

Abstract
Aberrant protein aggregation underlies a variety of age-related neurodegenerative disorders, including Alzheimer's and Parkinson's diseases. Little is known, however, about the molecular mechanisms that modulate the aggregation process in the cellular environment. Recently, MOAG-4/SERF has been identified as a class of evolutionarily conserved proteins that positively regulates aggregate formation. Here, by using nuclear magnetic resonance (NMR) spectroscopy, we examine the mechanism of action of MOAG-4 by characterizing its interaction with α-synuclein (α-Syn). NMR chemical shift perturbations demonstrate that a positively charged segment of MOAG-4 forms a transiently populated α-helix that interacts with the negatively charged C terminus of α-Syn. This process interferes with the intramolecular interactions between the N- and C-terminal regions of α-Syn, resulting in the protein populating less compact forms and aggregating more readily. These results provide a compelling example of the complex competition between molecular and cellular factors that protect against protein aggregation and those that promote it.
Keywords: α-synuclein (α-synuclein), neurodegenerative disease, nuclear magnetic resonance (NMR), protein aggregation, protein structure
Introduction
Insoluble filamentous aggregates, commonly referred to as amyloid fibrils, are associated with many human neurodegenerative disorders, and are typically composed of disease-specific aggregation-prone proteins, including amyloid β (Aβ)5 in Alzheimer's disease, and α-synuclein (α-Syn) in Parkinson's disease (1). The presence of these aggregation-prone proteins may lead to failure of protein homeostasis and loss of normal biological function (2–4). Although such aggregates are key pathological hallmarks of neurodegenerative disorders (5), much remains to be understood about the structural details of the molecular mechanisms leading to aggregate formation and those that protect against it, as the transient nature of species populated during the aggregation process makes it difficult to observe these in experiment.
It has been recently shown that a class of evolutionarily conserved proteins promotes aggregate formation (6). A genetic screen in Caenorhabditis elegans models led to the identification of a positive regulator of protein aggregation, called “modifier of aggregation 4” (MOAG-4). Inactivation of MOAG-4 suppresses protein aggregation and associated toxicity in C. elegans models for aggregation of polyglutamine, Aβ, and α-Syn (6). It has also been shown that a human ortholog of MOAG-4, small EDRK-rich factor (SERF) 1A, accelerates the aggregation of a broad range of amyloidogenic proteins in vitro (7). The ensuing biophysical analysis of the interaction between SERF1A and α-Syn suggested that the acceleration of α-Syn aggregation originates from a transient interaction in the initial stage of the process (7). However, structural details of MOAG-4/SERF proteins, and the molecular mechanism by which they promote aggregate formation have so far remained elusive.
To address this problem, we investigate here the effects of MOAG-4 on the aggregation of α-Syn by monitoring the kinetics of this process using thioflavin T (ThT), a dye known to exhibit enhanced fluorescence upon binding amyloid fibrils (8, 9). The aggregation assay was carried out at 37 °C under agitation (10). We found that MOAG-4 accelerates aggregate formation of α-Syn, which is consistent both with in vitro studies showing that aggregation of α-Syn is promoted by the human ortholog SERF1A (7), as well as with cellular studies attesting that loss of MOAG-4 suppresses aggregation and toxicity of α-Syn (6).
We then use NMR spectroscopy to determine the structural ensemble of MOAG-4, and provide structural evidence for the interaction between MOAG-4 and α-Syn. Although spontaneous (i.e. unseeded) in vitro formation of α-Syn amyloid fibrils at neutral pH under quiescent conditions is slow (11) and requires at least a few weeks (12), all NMR experiments were carried out at low temperature to avoid the possible formation of small oligomeric aggregates during the measurements (13). At pH 6.0 and 10 °C, the protein did not form detectable aggregates in the NMR tube during the NMR experiments. Our results show that MOAG-4 affects the intra-molecular interactions within α-Syn that bring together its N- and C-terminal regions. The presence of MOAG-4 is thus relevant as the transient interactions between the terminal regions of α-Syn result in rather compact conformations that appear to be relatively resistant to aggregation (14–18). In this way, MOAG-4 effectively competes with this self-protective mechanism, such that α-Syn takes on a less compact form, and may thereby become poised for aggregation and subsequent amyloid formation.
Results
MOAG-4 enhances α-Syn aggregate formation
To probe for a direct effect of MOAG-4 on α-Syn aggregation, we monitored the enhancement of ThT fluorescence in an aggregation kinetics assay (Fig. 1A). Amyloid formation of α-Syn was monitored in the absence or presence of an equimolar concentration of MOAG-4 at pH 6.0 and 37 °C. Some of the data displayed biphasic behavior, which cannot be readily explained by simple nucleation-dependent aggregation mechanisms. Although such complex behavior may be an intrinsic and relevant property of α-Syn aggregation induced by MOAG-4, it remains extremely challenging to extract from the data the underlying mechanistic information (19, 20). Thus, although we could not extract from the experimental data the kinetic parameters of the underlying microscopic processes, we found that, whereas α-Syn aggregation without MOAG-4 takes several days (10), MOAG-4 accelerated the process significantly, with a lag time of ∼3 h (Fig. 1A).
Figure 1.

MOAG-4 accelerates α-Syn aggregation. A, the aggregation kinetics of α-Syn (12.5 μm) in the presence (red) and absence (black) of an equal molar concentration of MOAG-4 was monitored by measuring ThT fluorescence at pH 6.0 and 37 °C. The data represent nine replicates, where one solution was divided into nine samples and the assay was run in parallel in the same plate. B, SDS-PAGE analysis of the soluble and insoluble fractions of the sample at different incubation times. The aggregation of α-Syn (200 μm) was made at pH 7.4 and 37 °C in the presence of an equal molar concentration of MOAG-4.
We then examined whether MOAG-4 is integrated into the α-Syn fibrils (Fig. 1B). The α-Syn aggregation experiment was performed at pH 7.4 and 37 °C in the presence of MOAG-4. α-Syn aggregation was slower at pH 7.4 than 6.0, in agreement with previous studies showing that a decrease in pH enhanced the kinetics of α-Syn amyloid fibril formation (21, 22). SDS-PAGE analysis confirmed that the insoluble fraction of the sample did not contain MOAG-4 (Fig. 1B), suggesting that the modulation of the α-Syn amyloidogenesis by MOAG-4 occurred during an early and transient interaction with α-Syn.
