incubated on ice for an additional hour. The resins were spun down, and 10 μl of supernatant was removed as the flow-through fraction followed by washing three to five times with 150 μl of PB buffer to remove unbound protein. The bound proteins were eluted with 17 μl of BP buffer supplemented with 0.5 m imidazole, and the fractions were separated by SDS-PAGE. The ration between the SDS-PAGE band intensities of Rad3 and Rad26 were quantified with ImageJ.
Bio-layer interferometry
The binding of Rad26 to Rad3 was measured on an Octet RED bio-layer interferometer (Pall ForteBio). Histidine-tagged Rad26 were immobilized on anti-His (HIS2) biosensors (Pall ForteBio) at a concentration of 50 nm for 400 s. Interactions with Rad3 was measured in dilutions series at protein concentrations ranging from 50 to 6.25 nm in PB buffer (20 mm potassium phosphate, pH 7.6, 200 mm NaCl, 40 mm imidazole, and 5 mm β-mercaptoethanol) at 30 °C for 400 s followed by dissociation in PB buffer for 400 s. Background measurements using biosensors immobilized with Rad26 into PB buffer were subtracted to account for baseline drift. Sensorgrams were aligned to the baseline and fitted globally over the entire measurement using ForteBio data analysis 7.0 (Pall ForteBio). The KD values and standard errors were calculated from three measurements, and the data plots were prepared with GraphPad Prism 6.0.
SAXS
Rad26 (residues 185–841) was purified by gel filtration in GF buffer, and the monodisperse peak fraction was directly used for SAXS measurements. The data were collected at the EMBL P12 beamline at PETRA III equipped with at 2 m Pilatus detector. Scattering from Rad26 at concentration of 0.76 mg/ml was recorded in a temperature-controlled cell at 20 °C at a wavelength of 1.224 Å. Normalization, radial averaging and buffer subtraction of the data were done at the beamline by the automated pipeline with a Porod volume estimate of 190 nm3 and an estimated molecular mass Mr (from I(0)) of 160 kDa (45). Data analysis and ab initio low resolution modeling was done in DAMMIN (46), using the data range 0 < s < 2.0 nm−1. Calculation of theoretical scattering profiles of atomic structures and their fits to the experimental data were done with CRYSOL (47). The plots were prepared with GraphPad Prism 6.0.
Single-particle EM
Purified Rad3-Rad26 complex was isolated by gel filtration and diluted in GF buffer to ∼5 μg/ml and negatively stained with uranyl acetate (2% w/v) on carbon-film grids. Single-particle of the specimens were recorded on an FEI Tecnia G2 spirit electron microscope at 120 KeV, equipped with a Tietz-CCD camera (TVIPS) using −1 μm defocus and 67,000× magnification with a pixel size of 3.15 Å. A total of 5168 particles of the Rad3-Rad26 complex were picked and subjected to reference-free 2D classification in the program Relion (48).
Author contributions
K. R. A. designed and carried out the experiments, analyzed the data, and wrote the paper.
Supplementary Material
Acknowledgments
I am grateful to the staff at the APS NE-CAT beamline for assistance with synchrotron data collection, Rasmus Kjeldsen Jensen for SAXS data collection and processing, Arne Möller for help with EM, Tristian Croll and Jesper Lykkegaard Karlsen for valuable input on crystal structure refinement, and Gregers Rom Andersen and Dugald Reid for critical comments on the manuscript.
This work was supported by the Danish Diabetes Academy, which is supported by the Novo Nordisk Foundation, and by the Sapere Aude grant from the Danish Council for Independent Research, and a grant from the Lundbeck Foundation. The author declares that he has no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5LOI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Table S1 and Figs. S1–S4.
- DSB
- double-strand break
- PIKK
- PI3K-related kinase
- ssDNA
- single-stranded DNA
- SAXS
- small-angle X-ray scattering
- NTD
- N-terminal domain
- CTD
- C-terminal domain
- TTT
- Tel2-Tti1-Tti2
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Hunter T. (1995) When is a lipid kinase not a lipid kinase?: when it is a protein kinase. Cell 83, 1–4 [DOI] [PubMed] [Google Scholar]
- 2. Keith C. T., and Schreiber S. L. (1995) PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 270, 50–51 [DOI] [PubMed] [Google Scholar]
- 3. Perry J., and Kleckner N. (2003) The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell 112, 151–155 [DOI] [PubMed] [Google Scholar]
- 4. Bosotti R., Isacchi A., and Sonnhammer E. L. (2000) FAT: a novel domain in PIK-related kinases. Trends Biochem. Sci. 25, 225–227 [DOI] [PubMed] [Google Scholar]
- 5. Cortez D., Guntuku S., Qin J., and Elledge S. J. (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716 [DOI] [PubMed] [Google Scholar]
- 6. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R. 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., and Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 7. Paulovich A. G., and Hartwell L. H. (1995) A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82, 841–847 [DOI] [PubMed] [Google Scholar]
- 8. Ball H. L., Ehrhardt M. R., Mordes D. A., Glick G. G., Chazin W. J., and Cortez D. (2007) Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol. Cell Biol. 27, 3367–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zou L., and Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 10. Lee J., Kumagai A., and Dunphy W. G. (2007) The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 282, 28036–28044 [DOI] [PubMed] [Google Scholar]
- 11. Mordes D. A., Glick G. G., Zhao R., and Cortez D. (2008) TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 22, 1478–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shomura Y., Dragovic Z., Chang H.-C., Tzvetkov N., Young J. C., Brodsky J. L., Guerriero V., Hartl F. U., and Bracher A. (2005) Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol. Cell 17, 367–379 [DOI] [PubMed] [Google Scholar]
