

Abstract
Excitation-contraction (EC) coupling in skeletal muscle requires a physical interaction between the voltage-gated calcium channel dihydropyridine receptor (DHPR) and the ryanodine receptor Ca2+ release channel. Although the exact molecular mechanism that initiates skeletal EC coupling is unresolved, it is clear that both the α1 and β subunits of DHPR are essential for this process. Here, we employed a series of techniques, including size-exclusion chromatography-multi-angle light scattering, differential scanning fluorimetry, and isothermal calorimetry, to characterize various biophysical properties of the skeletal DHPR β subunit β1a. Removal of the intrinsically disordered N and C termini and the hook region of β1a prevented oligomerization, allowing for its structural determination by X-ray crystallography. The structure had a topology similar to that of previously determined β isoforms, which consist of SH3 and guanylate kinase domains. However, transition melting temperatures derived from the differential scanning fluorimetry experiments indicated a significant difference in stability of ∼2–3 °C between the β1a and β2a constructs, and the addition of the DHPR α1s I-II loop (α-interaction domain) peptide stabilized both β isoforms by ∼6–8 °C. Similar to other β isoforms, β1a bound with nanomolar affinity to the α-interaction domain, but binding affinities were influenced by amino acid substitutions in the adjacent SH3 domain. These results suggest that intramolecular interactions between the SH3 and guanylate kinase domains play a role in the stability of β1a while also providing a conduit for allosteric signaling events.
Keywords: dihydropyridine receptor (DHPR), excitation-contraction coupling (E-C coupling), protein structure, skeletal muscle, X-ray crystallography
Introduction
Dihydropyridine receptors (DHPRs)3 are multi-subunit voltage-gated calcium channels that play an essential role in a variety of biological processes including muscle contraction, insulin secretion, and synaptic transmission. The DHPR is made up of a major, pore-forming, α1 subunit with associated α2, δ, and β subunits and, in skeletal muscle, α and γ subunits (see Fig. 1a). Key elements of the DHPR involve the voltage sensor and the Ca2+ ion pore, which are located in the α subunit (α1s), whereas other subunits are essential for correct α1S Ca2+ signaling, channel gating, and surface expression (1). DHPR α1s is the largest of the DHPR subunits consisting of four transmembrane repeats that are connected by a series of loops. These loops are linked to the cytoplasmic β subunit, which in the case of skeletal muscle interacts with ryanodine receptor and initiates skeletal EC coupling (2). The structure of the skeletal α1 subunit was initially solved to an overall resolution of 4.2 Å (3) and more recently to 3.6 Å (4) using cryo-electron microscopy. However, the β1a component of these structures was resolved by docking the structure of the β2a isoform (PDB code 4DEY) caused by the unavailability of the β1a structure. It would therefore be useful if the structure of the β1a were to be determined. Similar to other DHPR β subunits, β1a is made up of a core Src-3 (SH3)-guanylate kinase (GK) module, with the SH3 domain split by a hook region of unknown function (see Fig. 1b) (5–7). The SH3 and GK domains are conserved in the β subunits, but the hook and N- and C-terminal regions show greater sequence diversity. It is known that the β subunit is anchored to α1 through a high affinity (∼5–50 nm) interaction (8) through its GK domain via the I-II loop of α1 (α-interaction domain (AID)). Other lower affinity α1/β interactions confer isoform-specific functions on α1 (8).
Figure 1.
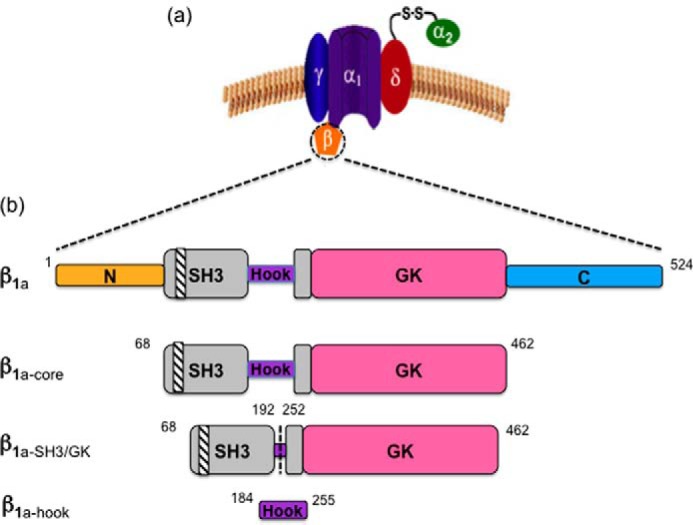
Schematic representation of the DHPR subunits. a, diagram outlining the skeletal DHPR subunits. b, the domain architecture of β1a illustrating a split SH3 domain (gray), which contains a polyproline binding site (diagonal stripes). The GK domain is shown in pink. The N and C termini (orange and cyan) and the hook region (purple) are intrinsically disordered. Modified β1a constructs (β1a-core, β1a-SH3/GK, and β1a-hook) are illustrated.
