

Abstract
NHE3 directly binds Na+/H+ exchanger regulatory factor (NHERF) family scaffolding proteins that are required for many aspects of NHE3 regulation. The NHERFs bind both to an internal region (amino acids 586–660) of the NHE3 C terminus and to the NHE3 C-terminal four amino acids. The internal NHERF-binding region contains both putative Class I (-592SAV-) and Class II (-595CLDM-) PDZ-binding motifs (PBMs). Point mutagenesis showed that only the Class II motif contributes to NHERF binding. In this study, the roles in regulation of NHE3 activity of these two PBMs were investigated, revealing the following findings. 1) Interaction occurred between these binding sites because mutation of either removed nearly all NHERF binding. 2) Mutations in either significantly reduced basal NHE3 activity. Total and percent plasma membrane (PM) NHE3 protein expression was reduced in the C-terminal but not in the internal PBD mutation. 3) cGMP- and Ca2+-mediated inhibition of NHE3 was impaired in both the internal and the C-terminal PBM mutations. 4) There was a significant reduction in half-life of the PM pool of NHE3 in only the internal PBM mutation but no change in total NHE3 half-life in either. 5) There were some differences in NHE3-associating proteins in the two PBM mutations. In conclusion, NHE3 binds to NHERF proteins via both an internal Class II PBM and C-terminal Class I PBM, which interact. The former determines NHE3 stability in the PM, and the latter determines total expression and percent PM expression.
Keywords: calcium, epithelial cell, PDZ domain, sodium transport, sodium-proton exchange, NHE3
Introduction
PDZ domains (80–90 amino acids) are modular protein-protein interaction domains that play a role in protein targeting and protein complex assembly (1–4). PDZ domains are generally classified based on the C-terminal binding motifs of their ligands. There are at least three types of PDZ domains defined by the amino acids generally at the 0 and −2 positions in the C-terminal ligand sequence: Class I (X(S/T)XΦ-COOH), Class II (XΦXΦ-COOH), and Class III (X(D/E)XΦ-COOH) where Φ represents any hydrophobic amino acid residue and X is any residue (5–9). In addition, there are several examples of PDZ domain interactions that occur via internal PDZ domain-binding motifs (10–14).
The intestinal and renal brush border (BB)2 Na+/H+ exchanger NHE3 is highly regulated during digestion. Our previous studies and those of others have demonstrated that four related multi-PDZ domain-containing proteins, NHERF1 (EBP50), NHERF2 (E3KARP), NHERF3 (PDZK1, CLAMP, CAP70), and NHERF4 (IKEPP) (15), play significant roles in the regulation of NHE3 activity. The NHERF family of PDZ proteins are closely related to each other, reside in locations at or near the BB in the renal proximal tubule and small intestinal enterocytes, and scaffold multiple transport proteins in addition to NHE3. Regulation of NHE3 activity involves two identified multiprotein complexes that form on the NHE3 intracellular regulatory domain. These complexes are highly dynamic as part of this regulation, and many of the proteins involved bind the NHERF scaffolds. NHE3 interacts with NHERF2 via an internal sequence (aa 586–660) within the cytoplasmic domain of NHE3, but binding to the C-terminal PDZ domain-binding motif (PBM) that is present in NHE3 was not observed (16). However, separate studies did demonstrate binding of NHERF1 and NHERF3 to NHE3 via this C-terminal PBM, although functional consequences of this binding indicated only a partial reduction of NHE3 inhibition by cAMP (17, 18). In attempting to resolve these differences, we identified an internal PDZ domain-binding site in the NHE3 C terminus as a Class II PDZ domain-binding motif (-595CLDM-) and have characterized the functional consequences of mutating both NHE3 PBMs.
Results
Putative internal Class I and Class II PDZ-binding motifs are present between aa 590 and 610 of the NHE3 C terminus
We showed previously that NHERF2 binds to an internal sequence of the NHE3 C terminus between aa 586 and 660 (16). However, the specific amino acids required for NHERF2 binding were not identified. Therefore, in vitro binding assays were performed to further define the binding site. GST-NHERF1 or GST-NHERF2 protein immobilized on glutathione-Sepharose® beads was used for “pulldown” assays with 35S-labeled His6-tagged NHE3 C-terminal truncations (C refers to C terminus) C589, C610, C660, C689, C711, and C832. Fig. 1A shows that NHERF1 and NHERF2 bind to all NHE3 C-terminal truncations C610 or larger but not to C589. The results from Fig. 1A show that a NHERF1- and NHERF2-binding domain of the NHE3 C terminus is located between aa 590 and 610, confirming but further localizing results from the previous report (16).
Figure 1.
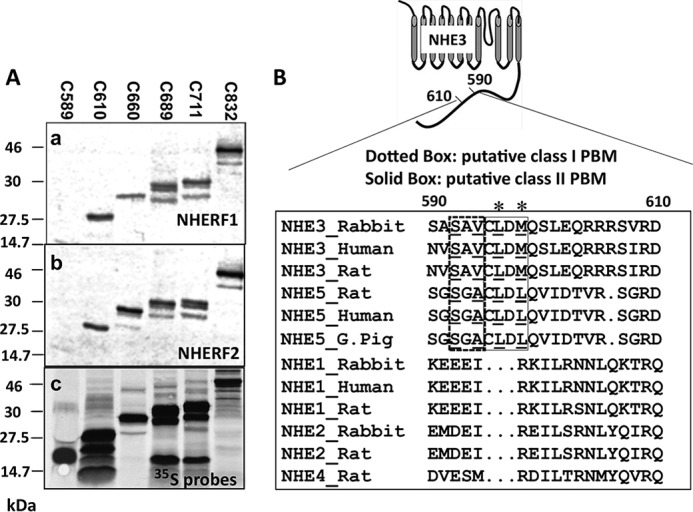
NHERF1 and NHERF2 bind to an internal region of the NHE3 C terminus (aa 590–610). A, the interactions between GST-NHERF1 (panel a) or GST-NHERF2 (panel b) and 35S-labeled (panel c) NHE3 C-terminal His6-tagged truncations (C refers to C terminus) C589 (aa 475–589), C610 (aa 475–610), C660 (aa 475–660), C689 (aa 475–689), C711(aa 475–711), and C832(aa 475–832) were determined with pulldown assays (see “Experimental Procedures”). 6 μl of 35S-labeled NHE3 C-terminal truncations and 3 μg of recombinant proteins (GST-NHERF1 or GST-NHERF2) were loaded for 10% SDS-PAGE. B, putative Class I (-592SAV-) and Class II (-595CLDM-) PBMs in NHE3 C terminus (aa 590–610): amino acid sequence alignment of conserved amino acids for NHERF2-binding sites in internal sequences of the mammalian NHE gene family 1–5 cytoplasmic domains. The putative internal Class I PBM (-592SAV-) is boxed (dotted line), and critical amino acids for Class I PBM are underlined. The putative internal Class II PBM (-595CLDM-) is boxed (solid line), and the conserved hydrophobic amino acids are underlined. Accession numbers for the sequences, top to bottom, are P26432 (NHE3_Rabbit), NP_004165 (NHE3_Human), NP_036786 (NHE3_Rat), NP_620213.1 (NHE5_Norway rat), AAI42672 (NHE5_Human), XP_005008798.1 (NHE5_G. Pig (domestic guinea pig)), P23791 (NHE1_Rabbit), NP_003038 (NHE1_Human), NP_036784 (NHE1_Norway rat), P50482 (NHE2_Rabbit), P48763 (NHE2_Rat), and NP_775121 (NHE4_Rat). Sequences were aligned with ClustalW. An asterisk indicates the hydrophobic amino acids of the class II PBM.
Two putative PDZ-binding motifs are found in this region: a Class I (-592SAV-) PBM and a Class II (-595CLDM-) PBM. Both Class I and Class II PBMs are conserved in the recycling PM-NHEs NHE3 and NHE5 but are not present in the resident plasma membrane NHEs NHE1, -2, and -4 (Fig. 1B).
Mutation of the Class II PBM NHE3CLDM/EK (or NHE3CLDM/QA) but not the Class I PBM NHE3SAV/AA abolished NHE3 binding to the NHERFs
Co-immunoprecipitation (co-IP) was used to test whether putative internal NHE3 Class I (-592SAV-) or Class II (-595CLDM-) PBMs were involved in the binding of PDZ proteins. Hemagglutinin (HA)-tagged NHE3 WT, putative Class I mutant NHE3SAV/AA, and Class II mutants NHE3CLDM/EK (called PBMIImut in this study) and NHE3CLDM/QA were engineered as shown in Fig. 2A and stably expressed in PS120/NHERF2 cells. Co-IP of the NHE3 WT and mutant constructs with NHERF2 was performed (Fig. 2B). NHE3 WT and NHE3SAV/AA precipitated NHERF2 similarly; however, NHE3CLDM/EK (or NHE3 PBMIImut) and NHE3CLDM/QA failed to bind NHERF2 (Fig. 2B). Thus the internal Class II PBM (-595CLDM-) was necessary for NHERF2 binding.
Figure 2.