MOAG-4 forms a mutable structure with α-helical propensity
The 1H-15N heteronuclear single-quantum correlation (HSQC) spectrum of MOAG-4 (Fig. 2A) has the typical appearance expected for an intrinsically disordered protein, with very limited amide chemical shift dispersion. To characterize the structure in more detail, sequential assignment of backbone and aliphatic side chain 1H, 13C, and 15N resonances was achieved using a set of three-dimensional (3D) triple-resonance experiments (see “Experimental procedures”). Upon attainment of the resonance assignments, an ensemble of MOAG-4 structures was determined using molecular dynamics simulations restrained with backbone NMR chemical shifts (23, 24). The corresponding structural ensemble (Fig. 2B) displays a protein structure with a very high degree of disorder. Despite the global disorderliness, virtually all members of the structural ensemble contain an α-helical segment for residues Asp45-Asn70. When superposing the structures on the coordinates for these residues, the remainder of the protein appears spatially comparatively less well defined. However, a second, lowly-populated, transient α-helix is present in these ensembles, comprising residues Asp8-Lys22 (Fig. 2C). Neither significantly populated compact states nor local β-sheet conformations were found. The experimental chemical shifts were well correlated with those back-calculated from the MOAG-4 ensemble that we determined. To validate the MOAG-4 ensemble, we further measured NOE intensities and compared these with the values back-calculated from the ensemble of structures. The good results of this validation attest the accuracy of the ensemble of structures that we describe. Taken together, MOAG-4 is a highly mutable protein containing transiently populated elements of secondary structure, but devoid of tertiary interactions.
Figure 2.

MOAG-4 forms a malleable structure with α-helical propensity. A, 1H-15N HSQC spectrum of MOAG-4 (left), and a zoomed region (right). The amino acid sequence of MOAG-4 is shown on the top, using the following color scheme: basic (blue), acidic (red), alanine (green). B, ensemble of MOAG-4 structures. The structures are superimposed on the α-helical residues Asp45-Asn70. C, the population of α-helical structure is calculated from the MOAG-4 ensemble.
As can be seen in Fig. 2A, the peak intensity for the residues between Asp45 and Asn70 is relatively low. The explanation for this observation can now be rationalized with help of the structure: the stretch Asp45-Asn70 forms a long helical structure, in which amide bond vectors pointing along the direction of the axis of the α-helix experience fast transverse spin relaxation due to slow overall tumbling motion.
Electrostatic interactions between MOAG-4 and α-Syn
To determine the binding affinity and the location of the binding site on MOAG-4, a series of 1H-15N HSQC spectra for uniformly 13C,15N-labeled MOAG-4 were recorded with different amounts of unlabeled α-Syn. Fig. 3A shows the chemical shift changes observed for MOAG-4 upon the binding of α-Syn. The fit to saturating binding curves yielded a dissociation constant (Kd) value of 10.8 ± 0.8 μm. The backbone chemical shifts of amide proton (1HN), nitrogen (15N), and carbonyl (13C′) nuclei of MOAG-4 were obtained from 3D HNCO experiments (25, 26) at [α-Syn]/[MOAG-4] = 0 and 8. The chemical shift perturbations of backbone 15N and 13C′ nuclei (Fig. 3B) indicate that binding to α-Syn induces significant changes in MOAG-4 for a contiguous stretch of residues between Arg3 and Gly26, corresponding to the first α-helical segment, a region that is rich in positively charged amino acid residues (net charge +6). The structural propensities of MOAG-4, free and bound to α-Syn, were calculated from the backbone 1HN, 15N, and 13C′ chemical shifts using the program ncSPC (27). The α-helical propensity of the N-terminal segment increases slightly upon binding to α-Syn, indicating a direct effect of α-Syn on the MOAG-4 conformational ensemble (Fig. 3C).
Figure 3.

The amino terminus of MOAG-4 interacts with α-Syn. A, changes in the 1H-15N HSQC spectra of MOAG-4 for increasing amounts of α-Syn. [α-Syn]/[MOAG-4] = 0, 0.2, 0.4, 0.7, 1, 2, and 4 (from red to blue). The inset in panel A is the binding curve for Gly26. The solid line was obtained by global fitting of the chemical shift changes to Equation 1. B, backbone 13C′ (black) and 15N (magenta) chemical shift changes of MOAG-4 upon binding to α-Syn. The precision error is less than 0.03 ppm. C, structural propensity of MOAG-4 in the absence (red) and presence (blue) of α-Syn obtained from the backbone 1HN, 15N, and 13C′ chemical shifts.
Next, we examined the structural changes of α-Syn upon interaction with MOAG-4 by monitoring the corresponding chemical shift changes. Fig. 4A shows a series of BEST-TROSY 1H-15N correlation spectra (28) of uniformly 13C,15N-labeled α-Syn recorded with different amounts of unlabeled MOAG-4. Spectral assignment of α-Syn was taken from BMRB entry ID 18857, and confirmed using BEST-TROSY 3D HNCO and HN(CA)CO experiments (29, 30) before and after the titration. Chemical shift perturbations for backbone 15N and 13C′ upon binding to MOAG-4 (Fig. 4B) suggest that MOAG-4 interacts with the C terminus of α-Syn. This result parallels the previous finding that the interaction of α-Syn with SERF1A is localized to the C terminus of α-Syn (7). The C-terminal region of α-Syn (residues Glu110-Ala140) is acidic and negatively charged (net charge at neutral pH of −12), constituting a favorable binding location for positively charged proteins, such as MOAG-4 and SERF1A. Although 15N chemical shift changes are also observed for the N-terminal region, and to a lesser extent for the NAC (non-Aβ component of Alzheimer's disease amyloid) region, as we will show below, we found that these collateral effects on structure are likely to result from the loss of pre-existing interactions within the α-Syn ensemble. Fig. 4C shows the structural propensity of α-Syn in the absence and presence of MOAG-4, calculated from the backbone 1HN, 15N, and 13C′ chemical shifts using the program ncSPC (27). We found that, consistent with previous studies (27, 31), deviations of measured chemical shifts from their random coil values are very small throughout the sequence. We did not observe any significant differences in the structural propensities of α-Syn in the presence and absence of MOAG-4, but the conformational exchange of α-Syn with a less compact form upon binding to MOAG-4 was confirmed by an increase of backbone flexibility (see below).