- 13. Ball H. L., and Cortez D. (2005) ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J. Biol. Chem. 280, 31390–31396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Falck J., Coates J., and Jackson S. P. (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611 [DOI] [PubMed] [Google Scholar]
- 15. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 16. Sawicka M., Wanrooij P. H., Darbari V. C., Tannous E., Hailemariam S., Bose D., Makarova A. V., Burgers P. M., and Zhang X. (2016) The dimeric architecture of checkpoint kinases Mec1ATR and Tel1ATM reveal a common structural organization. J. Biol. Chem. 291, 13436–13447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Itakura E., Sawada I., and Matsuura A. (2005) Dimerization of the ATRIP protein through the coiled-coil motif and its implication to the maintenance of stalled replication forks. Mol. Biol. Cell 16, 5551–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim S.-M., Kumagai A., Lee J., and Dunphy W. G. (2005) Phosphorylation of Chk1 by ATM- and Rad3-related (ATR) in Xenopus egg extracts requires binding of ATRIP to ATR but not the stable DNA-binding or coiled-coil domains of ATRIP. J. Biol. Chem. 280, 38355–38364 [DOI] [PubMed] [Google Scholar]
- 19. Bakkenist C. J., and Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 20. Williams D. R., Lee K.-J., Shi J., Chen D. J., and Stewart P. L. (2008) Cryo-EM structure of the DNA-dependent protein kinase catalytic subunit at subnanometer resolution reveals α helices and insight into DNA binding. Structure 16, 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammel M., Yu Y., Mahaney B. L., Cai B., Ye R., Phipps B. M., Rambo R. P., Hura G. L., Pelikan M., So S., Abolfath R. M., Chen D. J., Lees-Miller S. P., and Tainer J. A. (2010) Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J. Biol. Chem. 285, 1414–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walker J. R., Corpina R. A., and Goldberg J. (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412, 607–614 [DOI] [PubMed] [Google Scholar]
- 23. Yip C. K., Murata K., Walz T., Sabatini D. M., and Kang S. A. (2010) Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 38, 768–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaubitz C., and Loewith R. (2012) Amino acid signaling in high definition. Structure 20, 1993–1994 [DOI] [PubMed] [Google Scholar]
- 25. Aylett C. H., Sauer E., Imseng S., Boehringer D., Hall M. N., Ban N., and Maier T. (2016) Architecture of human mTOR complex 1. Science 351, 48–52 [DOI] [PubMed] [Google Scholar]
- 26. Baretić D., Berndt A., Ohashi Y., Johnson C. M., and Williams R. L. (2016) Tor forms a dimer through an N-terminal helical solenoid with a complex topology. Nat. Commun. 7, 11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Unsal-Kaçmaz K., and Sancar A. (2004) Quaternary structure of ATR and effects of ATRIP and replication protein A on its DNA binding and kinase activities. Mol. Cell Biol. 24, 1292–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wright J. A., Keegan K. S., Herendeen D. R., Bentley N. J., Carr A. M., Hoekstra M. F., and Concannon P. (1998) Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc. Natl. Acad. Sci. U.S.A. 95, 7445–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Edwards R. J., Bentley N. J., and Carr A. M. (1999) A Rad3-Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nat. Cell Biol. 1, 393–398 [DOI] [PubMed] [Google Scholar]
- 30. Liu S., Ho C. K., Ouyang J., and Zou L. (2013) Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 2175–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ball H. L., Myers J. S., and Cortez D. (2005) ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 16, 2372–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. You Z., Chahwan C., Bailis J., Hunter T., and Russell P. (2005) ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell Biol. 25, 5363–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin S., Kharbanda S., Mayer B., Kufe D., and Weaver D. T. (1997) Binding of Ku and c-Abl at the kinase homology region of DNA-dependent protein kinase catalytic subunit. J. Biol. Chem. 272, 24763–24766 [DOI] [PubMed] [Google Scholar]
- 34. Rivera-Calzada A., Maman J. P., Spagnolo L., Pearl L. H., and Llorca O. (2005) Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure 13, 243–255 [DOI] [PubMed] [Google Scholar]
- 35. Takai H., Wang R. C., Takai K. K., Yang H., and de Lange T. (2007) Tel2 regulates the stability of PI3K-related protein kinases. Cell 131, 1248–1259 [DOI] [PubMed] [Google Scholar]
- 36. Takai H., Xie Y., de Lange T., and Pavletich N. P. (2010) Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 24, 2019–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hurov K. E., Cotta-Ramusino C., and Elledge S. J. (2010) A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 24, 1939–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andersen K. R., Leksa N. C., and Schwartz T. U. (2013) Optimized E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins 81, 1857–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Croll T. I., Smith B. J., Margetts M. B., Whittaker J., Weiss M. A., Ward C. W., and Lawrence M. C. (2016) Higher-resolution structure of the human insulin receptor ectodomain: multi-modal inclusion of the insert domain. Structure 24, 469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashkenazy H., Erez E., Martz E., Pupko T., and Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Franke D., Kikhney A. G., and Svergun D. I. (2012) Automated acquisition and analysis of small angle X-ray scattering data. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 689, 52–59 [Google Scholar]
- 46. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svergun D., Barberato C., and Koch M. H. (1995) CRYSOL–a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 48. Scheres S. H. (2012) A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.