Mutation in putative Class I (-592SAV-) and Class II (-595CLDM-) internal PBMs and testing of their NHERF2 binding. A, one Class I PBM mutant, NHE3SAV/AA (592SAV → 592AAA), and two Class II PBM mutants, NHE3CLDM/EK (-595CLDM- → -595CEDK-) and NHE3CLDM/QA (-595CLDM- → -595CQDA-), were constructed. B, in vivo binding assays were performed. HA-tagged NHE3 constructs were immobilized by monoclonal anti-HA affinity matrix or by monoclonal anti-VSV-G antibodies immobilized on agarose as a negative control and co-precipitated with lysates from NHE3_WT, NHE3SAV/AA, NHE3CLDM/EK, and NHE3CLDM/QA in PS120/NHERF2 stable cell lines. NHE3_WT and NHE3SAV/AA precipitated NHERF2, but neither NHE3CLDM/EK nor NHE3CLDM/QA bound NHERF2. Monoclonal anti-HA antibody and polyclonal anti-NHERF2 antibody (α-2570) were used to detect HA-tagged NHE3 constructs and NHERF2, respectively. A representative experiment of three similar experiments is shown. IB, immunoblotting.
NHERF2 largely fails to co-immunoprecipitate both NHE3 PBMIImut and NHE3 Δ4
We next evaluated the role of the NHE3 C-terminal Class I PBM (-829STHM) in NHE3 association with NHERF2 in comparison with that of NHE3 PBMIImut. Mutation of this C-terminal domain had been shown to reduce but not abolish cAMP inhibition of NHE3 activity (17). To evaluate the role of the NHE3 C-terminal as well as the NHE3 internal PBM on NHE3, we engineered and studied the NHE3 constructs in Fig. 3A (NHE3 WT, NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4).
Figure 3.

NHERF2 co-precipitates NHE3 WT but has reduced association with NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4. A, to compare the roles of internal PBM (-595CLDM-) and C-terminal putative Class I PBM (-829STHM), the HA-tagged NHE3 constructs NHE3 WT, NHE3 PBMIImut, NHE3 Δ4 (C-terminal four amino acids, STHM, deleted), and NHE3 PBMIImut + Δ4 were designed in pcDNA3.1_Hygro vector and stably transfected into PS120/NHERF2 cells. B, immunoprecipitation was performed with anti-NHERF2 antibody (α-2570) from PS120/HA-NHE3 WT (as a negative control; first lane), PS120/NHERF2/HA-NHE3 WT (second lane), PS120/NHERF2/HA-NHE3 PBMIImut (third lane), PS120/NHERF2/HA-NHE3 Δ4 (fourth lane), and PS120/NHERF2/HA-NHE3 PBMIImut + Δ4 (fifth lane). NHERF2 co-immunoprecipitated NHE3 WT but not the negative control (first lane) and immunoprecipitated reduced amounts of NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4. Input and bound NHE3 were detected with monoclonal anti-HA antibody. NHERF2 was detected by polyclonal anti-NHERF2 antibody (α-2570). A representative experiment of three similar experiments is shown above, and quantitation of the amount of NHE3 immunoprecipitated by NHERF2 normalized to the amount of NHERF2 immunoprecipitated is show below. Results are mean ± S.D., n = 3. Error bars represent S.D. Means are shown in this and subsequent figures as open symbols, whereas individual experiments are shown as filled symbols. p values compare results between mutants. C, pulldown assays were performed to determine the interactions between NHE3 (WT, PBMIImut, Δ4, and PBMIImut + Δ4) and NHERFs (NHERF1 (NF1), NHERF2 (NF2), and NHERF3 (NF3)). GST or GST-tagged NHERF1, NHERF2, and NHERF3 were used to pull down HA-tagged NHE3 WT, PBMIImut, Δ4, and PBMIImut + Δ4 in lysates from PS120/NHERF2 cells. The left panel shows the total cell lysates of GST and GST-NHERF1, -NHERF2, and -NHERF3 used for pulldown assays. The right panel shows the pulldown results. A representative experiment of three similar experiments is shown. NS, not significant; IB, immunoblotting.
Co-IP studies between NHERF2 and wild-type and mutated HA-NHE3 (NHE3 WT, NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4) are shown in Fig. 3B. NHE3 WT was co-precipitated by NHERF2; however, NHERF2 had reduced co-precipitation of NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4 with only 10–20% of the association present in NHE3 WT with all three mutations. PS120/HA-NHE3 cells, which do not express NHERF2, were used as a negative control for IP.
These co-precipitation studies were confirmed by pulldown assays between GST-tagged NHERF1, NHERF2, and NHERF3 and HA-tagged NHE3 WT, NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4 (Fig. 3C). The inputs of fusion proteins of GST, GST-NHERF1, GST-NHERF2, and GST-NHERF3 are shown in Fig. 3C, left panel. GST-tagged NHERF1, NHERF2, and NHERF3 “pulled down” NHE3 WT; however, these NHERFs had markedly reduced but still present binding to NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4 (Fig. 3C, right panel). The band of NHE3 in the GST-NHERF3 column was pushed down with loading of NHERF3 because of the similar sizes of NHERF3 and NHE3. These results showed that PDZ domain-containing proteins NHERF1, NHERF2, and NHERF3 bind to NHE3 via both internal Class II (-595CLDM-) and C-terminal Class I (-829STHM) PBMs, but mutating either greatly reduces, but does not abolish, total NHE3-NHERF2 association.
NHE3 activity is decreased in both internal Class II and C-terminal Class I PBM mutations, and the effects are additive
The basal activities of NHE3 WT, NHE3SAV/AA, NHE3 PBMIImut, and NHE3CLDM/QA were measured by Hill plot analysis, demonstrating that the basal activity of NHE3SAV/AA was similar to wild type. However, the basal activities of Class II PBM mutants NHE3 PBMIImut and NHE3CLDM/QA were reduced, significantly for NHE3 PBMIImut (Fig. 4A) (NHE3 WT, Vmax = 1329 ± 263 μm/s, K′(H+) = 0.35 ± 0.04 μm; NHE3SAV/AA, Vmax = 1132 ± 187 μm/s, K′(H+) = 0.39 ± 0.03 μm; NHE3CLDM/EK (or PBMIImut), Vmax = 468 ± 83 μm/s, K′(H+) = 0.38 ± 0.04 μm); NHE3CLDM/QA, Vmax = 713 ± 139 μm/s, K′(H+) = 0.44 ± 0.24 μm).
Figure 4.

NHE3 basal transport activity is reduced by both internal Class II PBM mutants NHE3CLDM/EK (or NHE3 PBMIImut) and C-terminal NHE3 Δ4. A, basal transport activity of NHE3 is decreased in internal Class II PBM mutants NHE3CLDM/EK (or NHE3 PBMIImut) and NHE3CLDM/QA but not in the putative internal Class I PBM mutant NHE3SAV/AA. Basal NHE3 activity was determined using BCECF as described under “Experimental Procedures.” Shown are the results of a single experiment, which was repeated at least four times with Vmax mean ± S.D. shown in the inset. Error bars represent S.D. p values compare the basal activities of NHE3 PBMIImut and NHE3CLDM/QA with NHE3 WT (unpaired t test). B, basal transport activity of NHE3 (Vmax) was decreased in both internal Class II PBM mutation NHE3 PBMIImut and C-terminal Class I PBM mutation NHE3 Δ4 and was further decreased in the combination NHE3 PBMIImut + Δ4. Shown are the results of a single experiment, which was repeated at least five times with Vmax mean ± S.D. shown in the inset. Error bars represent S.D. p values compare the basal activities of NHE3 PBMIImut and NHE3 Δ4 with NHE3 WT and of NHE3 PBMIImut + Δ4 with NHE3 Δ4 (unpaired t test). NS, not significant.
NHE3 activity was measured for NHE3 WT, NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4 (Fig. 4B). Basal activity of NHE3 PBMIImut was significantly reduced compared with NHE3 WT. This result was consistent with NHE3 PBMIImut and NHE3CLDM/QA in Fig. 4A. The basal activity of NHE3 Δ4 was also dramatically reduced compared with NHE3 WT. The double mutation of NHE3 PBMIImut +Δ4 had further diminished NHE3 activity compared with the single mutation (Fig. 4B). The measured parameters for transport activity were Vmax = 1185 ± 220 μm/s, K′(H+) = 0.36 ± 0.04 μm for wild-type NHE3 (■); Vmax = 431 ± 65 μm/s, K′(H+) = 0.38 ± 0.04 μm for NHE3 PBMIImut (●); Vmax = 308 ± 85 μm/s, K′(H+) = 0.33 ± 0.01 μm for NHE3 Δ4 (▴); and Vmax = 150 ± 50 μm/s, K′(H+) = 0.72 ± 0.2 μm for NHE3 PBMIImut + Δ4 (♦) (Fig. 4B).