Figure 4.

The structure of α-Syn is affected in several regions upon interaction with MOAG-4. A, BEST-TROSY 1H-15N correlation spectra of α-Syn (45 μm) with an increasing amount of unlabeled MOAG-4. [MOAG-4]/[α-Syn] = 0, 0.3, 0.8, 1.8, and 5.2 (from red to blue). The cross-peak for Ala140, indicated by a dashed circle, is aliased in the 15N dimension. B, backbone 13C′ (black) and 15N (magenta) chemical shift changes of α-Syn upon binding to MOAG-4. The precision error is less than 0.03 ppm. C, structural propensity of α-Syn in the absence (red) and presence (blue) of MOAG-4 obtained from the backbone 1HN, 15N, and 13C′ chemical shifts.
α-Syn becomes less compact upon binding to MOAG-4
To examine the conformational change of α-Syn upon binding to MOAG-4, we measured the backbone 15N transverse relaxation rate constants (R2) of α-Syn in the presence and absence of MOAG-4 (Fig. 5A), and obtained the changes of the backbone 15N R2 values induced by MOAG-4 (Fig. 5C). The R2 values for the C terminus of α-Syn are larger in the presence of MOAG-4 than those without MOAG-4, indicative of restricted motion by addition of MOAG-4. These effects can be attributed to direct binding of α-Syn and MOAG-4 mediated by polyvalent electrostatic interactions. At the same time, a reduction in the R2 rates was observed for the N-terminal region (residues Val3-Thr59), indicating that the N terminus of α-Syn becomes more dynamic upon binding.
Figure 5.

Intra-molecular electrostatic interactions in α-Syn are released by addition of MOAG-4 or salt. A, backbone 15N R2 relaxation rates of α-Syn in the absence (black) and presence (red) of MOAG-4. B, backbone 15N R2 relaxation rates of α-Syn without (black) and with (blue) 100 mm NaCl. C, changes in backbone 15N R2 rates of α-Syn upon binding to MOAG-4 (ΔR2,MOAG-4). D, changes in backbone 15N R2 rates of α-Syn upon addition of 100 mm NaCl (ΔR2,NaCl). E, correlation between ΔR2,MOAG-4 and ΔR2,NaCl.
These results offer insight into the mechanism by which MOAG-4 enhances α-Syn aggregation. It has been proposed that, although α-Syn is highly dynamic as typical of intrinsically disordered proteins, the ensemble of α-Syn in the native state is more compact than that expected for a random coil polypeptide chain due to the presence of long-range transient intra-molecular interactions (14–18, 32, 33), originating from an electrostatic attraction between the slight positive charge in the central region (residues Gly31-Leu100) and the negatively charged C-terminal region (34). These intra-molecular interactions and the associated compaction of α-Syn shield the aggregation-prone central region of the protein from forming inter-molecular interactions under physiological conditions in the cell, thereby protecting it against aggregation (35).
NaCl reduces intra-molecular interactions within α-Syn and enhances aggregation
To test whether the action of MOAG-4 could be emulated by screening the electrostatic interaction between the N and C termini by salt, we measured the backbone 15N R2 relaxation rates at pH 6.0 and 10 °C in the presence and absence of 100 mm NaCl (Fig. 5B). In full agreement with a previous report (36), addition of NaCl reduced the backbone 15N R2 relaxation rates in the N- and C-terminal regions (Fig. 5D), indicating that the screening of electrostatic interactions increased mobility in both domains. A positive correlation between the changes in the 15N R2 relaxation rates at the N terminus of α-Syn by addition of NaCl and those upon interaction with MOAG-4 (Fig. 5E) indicates that the interaction of MOAG-4 with the C terminus of α-Syn abrogates pre-existing intra-molecular interactions, increasing the dynamics at the N terminus of α-Syn. These changes, in turn, affect the chemical shifts for regions outside the C terminus (Fig. 4), explaining the pervasive pattern observed throughout the protein sequence. The study of Bai et al. (36) shows clearly how paramagnetic relaxation enhancement effects can remain restricted to two regions of the protein, whereas leading to chemical shift changes that are transmitted also to regions other than those in direct contact.
Previous studies have shown that increasing the salt concentration accelerates the formation of α-Syn amyloid fibrils (37, 38). To examine the effect of NaCl on the formation of α-Syn amyloid fibrils, the aggregation kinetics of α-Syn at 100 mm NaCl was monitored by measuring ThT fluorescence at pH 6.0 and 37 °C. Overall, and consistent with previous studies (37, 38), the introduction of salt increases α-Syn aggregation (Fig. 6), in a way similar to that observed for MOAG-4 (Fig. 1A), suggesting a fundamental link between the modulation of intra-molecular electrostatic interactions and α-Syn aggregation.
Figure 6.
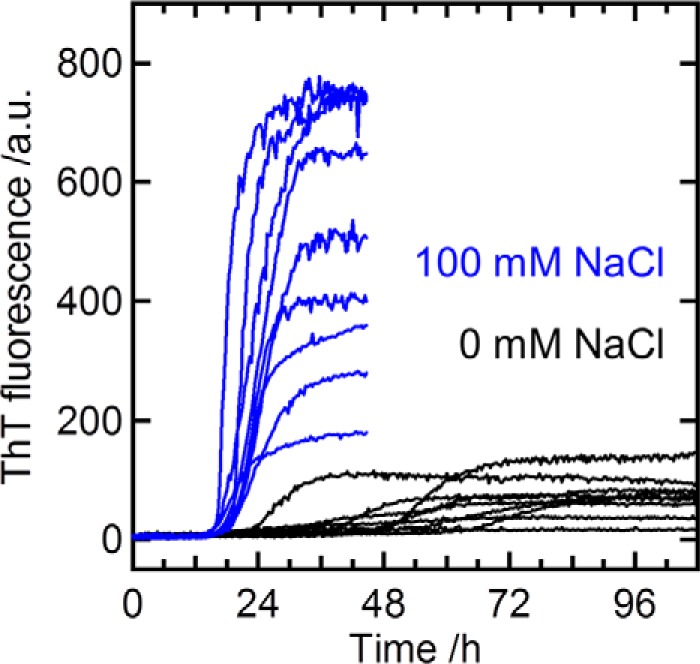
Salt accelerates α-Syn aggregation. Kinetics of α-Syn (12.5 μm) aggregation in the absence (black, the same data as in Fig. 1A) and presence (blue) of 100 mm NaCl. α-Syn aggregation was monitored by measuring ThT fluorescence at 37 °C. The data represent nine replicates, where one solution was divided into nine samples and the assay was run in parallel in the same plate.
Discussion
In vitro at neutral pH, and under quiescent conditions, spontaneous aggregation of α-Syn requires at least a few weeks (12). In this study, soluble α-Syn persists without aggregate formation during NMR measurement at 10 °C (Figs. 3–5). On the other hand, aggregation is accelerated upon agitation of the protein solution (10, 12), where the lag time of α-Syn fibril formation is reduced from weeks/months to hours/days (10) (Figs. 1 and 6). Although the lag phase preceding assembly was several days in the absence of MOAG-4 or NaCl, α-Syn readily self-associated with a lag time of ∼3 and ∼16 h in the presence of an equimolar amount of MOAG-4 and 100 mm NaCl, respectively.
To understand the role of MOAG-4 on α-Syn aggregation, we note that it is well known that electrostatic effects play key roles in the formation of amyloid fibrils (39–42). The effects of these interactions on protein aggregation have been extensively studied for a variety of amyloidogenic proteins, including insulin (43), α-Syn (37, 38), β2-microglobulin (44), glucagon (45), islet amyloid polypeptide (46), and the mouse prion protein (47). The C terminus of α-Syn is rich in negatively charged amino acid residues, and α-Syn remains soluble for an extended period of time because of inter-molecular electrostatic repulsion. The shielding of charge repulsion by NaCl decreases the free energy barrier of nucleation (39, 40), and enhances the kinetics of α-Syn aggregation (Fig. 6). Neutralization of charge repulsion can also be brought about by polyamine compounds (14, 48, 49) and metal cations (50–52), and these have been shown to enhance the kinetics of α-Syn aggregation. Furthermore, C terminally truncated recombinant α-Syn readily aggregates (53). These observations indicate that the negatively charged C terminus plays an inhibitory role in the formation of α-Syn amyloid fibrils. More recently, it was observed that interactions between the N and C termini persist in cells, despite the high ionic strength of the cytoplasm (35). It was also suggested that hydrophobic residues of the central amyloidogenic region of α-Syn are shielded from interactions with cellular components (35), although experiments with paramagnetic co-solute molecules in vitro have revealed solvent accessibility of the entire chain (54, 55).
Furthermore, it has been suggested that the amyloid-modifying properties of MOAG-4/SERF proteins are evolutionary conserved (6). The binding affinity of MOAG-4 to α-Syn obtained in this study (Kd = 10.8 ± 0.8 μm) is comparable with the Kd value for the binding of SERF1A, a human ortholog of MOAG-4, to α-Syn (7). The inter-molecular interaction with SERF1A is localized to the C terminus of α-Syn, and the C terminally truncated mutant, α-Syn(1–110), showed a very weak binding affinity to SERF1A (Kd > 1 mm) (7). Furthermore, the aggregation kinetics of α-Syn(1–110) was not affected by SERF1A (7). Therefore, the chemical shift perturbations (Fig. 4B) and a reduction in the backbone 15N R2 rates (Fig. 5C) observed for the N-terminal region of α-Syn upon binding to MOAG-4 is likely due to a combination of effects, as explained below.
Our experimental data suggest that the acceleration of α-Syn aggregation induced by MOAG-4 is due to electrostatic interactions between the two oppositely charged proteins (Fig. 7). Although α-Syn is typified as an intrinsically disordered protein, the long-range nature of electrostatic interactions allows transient intra-molecular contacts within α-Syn to occur (14–18, 32–36). As a result, the ensemble of conformations in the native state, although disordered, remains rather compact. Direct binding of MOAG-4 to the negatively charged C terminus of α-Syn could be facilitated because positively charged residues are distributed evenly throughout the first helical segment of MOAG-4. At the same time, the chemical shift perturbations and a reduction in the backbone 15N R2 rates were collaterally observed for the N-terminal region of α-Syn (Figs. 4B and 5C). These results reveal that the highly positively charged first α-helical segment of MOAG-4 binds the negatively charged C terminus of α-Syn, effectively competing with pre-existing intra-molecular interactions within α-Syn, thereby reducing the compactness of the protein.
Figure 7.
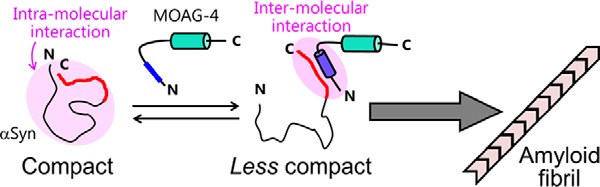
Enhancement of aggregation kinetics via loss of intra-molecular electrostatic interactions. The negatively charged C terminus of α-Syn and positively charged segment of MOAG-4 are shown in red and blue, respectively.
Our NMR data and aggregation assay demonstrate that MOAG-4 directly binds to α-Syn and enhances its aggregation, whereas the resulting insoluble fraction did not contain MOAG-4 (Fig. 1B). The aggregation-promoting activity of SERF1A results from early and transient interactions with α-Syn (7), which act catalytically. Electron microscopic images of the mature α-Syn amyloid fibrils obtained in the absence and presence of SERF1A did not show morphological differences (7). Although it still remains to be elucidated whether the reduction in compactness of α-Syn is required for the formation of α-Syn amyloid fibrils (32, 33), our results now shine light on the structures of the molecular species involved in the early and transient interaction between MOAG-4 and α-Syn, and the mechanism by which electrostatic interactions may prevent or drive protein aggregation. We also note that the type of interactions between MOAG-4 and α-Syn detected in the NMR measurements that we reported are likely to be conserved under the conditions used in the aggregation assays.