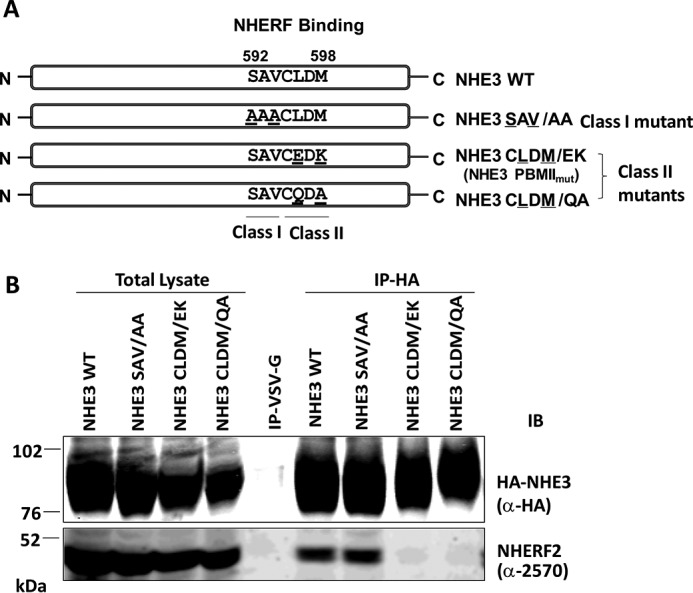
Surface expression of NHE3 PBMIImut was similar to wild-type NHE3, but both mutations NHE3 Δ4 and NHE3 PBMIImut + Δ4 were significantly decreased
Because the transport activities of NHE3 PBMIImut and NHE3 Δ4 were significantly decreased, we also determined whether this decrease was caused by decreases in plasma membrane expression. The surface expression of NHE3 PBMIImut was not significantly changed compared with wild-type NHE3 (Fig. 5, A and B); however, the surface amounts of NHE3 Δ4 and NHE3 PBMIImut + Δ4 were significantly reduced compared with NHE3 WT (Fig. 5, A and B). Quantitative cell-surface biotinylation results as the percentage of total NHE3 expression are shown in Fig. 5B.
Figure 5.
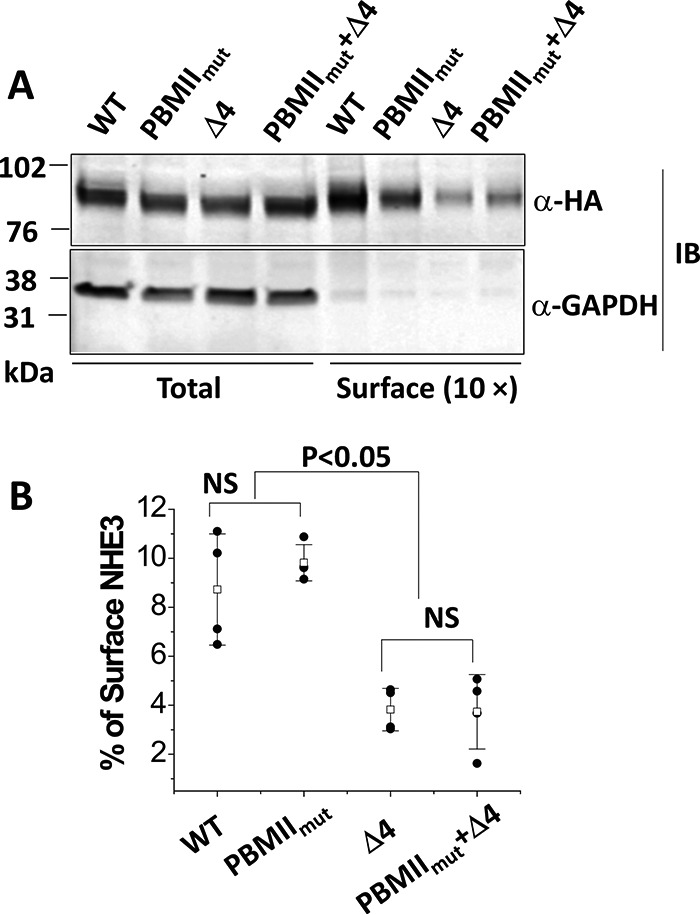
Cell surface expression of NHE3 WTΔ4 and NHE3 PBMIImut + Δ4 was dramatically decreased, whereas the NHE3 PBMIImut mutant was expressed on the plasma membrane similarly to NHE3 WT. A, representative Western blots of NHE3 WT, NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4 were quantified with total and surface fractions probed with monoclonal anti-HA antibody and visualized with the Odyssey system. The density of each band was determined by Image Studio software. B, summary of surface levels (as percentage of total) of NHE3 WT, PBMIImut, Δ4, and PBMIImut + Δ4. Experiments were repeated three times, and results are shown as mean ± S.D. Error bars represent S.D. NS, not significant.
Serum normally stimulates NHE3 activity in both internal NHE3 PBMIImut and NHE3 Δ4
The role of the internal and C-terminal PBMs in the acute stimulation of NHE3 activity was evaluated. We previously reported that dialyzed serum (10% FBS) stimulates the NHE3 transport activity by increasing its Vmax in an NHERF2-independent manner (19, 20). Whether the serum stimulatory effect on NHE3 was affected in NHE3 PBMIImut and NHE3 Δ4 was determined. As shown in Fig. 6, 10% FBS stimulated NHE3 activity similarly to NHE3 WT in both the internal Class II and C-terminal Class I PBM mutations.
Figure 6.
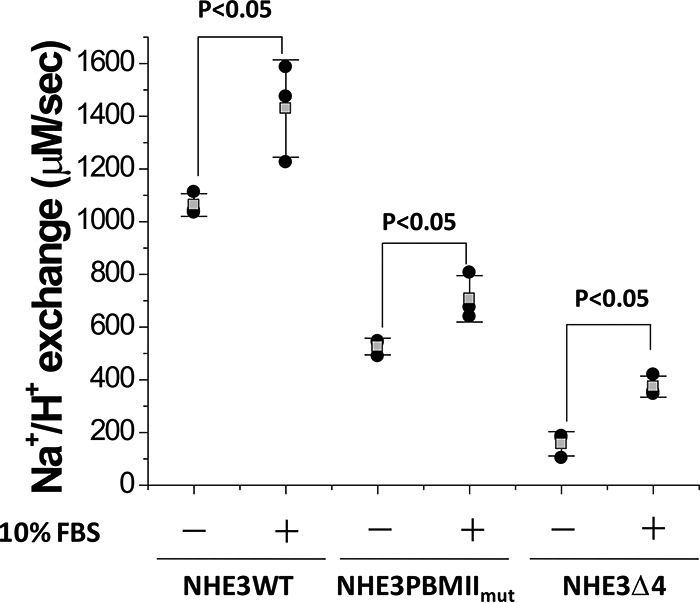
Serum stimulation occurs in NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4. Serum (10% FBS) similarly stimulated wild-type NHE3 activity (Vmax) and that in NHE3 PBMIImut and NHE3 Δ4. The experiment was repeated three times, and results are shown as mean ± S.D. Error bars represent S.D. p values are comparisons of the 10% FBS effect in NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4, respectively (unpaired t test).
Ca2+ and cGMP inhibition of NHE3 activity was reduced in both NHE3 PBMIImut and NHE3 Δ4
Because both Ca2+ and cGMP regulation of NHE3 activity require NHERF2 (not NHERF1) binding to NHE3 (21–23), we measured the effect of 4-Br-A23187 (Ca2+ ionophore) and 8-pCPT-cGMP on NHE3 WT, NHE3 PBMIImut, NHE3 Δ4, and NHE3 PBMIImut + Δ4 in PS120/NHERF2 cells. Because PS120 cells do not contain endogenous cGKII, adenoviral infection was used to express cGKII for the 8-pCPT-cGMP study as described previously (23, 24). 4-Br-A23187 and cGMP/cGKII inhibited NHE3 WT but not NHE3 PBMIImut and NHE3 Δ4 (Fig. 7, A and B). These results showed that the NHERF2-dependent Ca2+ and cGMP/cGKII inhibition of NHE3 activity requires both an intact internal Class II PBM and the C-terminal Class I PBM.
Figure 7.

NHERF2-dependent NHE3 inhibition by elevated Ca2+ and cGMP/cGKII do not occur in the NHE3 PBMIImut and NHE3 Δ4 mutations. The NHERF2-dependent inhibitory effect of elevated Ca2+ (A) and cGMP (B) on NHE3 activity (Vmax) was studied. 4-Br-A23187 (0.5 μm; 5 min) inhibits NHE3 activity in NHE3 WT but not in NHE3 PBMIImut and NHE3 Δ4 in PS120/NHERF2 cells. 8-pCPT-cGMP (100 μm; 30 min) inhibits NHE3 activity (Vmax) in NHE3 WT but not in NHE3 PBMIImut and NHE3 Δ4 in PS120/NHERF2/cGKII cells. p values are comparisons of the 4-Br-A23187 or 8-pCPT-cGMP effect with untreated control (unpaired t tests). Experiments in A and B were repeated three times, and results are shown as mean ± S.D. Error bars represent S.D. NS, not significant.
Half-lives of plasma membrane NHE3 PBMIImut and NHE3 Δ4 are shortened
Both NHE3 PBMIImut and NHE3 Δ4 were decreased in their activity compared with NHE3 WT, but the surface amount was not significantly decreased in NHE3 PBMIImut, whereas it was significantly decreased in NHE3 Δ4 (Fig. 5). Therefore we measured plasma membrane half-life of NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 using a biotinylation method as we described previously (25) (Fig. 8, A and B). The amount of the initial surface biotinylated NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 proteins remaining in the cells 0–30 h after initial biotinylation was determined. The results were normalized with the initial surface NHE3s. Plasma membrane NHE3 PBMIImut decreased rapidly compared with that of NHE3 WT and NHE3 Δ4 during the early (0–6 h) times studied (Fig. 8B; intensities were compared at 3 h (inset)). The half-lives were 12.0 ± 2.7 h for NHE3 WT, which is similar to our previous findings (25, 26); 4.6 ± 0.5 h for NHE3 PBMIImut (significantly shortened compared with WT); and 16.5 ± 1.4 h for NHE3 Δ4 (not significantly different from WT). These data indicate that the mutation of the NHE3 internal (NHE3 PBMIImut) but not the C-terminal (NHE3 Δ4) PBM decreases the early component of the NHE3 plasma membrane half-life, which probably relates to the NHE3 initially mobilized from the plasma membrane. Of interest, there was a second component of the half-life of the NHE3 PBMIImut, which when calculated from 6 to 30 h was longer than WT (16.0 ± 4.1 h for WT and 21.5 ± 3.8 h for PBMIImut). We suggest this relates to a plasma membrane pool of NHE3 that was less or not involved in NHE3 regulation by trafficking. The half-lives of the total NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were also measured using 35S pulse-chase labeling (25). Even though the surface half-life of NHE3 PBMIImut was much shorter than wild type, the total half-lives of NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were not significantly different (Fig. 8, C and D). The relative intensities of NHE3 PBMIImut and NHE3 Δ4 were not significantly changed compared with NHE3 WT at 4 h (Fig. 8D, inset). The half-lives were 15.8 ± 1.3 h for NHE3 WT, 15.3 ± 0.8 h for NHE3 PBMIImut, and 21.0 ± 5.6 h for NHE3 Δ4 (none significantly different). This indicates that the internal PBM mutation NHE3 PBMIImut alters the NHE3 half-life by an effect at the plasma membrane but not intracellularly; however, the C-terminal PBM mutant NHE3 Δ4 does not affect half-life at either plasma membrane or total NHE3 pools.
Figure 8.

The half-life of plasma membrane NHE3 PBMIImut but not that of NHE3 Δ4 is decreased compared with NHE3 WT, but the total half-lives of NHE3 PBMIImut and NHE3 Δ4 are similar to NHE3 WT. A, half-lives of plasma membrane NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 in PS120/NHERF2 cells were estimated using cell-surface biotinylation. Plasma membrane NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were biotinylated at 4 °C, and the biotin-labeled NHE3 was collected at varying times (from 0 to 30 h) after incubation at 37 °C. The biotinylated NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were quantified using the Odyssey system. Results from a single experiment are shown. B, graph shows quantitation of biotinylated surface (S) NHE3s normalized by total NHE3s (S-NHEs/Tot-NHE3s) for NHE3 WT (■), NHE3 PBMIImut (●), and NHE3 Δ4 (▴). The percentage of biotinylated NHE3 PBMIImut remaining was significantly decreased at 3 and 6 h compared with NHE3 WT (*, p < 0.05), but there was no significant change in NHE3 Δ4 compared with NHE3 WT (inset shows results at 3 h). The plasma membrane half-lives are shown in the bottom left. Results are mean ± S.D., n = 3. Error bars represent S.D. C, total half-lives of NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were determined with 35S pulse-chase experiments (see “Experimental Procedures”). PS120/NHERF2 cells expressing NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were pulse-labeled with Met/Cys-free DMEM containing 0.2 mCi/ml [35S]methionine/cysteine cell-labeling mixture for 4 h and chased in medium containing methionine/cysteine at varying time points (from 0 to 41 h). After solubilization, proteins were immunoprecipitated with monoclonal anti-≤A affinity matrix and subjected to SDS-PAGE. Proteins were transferred to nitrocellulose, and 35S-labeled NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were measured with a Storm 860 phosphorimaging system and analyzed with ImageQuant. Results of a single experiment are shown. D, graph showing quantification of autoradiographs for NHE3 WT (■), NHE3 PBMIImut (●), and NHE3 Δ4 (▴). Results are mean ± S.D., n = 3. Error bars represent S.D. There were no significant changes in relative intensities between NHE3 PBMIImut and NHE3 Δ4 compared with NHE3 WT at 4 h (inset). The half-lives are shown in the bottom left. NS, not significant.
This indicates that the internal PBM mutation shortens the presence in the plasma membrane of a pool of NHE3, which we suggest is a rapidly cycling probably as part of acute NHE3 regulation, but there is an additional slower cycling pool in the plasma membrane. In contrast, the C-terminal PBM mutant NHE3 Δ4 does affect the half-life either at the plasma membrane or the total NHE3 pool.
Lateral mobility of NHE3 PBMIImut and NHE3 Δ4 are increased as assessed by fluorescence recovery after photobleaching (FRAP)
We have shown that the lateral mobility changes of surface NHE3 caused by multiple acute regulators of NHE3 activity are NHERF2-dependent (including high concentration LPA exposure (23), LPA/LPA5R (27), Ca2+ ionophore A23187 (28), and d-glucose (29)) and that the NHE3 mobile fraction is increased by conditions associated with NHE3 dissociation from NHERF2. NHE3 mobility was also increased in the NHERF2 and NHERF1 knockdown Caco-2 cells (30). Because both NHE3 PBMIImut and NHE3 Δ4 mutations decrease their NHERF2 binding, FRAP was performed to test whether their mobile fractions were also changed. The mobile fractions of both NHE3 PBMIImut (●) and NHE3 Δ4 (▴) were increased compared with NHE3 WT (■) (Fig. 9). These results are consistent with the hypothesis that conditions in which NHE3/NHERF2 binding is decreased are associated with an increased NHE3 mobile fraction.
Figure 9.
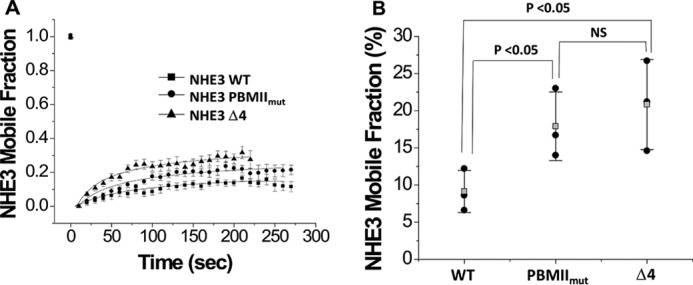
The lateral mobile fractions of NHE3 PBMIImut and NHE3 Δ4 were significantly increased compared with wild-type NHE3. A, OK cells depleted in NHE3 expression by the acid suicide technique were transiently transfected with HA-tagged NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 and labeled with monoclonal HA antibody as primary antibody and Alexa Fluor 488 anti-mouse as secondary antibody at 4 °C. FRAP was measured for NHE3 WT (■), NHE3 PBMIImut (●), and NHE3 Δ4 (▴) as described under “Experimental Procedures”. Data were collected as 30 images every 10 s. B, the mobile fractions (%) of NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were determined at the 200-s time point. Each experiment analyzed FRAP results from more than five regions of interest from multiple cells. Experiments were repeated three times, and results are shown as mean ± S.D. Error bars represent S.D. p values were determined by unpaired t tests. NS, not significant; Mf, mobile fraction.
Differential binding to the NHE3 C termini of NHE3 PBMIImut and NHE3 Δ4
Because both NHE3 PBMIImut and NHE3 Δ4 markedly and similarly reduced the NHERF binding but had multiple different effects on NHE3 activity, we hypothesized that some of these differences might be related to different changes in proteins binding to the NHE3 C terminus. We considered that these changes might be related to proteins directly binding the NHE3 C terminus or indirectly binding via association via interacting with the NHERF proteins. A co-IP approach was initially used, comparing the amount of calmodulin (CaM) co-precipitated with NHE3 WT and the two PBM mutants. As shown in Fig. 10A, less CaM was co-precipitated with NHE3 Δ4 than NHE3 WT and NHE3 PBMIImut. Similarly, GST-SNX27 (full length) was previously reported to pull down less NHE3 Δ4 than NHE3 WT (32); however, it pulled down similar amounts of NHE3 WT and NHE3 PBMIImut (Fig. 10B). That not all NHE3-binding proteins were affected differently by the two PBM mutants was demonstrated using a pulldown approach. GST-CK2 fusion protein pulled down similar amounts of NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 (Fig. 11).
Figure 10.

CaM has reduced binding to NHE3 Δ4 but CaM and SNX27 have normal binding to NHE3 PBMIImut. A, HA-tagged NHE3 constructs were immobilized by monoclonal anti-HA affinity matrix and co-precipitated with lysates of PS120, NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 in PS120/NHERF2 cells. CaM had reduced binding to NHE3 Δ4 but bound like NHE3 WT to NHE3 PBMIImut. Samples were detected by Western blotting (IB) with monoclonal anti-CaM antibody (Sigma) and monoclonal HA antibody (Sigma). A representative experiment is show, and these experiments were repeated three times with similar results. B, NHE3 PBMIImut bound SNX27 like NHE3 WT. We previously reported that NHE3 Δ4 had minimal SNX27 binding (32). GST or GST-tagged SNX27 was immobilized by GSH beads used to perform in vitro pulldown assays with the HA-tagged NHE3 WT and NHE3 PBMIImut. Samples were analyzed by Western blotting (IB) with antibodies against GST (Cell Signaling Technology) and HA (Sigma).