In closing, we observe that the competition between inter-molecular and intra-molecular electrostatics may offer ways for cells to cope with aggregation-prone proteins in general. By protecting the hydrophobic regions in proteins through intra-molecular interactions leading to the formation of compact forms, the fraction of proteins susceptible to aggregation can be maintained at a manageable level. In this view, proteins like MOAG-4/SERF would be employed to reduce the self-protection provided by such a compaction, and effectively convert disordered proteins into aggregates when concentrations reach dangerous levels. In addition, proteins like MOAG-4/SERF can use this mechanism to support other physiological roles, as in the generation of functional amyloids.
Experimental procedures
Sample preparation
Expression and purification of human α-Syn was described previously (56). For expression of uniformly 13C,15N-labeled and 15N-labeled α-Syn, Escherichia coli cells were grown in M9 minimal medium supplemented with 15N-labeled NH4Cl and 13C-labeled d-glucose as sole source for 15N and 13C, respectively. Uniformly 13C,15N-labeled and unlabeled MOAG-4 was obtained from Giotto Biotech (Florence, Italy).
ThT fluorescence assay
Aggregation of α-Syn was performed at a concentration 12.5 μm. Lyophilized α-Syn was dissolved in 10 mm sodium phosphate (pH 6.0) containing 40 μm ThT. A clear-bottom 96-well plate with a final volume of 150 μl of assay solution in each well was loaded into a Genios Pro (Tecan) fluorescence plate reader and incubated at 37 °C (10). ThT fluorescence was measured with excitation and emission at 448 and 485 nm, respectively. A 3-mm glass bead was added to each of the wells, and the plate was subjected to cycles of an orbital shaking at 300 rpm for 16 min followed by quiescence for 4 min. Because amyloid fibril formation often varies across sample replicates and low reproducibility of the kinetic assays is attributed to the inherently stochastic nature of the aggregation process (57–59), the experiment was performed in nine replicates, where one solution was divided into nine samples and the assay was run in parallel in the same plate. Binding of MOAG-4 to ThT was negligible.
SDS-PAGE analysis
A 250-μl volume of 200 μm α-Syn in 50 mm Tris-HCl, 150 mm NaCl, and 3 mm NaN3 (pH 7.4) was mixed with 250 μl of 200 μm MOAG-4 in the same buffer, taped to the rack of an Eppendorf themomixer 5436, and agitated at 37 °C and 1,400 rpm. After given time points, 35-μl samples were taken and frozen immediately at −20 °C. Each sample was then centrifuged at 20,000 × g, and the insoluble fraction was washed twice and resuspended in 50 μl of 50 mm Tris-HCl, 150 mm NaCl, and 3 mm NaN3 (pH 7.4). The soluble and insoluble samples (5 μl) were run on a 12% NuPAGE SDS-PAGE gel (Life Technologies, Germany), followed by Coomassie staining.
NMR assignment experiments
For resonance assignment of MOAG-4, all NMR spectra were recorded at 10 °C on a Bruker Avance III spectrometer at 1H frequency of 500 MHz. Backbone resonance assignments of uniformly 13C,15N-labeled MOAG-4 (0.4 mm) in 10 mm sodium phosphate (pH 6.0) containing 7%(v/v) D2O for field lock and 50 μm 4,4-dimethyl-4-silapentane-1-sulfonate (DSS) as chemical shift reference (60) were made by using 3D HNCO (25, 26), HN(CA)CO (61), HNCACB (26), HN(CA)NNH (62), (HN)CO(CO)NH (63, 64), and (H)N(COCO)NH (64, 65) spectra. In the (HN)CO(CO)NH and (H)N(COCO)NH experiments, the mixing period of the MOCCA-XY16 scheme (66) was 390 ms for 13C′ magnetization transfer via 3JC'C' couplings. Furthermore, 3D (H)CC(CO)NH and H(CC)(CO)NH spectra were recorded for assignment of aliphatic 1H and 13C side chain resonances and identification of the amino acid types from their chemical shifts (67). In the (H)CC(CO)NH and H(CC)(CO)NH experiments, the mixing period was 15 ms for aliphatic 13C magnetization transfer. Spectra were processed using nmrPipe (68) and analyzed with Sparky (Goddard and Kneller SPARKY 3, University of California, San Francisco).
Structural calculation and NOESY experiments
The ensemble of MOAG-4 structures was obtained using the replica-averaged metadynamics method (23) with chemical shift restraints implemented with the program CamShift (24). Back-calculations of the backbone (13Cα, 13Cβ, 13C′, 1Hα, 1HN, and 15N) and side chain (1Hβ, 1Hγ, 1Hδ, and 1Hϵ) chemical shifts from the MOAG-4 ensemble were performed using the programs SPARTA+ (69) and SHIFTX2 (70), respectively. The population of α-helical structure was calculated using the DSSP software (71). Two types of 15N-resolved 3D 1H-1H NOESY spectra (i.e. HSQC-NOESY-HSQC (3D 15N-15N-1H) and NOESY-HSQC (3D 1H-15N-1H)) were recorded to collect NOEs for structural validation (72). These experiments were performed at a MOAG-4 concentration of 400 μm on a Bruker Avance III 950 MHz spectrometer at 10 °C.
Titration of MOAG-4 with α-Syn
NMR titration experiments (73) were performed at 10 °C on a Bruker Avance III 950 MHz spectrometer equipped with a TCI cryogenic probehead. A series of 2D 1H-15N HSQC NMR spectra of uniformly 13C,15N-labeled MOAG-4 (100 μm) in 10 mm sodium phosphate (pH 6.0), containing 2% (v/v) D2O and 40 μm DSS, were recorded with increasing amounts of unlabeled α-Syn. A 3D HNCO spectrum was recorded after the titration with α-Syn, where [α-Syn]/[MOAG-4] = 8, to obtain the 1HN, 15N and 13C′ chemical shifts. The binding curves were obtained from chemical shift changes during the titration in a fast-exchange manner, and fitted to the following equation,
| (Eq. 1) |
where Δδobs is the observed chemical shift change from the free state, and Δδmax is the maximum chemical shift change upon saturation. Kd is the dissociation constant. Nonlinear least-squares fitting was performed with Igor Pro software.
15N R2 relaxation experiment
The transverse relaxation rate constant, R2, of α-Syn was obtained from an 15N Hahn-echo pulse sequence (74). Two spectra were recorded, one without the relaxation delay and the other with the relaxation delay. The R2 value was obtained by the following equation,
| (Eq. 2) |
where I(τ) values are resonance intensities at the relaxation delay τ. Temperature compensation was included to minimize the effects of heating caused by 1H decoupling during the 15N chemical shift evolution and relaxation delay periods (75). Proton x and y purge pulses were applied prior to the recycling delay to ensure that the amount of water magnetization present at the beginning of each scan is always identical (76). The spectra were recorded at 10 °C on a Bruker Avance III 950 MHz spectrometer equipped with a TCI cryogenic probehead (with/without MOAG-4; Fig. 5, A and C) or a Bruker Avance III 500 MHz spectrometer (with/without NaCl; Fig. 5, B and D).
Author contributions
F. A. A. M. and E. A. A. N. designed the experiments. E. A. A. N., A. M.-C., S. F. F., and C. B. A. provided materials. Y. Y., M. A. H., and S. F. F. conducted the experiments. Y. Y., M. A. H., P. K., M. V., and F. A. A. M. analyzed the data. Y. Y., E. A. A. N., and F. A. A. M. wrote the paper. All authors agreed on its contents.
Acknowledgments
This work was performed using the NMR spectrometers at the Danish Center for Ultra-High Field NMR Spectroscopy. Prof. Daniel E. Otzen (Aarhus University) is acknowledged for the use of the fluorescence plate reader. We thank Dr. Dennis W. Juhl (Aarhus University) for valuable comments, Tania A. Nielsen and Dr. Mengjun Xue (Aarhus University) for sample preparation, and Cagla Sahin (Aarhus University) for assistance with the setup of the fluorescence plate reader.
The authors declare that they have no conflicts of interest with the contents of this article.
Resonance assignment of MOAG-4 at pH 6.0 is available from the BioMagResBank (http://bmrb.wisc.edu/) with accession number 27058.
- Aβ
- amyloid β
- α-Syn
- α-synuclein
- MOAG-4
- modifier of aggregation 4
- SERF
- small EDRK-rich factor
- ThT
- thioflavin T
- HSQC
- heteronuclear single-quantum correlation
- DSS
- 4,4-dimethyl-4-silapentane-1-sulfonate.
References
- 1. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2. Hipp M. S., Park S. H., and Hartl F. U. (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514 [DOI] [PubMed] [Google Scholar]
- 3. Knowles T. P., Vendruscolo M., and Dobson C. M. (2014) The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 [DOI] [PubMed] [Google Scholar]
- 4. Sin O., and Nollen E. A. (2015) Regulation of protein homeostasis in neurodegenerative diseases: the role of coding and non-coding genes. Cell. Mol. Life Sci. 72, 4027–4047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. López-Otín C., Blasco M. A., Partridge L., Serrano M., and Kroemer G. (2013) The hallmarks of aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Ham T. J., Holmberg M. A., van der Goot A. T., Teuling E., Garcia-Arencibia M., Kim H. E., Du D., Thijssen K. L., Wiersma M., Burggraaff R., van Bergeijk P., van Rheenen J., Jerre van Veluw G., Hofstra R. M., Rubinsztein D. C., and Nollen E. A. (2010) Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612 [DOI] [PubMed] [Google Scholar]
- 7. Falsone S. F., Meyer N. H., Schrank E., Leitinger G., Pham C. L., Fodero-Tavoletti M. T., Holmberg M., Dulle M., Scicluna B., Gesslbauer B., Rückert H. M., Wagner G. E., Merle D. A., Nollen E. A., Kungl A. J., Hill A. F., Cappai R., and Zangger K. (2012) SERF protein is a direct modifier of amyloid fiber assembly. Cell Rep. 2, 358–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naiki H., Higuchi K., Hosokawa M., and Takeda T. (1989) Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T. Anal. Biochem. 177, 244–249 [DOI] [PubMed] [Google Scholar]
- 9. LeVine H., 3rd. (1999) Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309, 274–284 [DOI] [PubMed] [Google Scholar]
- 10. Giehm L., and Otzen D. E. (2010) Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal. Biochem. 400, 270–281 [DOI] [PubMed] [Google Scholar]
- 11. Buell A. K., Galvagnion C., Gaspar R., Sparr E., Vendruscolo M., Knowles T. P., Linse S., and Dobson C. M. (2014) Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 111, 7671–7676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conway K. A., Harper J. D., and Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 13. Ahmad B., Chen Y., and Lapidus L. J. (2012) Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc. Natl. Acad. Sci. U.S.A. 109, 2336–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bertoncini C. W., Jung Y. S., Fernandez C. O., Hoyer W., Griesinger C., Jovin T. M., and Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. U.S.A. 102, 1430–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dedmon M. M., Lindorff-Larsen K., Christodoulou J., Vendruscolo M., and Dobson C. M. (2005) Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127, 476–477 [DOI] [PubMed] [Google Scholar]
- 16. Allison J. R., Varnai P., Dobson C. M., and Vendruscolo M. (2009) Determination of the free energy landscape of α-synuclein using spin label nuclear magnetic resonance measurements. J. Am. Chem. Soc. 131, 18314–18326 [DOI] [PubMed] [Google Scholar]
- 17. Bertoncini C. W., Fernandez C. O., Griesinger C., Jovin T. M., and Zweckstetter M. (2005) Familial mutants of α-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 280, 30649–30652 [DOI] [PubMed] [Google Scholar]
- 18. Cho M. K., Nodet G., Kim H. Y., Jensen M. R., Bernado P., Fernandez C. O., Becker S., Blackledge M., and Zweckstetter M. (2009) Structural characterization of α-synuclein in an aggregation prone state. Protein Sci. 18, 1840–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meisl G., Kirkegaard J. B., Arosio P., Michaels T. C., Vendruscolo M., Dobson C. M., Linse S., and Knowles T. P. (2016) Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 11, 252–272 [DOI] [PubMed] [Google Scholar]