Figure 11.
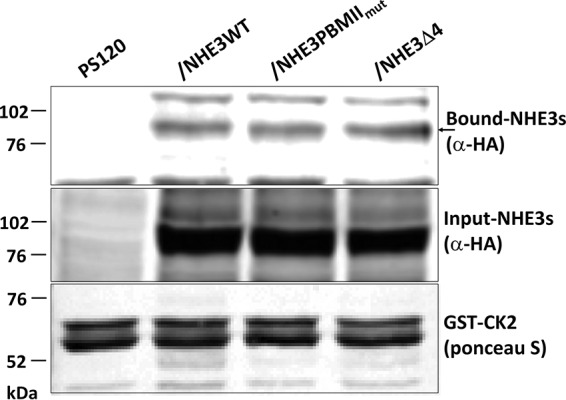
NHE3 PBMIImut and NHE3 Δ4 bind to casein kinase 2 (CK2) similarly to NHE3 WT. GST-tagged CK2 was used to pull down PS120/NHERF2 total cell lysates comparing PS120 cells (negative control), HA-tagged NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4. Total expression and bound NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were detected with monoclonal anti-HA antibody. The amounts of NHE3 PBMIImut and NHE3 Δ4 pulled down with GST-CK2 were similar to NHE3 WT. The arrow indicates the bound NHE3s. 5 μg of GST-CK2 was immobilized to 10 μl of glutathione-Sepharose 4B beads to pull down total lysates (1 mg/ml) of PS120 (negative) and NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 in PS120/NHERF2 cells. A representative experiment is shown, and these experiments were repeated three times with similar results.
Discussion
In this study, we demonstrate that NHE3 uses two separate domains in its intracellular C terminus to interact with members of the NHERF family of multi-PDZ domain-containing scaffolds and that these influence each other but also have separate effects on NHE3 regulation. This detailed study was undertaken because of previous reports of NHE3 regulation that appeared to be contradictory relating to NHE3-NHERF interactions. The NHE3 C-terminal Class I PDZ-binding motif had been shown to bind to NHERF1 and NHERF3, whereas mutating the C-terminal four amino acids of NHE3 to remove this motif caused only a partial reduction of cAMP inhibition of NHE3 (17, 18). In contrast, NHERF2 had been shown to bind to an internal NHE3 C-terminal site (16) with NHERF binding needed for cAMP inhibition of NHE3. Yeast two-hybrid analyses previously demonstrated that the C-terminal Class I PDZ-binding motif sequence of NHE3 was necessary for its association with NHERF1 (17). In this yeast two-hybrid assay, NHE3 bound both NHERF1 PDZ-I and PDZ-II when presented as isolated domains, but mutations of the individual PDZ domains in the full-length NHERF1 suggested a significant preference of NHE3 for the PDZ-II domain. However, based on in vitro binding assays, there was still NHE3/NHERF1 binding despite mutation of the C-terminal PDZ-binding motif, suggesting the presence of at least one additional PDZ-binding motif in the NHE3 C terminus.
As summarized in Table 1 and shown in Figs. 2 and 3, mutating each of the two functionally important PDZ-binding domains greatly reduced NHE3 and NHERF binding. We examined this binding in most detail for NHERF2 because we have shown that NHE3 regulation is most dependent on NHERF2, although NHERF1 and especially NHERF3 are functionally important as well. Mutation of the internal Class II PBM and the C-terminal Class I PBM similarly reduced NHERF2 binding by 80–90% in a non-additive manner. The simplest explanation for this finding is that NHERF binding to the two NHE3 PBMs is cooperative with NHERFs simultaneously interacting with both such that changes in one decreases association with the other. We have recently shown that NHERF3-NHERF2 heterodimers are involved with setting basal NHE3 activity and necessary for Ca2+ inhibition of NHE3 (31), and we suggest that involvement of such a heterodimer might explain the interaction of the two PBMs in the NHE3 C terminus.
Table 1.
Summary of NHE3 effects of PBM mutations from this study
All changes are relative to WT. NC, no change relative to WT; X, WT response prevented.
| Effect on NHE3 | NHE3 PBMIImut mutant (internal Class II PBM) | NHE3 Δ4 mutant (C-terminal Class I PBM) |
|---|---|---|
| Basal activity | Reduced | Reduced |
| Surface expression (%) | NC | Reduced |
| t½ PM | Decreased | NC |
| t½ total | NC | NC |
| Total amount | NC | Decreased |
| Stimulation by serum (not NHERF-dependent) | NC | NC |
| Inhibition by Ca2+ | X | X |
| Inhibition by cGMP | X | X |
| Basal Mf | Increased | Increased |
| Co-IP (NHERF2/NHE3) | X | X |
| SNX27, CaM-binding | NC | Decreaseda |
a Singh et al. (32) for SNX27.
The dependence of Ca2+ and cGMP/cGKII inhibition of NHE3 on both internal and C-terminal PBMs is consistent with the previously demonstrated dependence of both processes on NHERF2-NHE3 binding. However, given the marked and similar reduction in NHERF binding to NHE3 of the two PBM mutants, we considered what could explain their differential effects on multiple aspects of NHE3 activity. Because the NHERFs scaffold multiple proteins simultaneously with their multiple PDZ domains, we tested the hypothesis that the two mutations might have different effects on NHE3-associating proteins. That this was the case for some but not all NHE3-associating proteins is shown in Figs. 10 and 11 with NHE3 Δ4 but not NHE3 PBMIImut binding less CaM and SNX27 (32) than NHE3 WT but all binding similar amounts of CK2. Thus, in considering differences in effects of the two mutations, changes in NHERF binding must be considered along with the potential role of differences in additional NHE3-associating proteins.
The reduced basal NHE3 activity with no change in amount of surface expression with mutation of the internal PBM is also consistent with an effect on the NHE3 signaling complex formed in this area in which CK2 binds to stimulate basal NHE3 activity and CaMKII binds to inhibit basal NHE3 (30–33). Of note is that the CaMKII inhibition of NHE3 activity is not associated with a change in NHE3 plasma membrane expression (33). This Class II PBD mutation also reduced the half-life of NHE3 initially on the plasma membrane, particularly the early component, which suggests an effect on the regulated plasma membrane pool of NHE3, and increased the NHE3 plasma membrane mobile fraction. Both effects are consistent with reduced NHERF2 binding. This mutant also had a plasma membrane component with a longer half-life, which we speculate indicates the presence of a pool of NHE3 that does not traffic readily. Furthermore, the normal total plasma membrane expression of NHE3 was not altered by this mutation, which is probably due to the normal interaction with SNX27 (as we reported previously (32)), which is needed for basal NHE3 exocytosis.
The role of the C-terminal Class I PDZ domain-binding sequence of NHE3 is interpreted as being necessary for increasing basal NHE3 activity and basal BB expression because of its role in SNX27 binding (32). However, this domain does not appear to play a role in NHE3 stability once NHE3 is in the BB because its absence did not alter the plasma membrane half-life or acute NHE3 inhibition by endocytosis stimulated by cGMP/cGKII or elevated Ca2+. The Class I C-terminal PDZ domain was involved in affecting the association of NHE3 with the microvillar cytoskeleton and when mutated increased the NHE3 mobile fraction. Not understood is the role of this domain in establishing total NHE3 expression, which was reduced when it was mutated.
The increase in NHE3 mobile fraction from ∼10 to ∼30% with either the internal or the C-terminal PBM mutations is much less than occurred with truncation of the NHE3 C terminus to aa 585 (∼65%), which eliminated both these binding sites. We attribute the larger increase to dissociation of NHE3 from multiple binding partners in addition to the NHERF family that we have speculated take part in limiting NHE3 mobility under basal conditions (34); however, the current results indicate that these additional proteins must bind at sites distinct from the NHERF-binding sites.
The demonstration of two NHERF family PDZ domain-containing proteins binding to a single protein at the same time has not been reported previously, although NHE3 was shown to bind the single PDZ domain-containing Shank2 in addition to the NHERFs (35). We could not identify other examples of a membrane protein that binds PDZ domains by both C-terminal and internal sequences, and we suggest this is an indication of the extensive regulation that NHE3 undergoes physiologically as part of digestion. Conversely, there is an example of the PDZ domain-containing protein Par-6 that can bind both internal and C-terminal PDZ domain-binding sequences. The PDZ domain of Par-6 can bind both a C-terminal PBM (from the transmembrane receptor Crumbs) and an internal sequence (from the protein Pals1/Stardust). The structure of the Pals1·Par-6 PDZ complex reveals that the PBM is deformed to allow for internal binding (12).
In addition, there are several examples of PDZ domain interactions via internal PDZ-binding motifs (10–14). The best characterized example of this internal PDZ domain-binding motif is the interaction of neuronal nitric-oxide synthase (nNOS) with the PDZ domain of PSD-95 or syntrophin (10) for which there is some structural information. The nNOS PDZ-binding motif has two opposite interaction surfaces: one face has the canonical peptide-binding groove, whereas the other has a β-hairpin “finger.” This nNOS β-hairpin finger docks in the syntrophin PDZ domain peptide-binding groove by mimicking a C-terminal peptide ligand. Another example of internal PBM sites, although lacking structural information, is Idax, a negative regulator of Wnt signaling in Drosophila, that acts by binding the PDZ domain of Dishevelled using its internal sequence KTXXXI. Also, an internal PDZ domain-binding sequence serves as a novel endocytic recycling motif for the endothelin ETA receptor, which belongs to Class A G protein-coupled receptors. This internal sequence is predicted to consist of an anti-parallel β-strand structure that resembles that in nNOS (13).
We showed that although there are contiguous putative Class I and II PDZ domain-binding motifs in the NHE3 C-terminal domain, it was the Class II PDZ domain-binding motif (-595CLDM-) that is involved in NHERF1–3 binding. This sequence is fully conserved in recycling plasma membrane NHEs (NHE3 and NHE5) and across species in these isoforms. Even though there are multiple examples of internal Class I PDZ-binding motifs, as reviewed above, this NHE3 internal PDZ-binding motif is the first example of a Class II internal PDZ-binding motif. Our modeling of this NHE3 domain suggests either a β-sheet-turn-β-sheet (LINUS program (36, 37) or an α-helical-turn-α-helical structure (Rosetta program) (38). However, unlike the nNOS internal PDZ domain recognition sequence, the structure of the NHE3 Class II PDZ domain recognition sequence has not been solved.
Acute NHE3 regulation is important for both the stimulation and inhibition of Na+ absorption that is part of normal digestion and is involved in NHE3 inhibition seen in diarrhea, whereas the acute stimulation of NHE3 is a therapeutic goal for drug development to treat diarrhea. This regulation largely is dependent on NHERF binding to NHE3. This study has shown that this regulation involves two interacting PBM domains of the NHE3 C terminus via somewhat overlapping contributions to regulation. Some structural information is required to further understand how the two domains cooperate while sharing many similar but some different functional contributions to NHE3 activity.
Experimental procedures
The QuikChange site-directed mutagenesis kit was from Stratagene (La Jolla, CA). Nigericin and 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF) were from Molecular Probes (Eugene, OR). 8-pCPT-cGMP was from BIOLOG Life Science Institute (Bremen, Germany). 4-Bromo-A23187, a non-fluorescent analog of the calcium ionophore, was from BIOMOL (Plymouth meeting, PA). EZ-Link sulfo-NHS-SS-biotin and streptavidin-agarose beads were from Thermo Scientific/Pierce Protein Research Products (Rockford, IL). Monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000) antibody was from US Biological (Swampscott, MA). Polyclonal anti-NHERF1 (α-5199; 1:5000) and anti-NHERF2 (α-2570; 1:3000) antibodies were described previously (16). Monoclonal anti-HA affinity matrix was from Roche Diagnostics. Protease inhibitor mixture, wortmannin, monoclonal anti-HA antibody (1:1000), monoclonal anti-calmodulin antibody (1:500), monoclonal β-actin antibody (1:1000), and anti-VSV-G tag antibody conjugated to agarose were purchased from Sigma. Monoclonal anti-GST antibody (1:1000) was from Cell Signaling Technology (Danvers, MA). Nickel-nitrilotriacetic acid-agarose was from Qiagen (Valencia, CA). l-[35S]Methionine and glutathione-Sepharose 4B beads were from Amersham Biosciences. Anti-rabbit IRDye® 680 LT and anti-mouse IRDye® 800 CW were from LI-COR Biosciences (Lincoln, NE). The concentrations of GST fusion proteins were determined by the method of Bradford (42).
Construction of expression vectors for NHE3 C terminus putative PDZ-binding mutants
To study the role of putative Class I and Class II PDZ-binding motifs in aa 590–610 of the NHE3 C terminus, three mutant NHE3 constructs were tagged on the N terminus with three copies of the HA epitope (YPYDVPDYA) inserted into the first extracellular loop of rabbit NHE3 between Glu37 and Ile38 by PCR as described previously (19). The NHE3 C terminus internal PDZ domain-binding mutations, including Class I (-592SAV-) and putative Class II (-595CLDM-), were constructed using a site-directed mutagenesis kit from Stratagene according to the manufacturer's protocol. HA-NHE3 WT was used as a template, and mutations were made in the putative Class I PDZ-binding motif, HA-NHE3SAV/AA (592SAV → 592AAA) and putative Class II PDZ-binding motifs, HA-NHE3CLDM/EK (-595CLDM- → -595CEDK-) and HA-NHE3CLDM/QA (-595CLDM- → -595CQDA-). The primers were HA-NHE3SAV/AA (sense primer, GAGAGTGCCGCCGCCGCGTGCCT GGAC), HA-NHE3CLDM/QA (sense primer, GCCGTGTGCCAGGACGCGCAGTCGCTG), and HA-NHE3CLDM/EK (sense primer, GCCGTGTGCGAGGACAAGCAGTCGCTG). All were inserted into pcDNA3.1/G418+ vector using the HindIII/XbaI cloning sites.
To study the role of the C-terminal putative Class I PDZ-binding motif (-829STHM) of NHE3, two HA-NHE3 mutations were constructed, HA-NHE3 Δ4 (C-terminal four amino acids omitted) and HA-NHE3 PBMIImut + Δ4, and inserted into pcDNA3.1/Hygro+ vector. HA-NHE3 Δ4 (-829STHM) was made by PCR using sense primer ATAAGCTTGATGTCAGGGCGCGGGGG and antisense primer GCTCTAGATCACTCGGGGTGTTCAGCGCC. HA-NHE3 PBMIImut + Δ4 was made via mutagenesis of HA-NHE3 Δ4 using the HA-NHE3CLDM/EK (also called NHE3 PBMIImut) primer as shown above. The changed bases are underlined. PCRs were performed with 16 cycles of 95 °C, 30 s; 55 °C, 1 min; 68 °C, 16 min. The cDNAs were sequenced before further study.
Fusion proteins and in vitro transcription/translation products
35S-Labeled NHE3 C-terminal His6-tagged truncations NHE3-C589 (aa 475–589), NHE3-C610 (aa 475–610), NHE3-C660 (aa 475–660), NHE3-C689 (aa 475–689), NHE3-C711 (aa 475–711), and NHE3-C832 (aa 475–832) were used for pulldown assays using 6 μl of 35S-labeled NHE3 C-terminal truncations and 3 μg of recombinant GST-NHERF2.
His6-tagged NHERF1 and NHERF2 were made in pET30a vector (Novagen) as described previously (24). GST fusion proteins were made of rabbit NHERF1, human NHERF2, and rat NHERF3. GST-tagged NHERF1, NHERF2, and NHERF3 fusion proteins were made in pGEX 4T1 using EcoRI/XhoI cloning sites, GST-tagged SNX27 was cloned into the BamHI-EcoRI site of pGEX4T1 vector (32), and GST-tagged CK2 was made in the pGEX4T1 vector (39) (Promega). In vitro translated, [35S]methionine-labeled NHE3 C-terminal fragments were generated using the Promega TnT kit. The amount of each labeled protein used in binding assays was determined by normalizing the intensity of the autoradiography signal for each labeled protein. GST fusion proteins were expressed in BL21 Gold(DE3) cells (Stratagene) induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4 h. Fusion proteins were purified on GST-Sepharose according to the manufacture's recommendations (GST Gene Fusion System, Amersham Biosciences). The purity of each preparation was determined by Ponceau S staining of protein transferred to nitrocellulose following SDS-PAGE, and the concentration was measured using the Bradford protein assay.
PS120 cell culture and cDNA transfection
The cDNAs of wild-type HA-NHE3 in pcDNA3.1/G418+ vector and its putative PDZ-binding mutants HA-NHE3SAV/AA, HA-NHE3CLDM/EK, and HA-NHE3CLDM/QA in pcDNA3.1/G418+ vector (Invitrogen) were transfected into plasma membrane Na+/H+ exchanger-deficient PS120 fibroblasts. The cDNAs of HA-NHE3 WT, HA-NHE3 PBMIImut, HA-NHE3 Δ4, and HA-NHE3 PBMIImut + Δ4 in pcDNA3.1/Hygro+ vector (Invitrogen) were transfected into PS120 cells stably transfected with NHERF2 using Lipofectamine 2000 (Invitrogen) and selected with neomycin (400 μg/ml) or hygromycin (600 μg/ml). Transfected PS120 fibroblasts were maintained at 37 °C in a humidified atmosphere with 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. In addition, transfected cells were selected by an H+-killing procedure consisting of 50 mm NH4Cl, saline solution for 1 h followed by an isotonic 2 mm Na+ solution for 1 h (40). Surviving cells were then placed in normal culture medium and allowed to reach 30–50% confluence. The H+-killing process was initially repeated every 2–3 days until more than 50% of the cells survived and was then repeated every week.
In vitro pulldown assays
GST fusion proteins immobilized on GSH-Sepharose beads were incubated with PS120 cell lysates or in vitro transcription/translation products in lysis buffer (60 mm Hepes, 150 mm NaCl, 3 mm KCl, 5 mm trisodium EDTA, 3 mm EGTA, 1 mm Na3VO4, pH 7.4, 1% Triton X-100 with protease inhibitor mixture) for 2 h or overnight at 4 °C. Complexes were pelleted at 10,000 × g for 2 min and then washed three times in 1 ml of lysis buffer. Bound proteins were eluted from the beads in sample buffer for 5 min at 80 °C. Proteins were separated by 10 or 14% SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with specific antibodies.