- 20. Morris A. M., Watzky M. A., and Finke R. G. (2009) Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim. Biophys. Acta 1794, 375–397 [DOI] [PubMed] [Google Scholar]
- 21. Uversky V. N., Li J., and Fink A. L. (2001) Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem 276, 10737–10744 [DOI] [PubMed] [Google Scholar]
- 22. Hoyer W., Antony T., Cherny D., Heim G., Jovin T. M., and Subramaniam V. (2002) Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322, 383–393 [DOI] [PubMed] [Google Scholar]
- 23. Camilloni C., Cavalli A., and Vendruscolo M. (2013) Replica-averaged metadynamics. J. Chem. Theory Comput. 9, 5610–5617 [DOI] [PubMed] [Google Scholar]
- 24. Kohlhoff K. J., Robustelli P., Cavalli A., Salvatella X., and Vendruscolo M. (2009) Fast and accurate predictions of protein NMR chemical shifts from interatomic distances. J. Am. Chem. Soc. 131, 13894–13895 [DOI] [PubMed] [Google Scholar]
- 25. Kay L. E., Xu G. Y., and Yamazaki T. (1994) Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. A 109, 129–133 [Google Scholar]
- 26. Muhandiram D. R., and Kay L. E. (1994) Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. B 103, 203–216 [Google Scholar]
- 27. Tamiola K., and Mulder F. A. (2012) Using NMR chemical shifts to calculate the propensity for structural order and disorder in proteins. Biochem. Soc. Trans. 40, 1014–1020 [DOI] [PubMed] [Google Scholar]
- 28. Lescop E., Kern T., and Brutscher B. (2010) Guidelines for the use of band-selective radiofrequency pulses in hetero-nuclear NMR: example of longitudinal-relaxation-enhanced BEST-type 1H-15N correlation experiments. J. Magn. Reson. 203, 190–198 [DOI] [PubMed] [Google Scholar]
- 29. Lescop E., Schanda P., and Brutscher B. (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 187, 163–169 [DOI] [PubMed] [Google Scholar]
- 30. Salzmann M., Wider G., Pervushin K., Senn H., and Wüthrich K. (1999) TROSY-type triple-resonance experiments for sequential NMR assignments of large proteins. J. Am. Chem. Soc. 121, 844–848 [Google Scholar]
- 31. Nielsen J. T., and Mulder F. A. (2016) There is diversity in disorder: “in all chaos there is a cosmos, in all disorder a secret order. Front. Mol. Biosci. 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rospigliosi C. C., McClendon S., Schmid A. W., Ramlall T. F., Barré P., Lashuel H. A., and Eliezer D. (2009) E46K Parkinson's-linked mutation enhances C-terminal-to-N-terminal contacts in α-synuclein. J. Mol. Biol. 388, 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ullman O., Fisher C. K., and Stultz C. M. (2011) Explaining the structural plasticity of α-synuclein. J. Am. Chem. Soc. 133, 19536–19546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Croke R. L., Patil S. M., Quevreaux J., Kendall D. A., and Alexandrescu A. T. (2011) NMR determination of pKa values in α-synuclein. Protein Sci. 20, 256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Theillet F. X., Binolfi A., Bekei B., Martorana A., Rose H. M., Stuiver M., Verzini S., Lorenz D., van Rossum M., Goldfarb D., and Selenko P. (2016) Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50 [DOI] [PubMed] [Google Scholar]
- 36. Bai J., Cheng K., Liu M., and Li C. (2016) Impact of the α-synuclein initial ensemble structure on fibrillation pathways and kinetics. J. Phys. Chem. B 120, 3140–3147 [DOI] [PubMed] [Google Scholar]
- 37. Munishkina L. A., Henriques J., Uversky V. N., and Fink A. L. (2004) Role of protein-water interactions and electrostatics in α-synuclein fibril formation. Biochemistry 43, 3289–3300 [DOI] [PubMed] [Google Scholar]
- 38. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morel B., Varela L., Azuaga A. I., and Conejero-Lara F. (2010) Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys. J. 99, 3801–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoshimura Y., Lin Y., Yagi H., Lee Y. H., Kitayama H., Sakurai K., So M., Ogi H., Naiki H., and Goto Y. (2012) Distinguishing crystal-like amyloid fibrils and glass-like amorphous aggregates from their kinetics of formation. Proc. Natl. Acad. Sci. U.S.A. 109, 14446–14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buell A. K., Hung P., Salvatella X., Welland M. E., Dobson C. M., and Knowles T. P. (2013) Electrostatic effects in filamentous protein aggregation. Biophys. J. 104, 1116–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tartaglia G. G., Pawar A. P., Campioni S., Dobson C. M., Chiti F., and Vendruscolo M. (2008) Prediction of aggregation-prone regions in structured proteins. J. Mol. Biol. 380, 425–436 [DOI] [PubMed] [Google Scholar]
- 43. Nielsen L., Khurana R., Coats A., Frokjaer S., Brange J., Vyas S., Uversky V. N., and Fink A. L. (2001) Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 40, 6036–6046 [DOI] [PubMed] [Google Scholar]
- 44. Raman B., Chatani E., Kihara M., Ban T., Sakai M., Hasegawa K., Naiki H., Rao Ch. M., and Goto Y. (2005) Critical balance of electrostatic and hydrophobic interactions is required for β2-microglobulin amyloid fibril growth and stability. Biochemistry 44, 1288–1299 [DOI] [PubMed] [Google Scholar]
- 45. Pedersen J. S., Flink J. M., Dikov D., and Otzen D. E. (2006) Sulfates dramatically stabilize a salt-dependent type of glucagon fibrils. Biophys. J. 90, 4181–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marek P. J., Patsalo V., Green D. F., and Raleigh D. P. (2012) Ionic strength effects on amyloid formation by amylin are a complicated interplay among Debye screening, ion selectivity, and Hofmeister effects. Biochemistry 51, 8478–8490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jain S., and Udgaonkar J. B. (2010) Salt-induced modulation of the pathway of amyloid fibril formation by the mouse prion protein. Biochemistry 49, 7615–7624 [DOI] [PubMed] [Google Scholar]
- 48. Fernández C. O., Hoyer W., Zweckstetter M., Jares-Erijman E. A., Subramaniam V., Griesinger C., and Jovin T. M. (2004) NMR of α-synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 23, 2039–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Antony T., Hoyer W., Cherny D., Heim G., Jovin T. M., and Subramaniam V. (2003) Cellular polyamines promote the aggregation of α-synuclein. J. Biol. Chem. 278, 3235–3240 [DOI] [PubMed] [Google Scholar]