Immunoprecipitation and immunoblotting analysis
Co-immunoprecipitation experiments were performed using lysates from PS120 cells stably expressing HA-NHE3 WT or PDZ-binding mutants. Cell lysates were prepared in cell lysis buffer (60 mm Hepes, pH 7.4, 150 mm NaCl, 3 mm KCl, 5 mm trisodium EDTA, 3 mm EGTA, 1 mm Na3VO4, 1% Triton X-100 with protease inhibitor mixture). Aliquots (1 mg of protein) of lysates were incubated with anti-HA affinity matrix (Roche Applied Science) with gentle mixing on a rotator overnight at 4 °C. After being washed five times with cell lysis buffer, the bound proteins were eluted in Laemmli sample buffer, separated by SDS-PAGE, and transferred to nitrocellulose. The blots were probed with monoclonal anti-HA or polyclonal anti-NHERF2 antibodies for the primary labeling; fluorescent anti-rabbit IRDye® 680 LT and anti-mouse IRDye® 800 CW were used as secondary antibodies (1:10,000 dilution). Protein bands were then visualized and quantitated with the Li-COR Biosciences Odyssey system and Li-COR Biosciences Image Studio software for the IRDye secondary antibodies.
Cell-surface biotinylation
Transfected PS120 cells were grown to 70–80% confluence in 10-cm Petri dishes. The cells were then serum-starved for ∼ 4 h. All subsequent manipulations were performed at 4 °C. For surface labeling of NHE3, cells were incubated with 1 mm NHS-SS-biotin for 20 min (this step was repeated once), solubilized with the lysis buffer, and then incubated for 1 h with streptavidin-agarose beads. Western analysis and the quantification of surface fraction were performed as described previously (41).
Measurement of Na+/H+ exchange activity
The transfected PS120 cells grown to 70–80% confluence on glass coverslips were placed in serum-free medium for 4 h to arrest growth. NHE3 activity of transfected PS120 cells was measured using the intracellular pH-sensitive dye BCECF-AM as described previously (40).
Measurement of half-life of plasma membrane NHE3 using cell-surface biotinylation
A cell-surface biotinylation method was used to determine the half-life of plasma membrane NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 mutants as described previously (26).
Total NHE3 half-life determined by pulse-chase labeling
PS120 cells stably transfected with 3×HA-tagged NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were grown to 50% confluence and then exposed to depletion medium (DMEM without Met and Cys from Life Technologies) for 1 h at 37 °C. To pulse, cells were then incubated with Met/Cys-free DMEM containing 0.2 mCi/ml [35S]Met/Cys cell labeling mixture (GE Healthcare) or 0.2 mCi/ml EXPRESS35S Protein Labeling Mix,[35S]-, EasyTagTM (PerkinElmer Life Sciences) for 4 h. To chase, the labeling solution was aspirated, and the cells were rinsed four times with PBS. Cells were then incubated with DMEM containing 10% serum, 2 mm Met, and 2 mm Cys for different times (0, 4, 8, 12, 18, 24, and 41 h). After solubilization, proteins were immunoprecipitated with monoclonal anti-HA affinity matrix and subjected to SDS-PAGE. Proteins were transferred to nitrocellulose, and 35S-labeled NHE3 WT, NHE3 PBMIImut, and NHE3 Δ4 were detected by a Storm 860 phosphorimaging system and ImageQuant. The details were described previously (25, 26).
FRAP
To quantitate the lateral mobility of NHE3 at the apical domain of polarized OK cells, FRAP was used as previously reported with minor modifications (28, 34). OK cells were used to allow study of a polarized epithelial cell that could be easily transfected. OK cells were cultured on Transwell 12-well plates in DMEM (without phenol red) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a 5% CO2, 95% air atmosphere. The cells were then transfected using Lipofectamine 2000 as described previously (28). FRAP was performed on a stage heated to 37 °C using an LSM 510/Meta confocal microscope. Surface HA-NHE3 was labeled with anti-mouse HA antibody as primary antibody (Sigma) and goat anti-mouse Alexa Fluor® 488 (Life Technologies) as secondary antibody at 4 °C. The 488-nm line of a 400-milliwatt krypton/argon laser was used in conjunction with a 100× Zeiss 1.4-numerical aperture Plan Apochromat oil immersion objective with signal collected in the OK cell apical domain (0.3-μm optical sections starting at the outer limit of the apical domain). Mobile fractions were calculated as described previously (28).
Statistical analysis
Statistical analyses were performed by Student's t test for paired or unpaired comparisons. Results were presented as means ± S.D. A p value of <0.05 was considered significant.
Author contributions
B. C. helped design the experiments, performed the experiments, analyzed and interpreted the data, and wrote the paper. J. Y. helped design the experiments, performed experiments, and analyzed and interpreted the data. V. S. helped design the experiments, performed experiments, and analyzed and interpreted the data. N. C. Z. helped design the experiments and interpreted the data. R. I. S. performed experiments and analyzed and interpreted the data. T. C. performed experiments and analyzed and interpreted the data. M. C. performed experiments and analyzed and interpreted the data. C.-M. T. helped design and interpret the experiments. M. D. helped design the experiments, interpreted the data, and wrote the paper.
Acknowledgments
We acknowledge the expert editorial assistance of Kate DeSantis. Vladimir Yarov-Yarovay, Ph.D., Department of Physiology and Membrane Biology, University of California, Davis performed the Rosetta analysis.
This work was supported by NIDDK, National Institutes of Health Grants R01-DK26523, R01-DK61765, P01-DK072084, and P30-DK89502; The Hopkins Digestive Diseases Basic and Translational Research Core Center; and The Hopkins Center for Epithelial Disorders. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- BB
- brush border
- NHERF
- Na+/H+ exchanger regulatory factor
- aa
- amino acid(s)
- PBM
- PDZ domain-binding motif
- PM
- plasma membrane
- NHE
- Na+/H+ exchanger
- IP
- immunoprecipitation
- 8-pCPT-cGMP
- 8-(4-chlorophenylthio)-guanosine 3′,5′-cyclic monophosphate
- cGKII
- cyclic GMP kinase II
- LPA
- lysophosphatidic acid
- FRAP
- fluorescence recovery after photobleaching
- CaM
- calmodulin
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- nNOS
- neuronal nitric-oxide synthase
- BCECF
- 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein
- VSV-G
- vesicular stomatitis virus G protein
- OK
- opossum kidney.
References
- 1. Ponting C. P., and Phillips C. (1995) DHR domains in syntrophins, neuronal NO synthases and other intracellular proteins. Trends Biochem. Sci. 20, 102–103 [DOI] [PubMed] [Google Scholar]
- 2. Kennedy M. B. (1995) Origin of PDZ (DHR, GLGF) domains. Trends Biochem. Sci. 20, 350. [DOI] [PubMed] [Google Scholar]
- 3. Lee H. J., and Zheng J. (2010) PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal. 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luck K., Charbonnier S., and Travé G. (2012) The emerging contribution of sequence context to the specificity of protein interactions mediated by PDZ domains. FEBS Lett. 586, 2648–2661 [DOI] [PubMed] [Google Scholar]
- 5. Songyang Z., Fanning A. S., Fu C., Xu J., Marfatia S. M., Chishti A. H., Crompton A., Chan A. C., Anderson J. M., and Cantley L. C. (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275, 73–77 [DOI] [PubMed] [Google Scholar]
- 6. Nourry C., Grant S. G., and Borg J. P. (2003) PDZ domain proteins: plug and play! Sci. STKE 2003, RE7. [DOI] [PubMed] [Google Scholar]
- 7. Stricker N. L., Christopherson K. S., Yi B. A., Schatz P. J., Raab R. W., Dawes G., Bassett D. E. Jr., Bredt D. S., and Li M. (1997) PDZ domain of neuronal nitric oxide synthase recognizes novel C-terminal peptide sequences. Nat. Biotechnol. 15, 336–342 [DOI] [PubMed] [Google Scholar]
- 8. Vaccaro P., and Dente L. (2002) PDZ domains: troubles in classification. FEBS Lett. 512, 345–349 [DOI] [PubMed] [Google Scholar]
- 9. Boisguerin P., Leben R., Ay B., Radziwill G., Moelling K., Dong L., and Volkmer-Engert R. (2004) An improved method for the synthesis of cellulose membrane-bound peptides with free C termini is useful for PDZ domain binding studies. Chem. Biol. 11, 449–459 [DOI] [PubMed] [Google Scholar]
- 10. Hillier B. J., Christopherson K. S., Prehoda K. E., Bredt D. S., and Lim W. A. (1999) Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 284, 812–815 [PubMed] [Google Scholar]
- 11. London T. B., Lee H. J., Shao Y., and Zheng J. (2004) Interaction between the internal motif KTXXXI of Idax and mDvl PDZ domain. Biochem. Biophys. Res. Commun. 322, 326–332 [DOI] [PubMed] [Google Scholar]