- 50. Paik S. R., Shin H. J., Lee J. H., Chang C. S., and Kim J. (1999) Copper(II)-induced self-oligomerization of α-synuclein. Biochem. J. 340, 821–828 [PMC free article] [PubMed] [Google Scholar]
- 51. Uversky V. N., Li J., and Fink A. L. (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: a possible molecular link between Parkinson's disease and heavy metal exposure. J. Biol. Chem. 276, 44284–44296 [DOI] [PubMed] [Google Scholar]
- 52. Nielsen M. S., Vorum H., Lindersson E., and Jensen P. H. (2001) Ca2+ binding to α-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 276, 22680–22684 [DOI] [PubMed] [Google Scholar]
- 53. Crowther R. A., Jakes R., Spillantini M. G., and Goedert M. (1998) Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 436, 309–312 [DOI] [PubMed] [Google Scholar]
- 54. Theillet F. X., Binolfi A., Liokatis S., Verzini S., and Selenko P. (2011) Paramagnetic relaxation enhancement to improve sensitivity of fast NMR methods: application to intrinsically disordered proteins. J. Biomol. NMR 51, 487–495 [DOI] [PubMed] [Google Scholar]
- 55. Oktaviani N. A., Risør M. W., Lee Y. H., Megens R. P., de Jong D. H., Otten R., Scheek R. M., Enghild J. J., Nielsen N. C., Ikegami T., and Mulder F. A. (2015) Optimized co-solute paramagnetic relaxation enhancement for the rapid NMR analysis of highly fibrillogenic peptide. J. Biomol. NMR 62, 129–142 [DOI] [PubMed] [Google Scholar]
- 56. Giehm L., Svergun D. I., Otzen D. E., and Vestergaard B. (2011) Low-resolution structure of a vesicle disrupting α-synuclein oligomer that accumulates during fibrillation. Proc. Natl. Acad. Sci. U.S.A. 108, 3246–3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hortschansky P., Schroeckh V., Christopeit T., Zandomeneghi G., and Fändrich M. (2005) The aggregation kinetics of Alzheimer's β-amyloid peptide is controlled by stochastic nucleation. Protein Sci. 14, 1753–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Umemoto A., Yagi H., So M., and Goto Y. (2014) High-throughput analysis of ultrasonication-forced amyloid fibrillation reveals the mechanism underlying the large fluctuation in the lag time. J. Biol. Chem. 289, 27290–27299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abdolvahabi A., Shi Y., Chuprin A., Rasouli S., and Shaw B. F. (2016) Stochastic formation of fibrillar and amorphous superoxide dismutase oligomers linked to amyotrophic lateral sclerosis. ACS Chem. Neurosci. 7, 799–810 [DOI] [PubMed] [Google Scholar]
- 60. Markley J. L., Bax A., Arata Y., Hilbers C. W., Kaptein R., Sykes B. D., Wright P. E., and Wüthrich K. (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. J. Biomol. NMR 12, 1–23 [DOI] [PubMed] [Google Scholar]
- 61. Clubb R. T., Thanabal V., and Wagner G. (1992) A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, and 13C′ chemical shifts in 15N-13C-labeled proteins. J. Magn. Reson. 97, 213–217 [Google Scholar]
- 62. Weisemann R., Rüterjans H., and Bermel W. (1993) 3D triple-resonance NMR techniques for the sequential assignment of NH and 15N resonances in 15N- and 13C-labelled proteins. J. Biomol. NMR 3, 113–120 [DOI] [PubMed] [Google Scholar]
- 63. Grzesiek S., and Bax A. (1997) A three-dimensional NMR experiment with improved sensitivity for carbonyl-carbonyl J correlation in proteins. J. Biomol. NMR 9, 207–211 [DOI] [PubMed] [Google Scholar]
- 64. Yoshimura Y., Kulminskaya N. V., and Mulder F. A. (2015) Easy and unambiguous sequential assignments of intrinsically disordered proteins by correlating the backbone 15N or 13C′ chemical shifts of multiple contiguous residues in highly resolved 3D spectra. J. Biomol. NMR 61, 109–121 [DOI] [PubMed] [Google Scholar]
- 65. Liu A., Riek R., Wider G., von Schroetter C., Zahn R., and Wüthrich K. (2000) NMR experiments for resonance assignments of 13C, 15N doubly-labeled flexible polypeptides: application to the human prion protein hPrP(23–230). J. Biomol. NMR 16, 127–138 [DOI] [PubMed] [Google Scholar]
- 66. Felli I. C., Pierattelli R., Glaser S. J., and Luy B. (2009) Relaxation-optimised Hartmann-Hahn transfer using a specifically Tailored MOCCA-XY16 mixing sequence for carbonyl-carbonyl correlation spectroscopy in 13C direct detection NMR experiments. J. Biomol. NMR 43, 187–196 [DOI] [PubMed] [Google Scholar]
- 67. Logan T. M., Olejniczak E. T., Xu R. X., and Fesik S. W. (1993) A general method for assigning NMR spectra of denatured proteins using 3D HC(CO)NH-TOCSY triple resonance experiments. J. Biomol. NMR 3, 225–231 [DOI] [PubMed] [Google Scholar]
- 68. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 69. Shen Y., and Bax A. (2010) SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR 48, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Han B., Liu Y., Ginzinger S. W., and Wishart D. S. (2011) SHIFTX2: significantly improved protein chemical shift prediction. J. Biomol. NMR 50, 43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 72. Gsponer J., Christodoulou J., Cavalli A., Bui J. M., Richter B., Dobson C. M., and Vendruscolo M. (2008) A coupled equilibrium shift mechanism in calmodulin-mediated signal transduction. Structure 16, 736–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Williamson M. P. (2013) Using chemical shift perturbation to characterse ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 73, 1–16 [DOI] [PubMed] [Google Scholar]
- 74. Wang C., and Palmer A. G. (2003) Solution NMR methods for quantitative identification of chemical exchange in 15N-labeled proteins. Magn. Reson. Chem. 41, 866–876 [Google Scholar]
- 75. Wang A. C., and Bax A. (1993) Minimizing the effects of radio-frequency heating in multidimensional NMR experiments. J. Biomol. NMR 3, 715–720 [DOI] [PubMed] [Google Scholar]
- 76. Yuwen T., and Skrynnikov N. R. (2014) Proton-decoupled CPMG: A better experiment for measuring 15N R2 relaxation in disordered proteins. J. Magn. Reson. 241, 155–169 [DOI] [PubMed] [Google Scholar]