- 12. Penkert R. R., DiVittorio H. M., and Prehoda K. E. (2004) Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat. Struct. Mol. Biol. 11, 1122–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paasche J. D., Attramadal T., Kristiansen K., Oksvold M. P., Johansen H. K., Huitfeldt H. S., Dahl S. G., and Attramadal H. (2005) Subtype-specific sorting of the ETA endothelin receptor by a novel endocytic recycling signal for G protein-coupled receptors. Mol. Pharmacol. 67, 1581–1590 [DOI] [PubMed] [Google Scholar]
- 14. Zencir S., Banerjee M., Dobson M. J., Ayaydin F., Fodor E. A., Topcu Z., and Mohanty S. (2013) New partner proteins containing novel internal recognition motif for human glutaminase interacting protein (hGIP). Biochem. Biophys. Res. Commun. 432, 10–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donowitz M., Cha B., Zachos N. C., Brett C. L., Sharma A., Tse C. M., and Li X. (2005) NHERF family and NHE3 regulation. J. Physiol. 567, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yun C. H., Lamprecht G., Forster D. V., and Sidor A. (1998) NHE3 kinase A regulatory protein E3KARP binds the epithelial brush border Na+/H+ exchanger NHE3 and the cytoskeletal protein ezrin. J. Biol. Chem. 273, 25856–25863 [DOI] [PubMed] [Google Scholar]
- 17. Weinman E. J., Wang Y., Wang F., Greer C., Steplock D., and Shenolikar S. (2003) A C-terminal PDZ motif in NHE3 binds NHERF-1 and enhances cAMP inhibition of sodium-hydrogen exchange. Biochemistry 42, 12662–12668 [DOI] [PubMed] [Google Scholar]
- 18. Thomson R. B., Wang T., Thomson B. R., Tarrats L., Girardi A., Mentone S., Soleimani M., Kocher O., and Aronson P. S. (2005) Role of PDZK1 in membrane expression of renal brush border ion exchangers. Proc. Natl. Acad. Sci. U.S.A. 102, 13331–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levine S. A., Montrose M. H., Tse C. M., and Donowitz M. (1993) Kinetics and regulation of three cloned mammalian Na+/H+ exchangers stably expressed in a fibroblast cell line. J. Biol. Chem. 268, 25527–25535 [PubMed] [Google Scholar]
- 20. Tse C. M., Levine S. A., Yun C. H., Brant S. R., Pouyssegur J., Montrose M. H., and Donowitz M. (1993) Functional characteristics of a cloned epithelial Na+/H+ exchanger (NHE3): resistance to amiloride and inhibition by protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 90, 9110–9114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J. H., Lee-Kwon W., Park J. B., Ryu S. H., Yun C. H., and Donowitz M. (2002) Ca2+-dependent inhibition of Na+/H+ exchanger 3 (NHE3) requires an NHE3-E3KARP-α-actinin-4 complex for oligomerization and endocytosis. J. Biol. Chem. 277, 23714–23724 [DOI] [PubMed] [Google Scholar]
- 22. Cha B., Zhu X. C., Chen W., Jones M., Ryoo S., Zachos N. C., Chen T. E., Lin R., Sarker R., Kenworthy A. K., Tse M., Kovbasnjuk O., and Donowitz M. (2010) NHE3 mobility in brush borders increases upon NHERF2-dependent stimulation by lysophosphatidic acid. J. Cell Sci. 123, 2434–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen T., Kocinsky H. S., Cha B., Murtazina R., Yang J., Tse C. M., Singh V., Cole R., Aronson P. S., de Jonge H., Sarker R., and Donowitz M. (2015) Cyclic GMP kinase II (cGKII) inhibits NHE3 by altering its trafficking and phosphorylating NHE3 at three required sites: identification of a multifunctional phosphorylation site. J. Biol. Chem. 290, 1952–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cha B., Kim J. H., Hut H., Hogema B. M., Nadarja J., Zizak M., Cavet M., Lee-Kwon W., Lohmann S. M., Smolenski A., Tse C. M., Yun C., de Jonge H. R., and Donowitz M. (2005) cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J. Biol. Chem. 280, 16642–16650 [DOI] [PubMed] [Google Scholar]
- 25. Cha B., Tse M., Yun C., Kovbasnjuk O., Mohan S., Hubbard A., Arpin M., and Donowitz M. (2006) The NHE3 juxtamembrane cytoplasmic domain directly binds ezrin: dual role in NHE3 trafficking and mobility in the brush border. Mol. Biol. Cell 17, 2661–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cavet M. E., Akhter S., Murtazina R., Sanchez de Medina F., Tse C. M., and Donowitz M. (2001) Half-lives of plasma membrane Na+/H+ exchangers NHE1–3: plasma membrane NHE2 has a rapid rate of degradation. Am. J. Physiol. Cell Physiol. 281, C2039–C2048 [DOI] [PubMed] [Google Scholar]
- 27. Cha B., Chen T., Sarker R., Yang J., Raben D., Tse C. M., Kovbasnjuk O., and Donowitz M. (2014) Lysophosphatidic acid stimulation of NHE3 exocytosis in polarized epithelial cells occurs with release from NHERF2 via ERK-PLC-PKCδ signaling. Am. J. Physiol. Cell Physiol. 307, C55–C65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu X., Cha B., Zachos N. C., Sarker R., Chakraborty M., Chen T. E., Kovbasnjuk O., and Donowitz M. (2011) Elevated calcium acutely regulates dynamic interactions of NHERF2 and NHE3 proteins in opossum kidney (OK) cell microvilli. J. Biol. Chem. 286, 34486–34496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin R., Murtazina R., Cha B., Chakraborty M., Sarker R., Chen T. E., Lin Z., Hogema B. M., de Jonge H. R., Seidler U., Turner J. R., Li X., Kovbasnjuk O., and Donowitz M. (2011) D-Glucose acts via sodium/glucose cotransporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+ exchange regulatory factor 2-dependent process. Gastroenterology 140, 560–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sarker R., Valkhoff V. E., Zachos N. C., Lin R., Cha B., Chen T. E., Guggino S., Zizak M., de Jonge H., Hogema B., and Donowitz M. (2011) NHERF1 and NHERF2 are necessary for multiple but usually separate aspects of basal and acute regulation of NHE3 activity. Am. J. Physiol. Cell Physiol. 300, C771–C782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Donowitz M., Mohan S., Zhu C. X., Chen T. E., Lin R., Cha B., Zachos N. C., Murtazina R., Sarker R., and Li X. (2009) NHE3 regulatory complexes. J. Exp. Biol. 212, 1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh V., Yang J., Cha B., Chen T. E., Sarker R., Yin J., Avula L. R., Tse M., and Donowitz M. (2015) Sorting Nexin 27 regulates basal and stimulated brush border trafficking of NHE3. Mol. Biol. Cell 26, 2030–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zizak M., Chen T., Bartonicek D., Sarker R., Zachos N. C., Cha B., Kovbasnjuk O., Korac J., Mohan S., Cole R., Chen Y., Tse C. M., and Donowitz M. (2012) Calmodulin kinase II constitutively binds, phosphorylates, and inhibits brush border Na+/H+ exchanger 3 (NHE3) by a NHERF2 protein-dependent process. J. Biol. Chem. 287, 13442–13456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cha B., Kenworthy A., Murtazina R., and Donowitz M. (2004) The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J. Cell Sci. 117, 3353–3365 [DOI] [PubMed] [Google Scholar]
- 35. Han W., Kim K. H., Jo M. J., Lee J. H., Yang J., Doctor R. B., Moe O. W., Lee J., Kim E., and Lee M. G. (2006) Shank2 associates with and regulates Na+/H+ exchanger 3. J. Biol. Chem. 281, 1461–1469 [DOI] [PubMed] [Google Scholar]
- 36. Srinivasan R., and Rose G. D. (1995) LINUS: a hierarchic procedure to predict the fold of a protein. Proteins 22, 81–99 [DOI] [PubMed] [Google Scholar]
- 37. Srinivasan R., and Rose G. D. (2002) Ab initio prediction of protein structure using LINUS. Proteins 47, 489–495 [DOI] [PubMed] [Google Scholar]
- 38. Bradley P., Malmström L., Qian B., Schonbrun J., Chivian D., Kim D. E., Meiler J., Misura K. M., and Baker D. (2005) Free modeling with Rosetta in CASP6. Proteins 61, 128–134 [DOI] [PubMed] [Google Scholar]
- 39. Sarker R., Grønborg M., Cha B., Mohan S., Chen Y., Pandey A., Litchfield D., Donowitz M., and Li X. (2008) Casein kinase 2 binds to the C terminus of Na+/H+ exchanger 3 (NHE3) and stimulates NHE3 basal activity by phosphorylating a separate site in NHE3. Mol. Biol. Cell 19, 3859–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Levine S. A., Nath S. K., Yun C. H., Yip J. W., Montrose M., Donowitz M., and Tse C. M. (1995) Separate C-terminal domains of the epithelial specific brush border Na+/H+ exchanger isoform NHE3 are involved in stimulation and inhibition by protein kinases/growth factors. J. Biol. Chem. 270, 13716–13725 [DOI] [PubMed] [Google Scholar]
- 41. Akhter S., Cavet M. E., Tse C. M., and Donowitz M. (2000) C-terminal domains of Na+/H+ exchanger isoform 3 are involved in the basal and serum-stimulated membrane trafficking of the exchanger. Biochemistry 39, 1990–2000 [DOI] [PubMed] [Google Scholar]
- 42. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]