A number of studies have investigated the isoform-specific properties of the various β subunit domains. A dynamic exchange study of β subunits in situ revealed that the β1a formed a stable complex, whereas the other isoforms interacted in a dynamic fashion with their α1 subunits (9). It is also understood that a unique element of β1a lies in its C-terminal domain where a subset of residues is critical in the communication between the DHPR and ryanodine receptor (10). The importance of the β1a C-terminal domain has been subsequently confirmed in a number of studies in both mouse (11) and zebrafish myotubes (12); the latter study also identified the β1a SH3 domain as a distinctive determinant for voltage sensing in skeletal muscle. From these works, there is a growing body of evidence that the β1a isoform could possess diverse functional traits compared with the other three isoforms (β2a, β3, and β4), and the structural make-up of β1a may be a determining factor driving these novel functions.
Structural determination of full-length β subunits has not been not possible for two main reasons. First, β subunits have a tendency to aggregate at high concentrations (13), and second, the disordered C- and N-terminal domains and the hook region most likely impact upon their ability to crystallize. Therefore to overcome both of these obstacles and aid in the crystallization of β subunit proteins, it has been necessary to remove the disordered regions including the N- and C-terminal domains and in some cases the hook region. The core structures of three different β subunit isoforms (β2a, β3, and β4) have been solved (some with AID) with all three isoforms displaying very similar structures that resemble a family of membrane-associated guanylate kinase (MAGUK) proteins (5–7, 14).
In this study we have determined the structure of the DHPR β1a core by X-ray crystallography. We also demonstrate key differences in the biophysical properties of β1a compared with the β2a isoforms by examining their stability and affinity with AID. Although there is high sequence homology between these two isoforms, subtle amino acid differences may be responsible for many of these biophysical differences. The capacity of the SH3 domain and the GK domains to interact with each other is an inherent feature of all β subunits, and the degree and mode of this interaction may play a role in conferring binding to various partner molecules.
Results
Oligomeric state of β1a subunit constructs
The β subunit protein constructs used in this study are shown in Fig. 1b and were purified to homogeneity as described under “Experimental procedures.” All tags were cleaved prior to experiments being performed, and the minor variation in pH for the different experiment types did not in any way impact upon the conclusions reached.
As part of the initial characterization of protein constructs, the solution molecular masses of β1a constructs were determined using size-exclusion chromatography-multi-angle light scattering (SEC-MALS) and are summarized in Table 1. This included the full-length protein (β1a), a core construct in which the unstructured termini are absent (β1a-core), and a construct that consisted only of structured folded domains (SH3 and GK) linked by a residual hook (β1a-SH3/GK). β1a eluted as two peaks; the first as a large (megadalton), soluble aggregate eluting at 8 ml, whereas the smaller peak was consistent with the monomer size of ∼58 kDa (Fig. 2a). The β1a·core construct eluted as multiple peaks, with a range of molecular masses including a large soluble aggregate (Fig. 2b). The non-integer values observed for each oligomeric state may be indicative of a protein in slow exchange between multiple states in solution and could explain why, despite multiple attempts, the β1a·core did not crystallize. The smaller β1a-SH3/GK construct eluted as a single, predominantly monodispersed peak with an experimental mass close to its predicted value peak of ∼37 kDa (Fig. 2c). To determine whether the excised hook region was responsible for the oligomerization of β1a, the hook region (β1a-hook) was expressed and examined by several techniques including SEC-MALS, circular dichroism, and NMR. The SEC-MALS profile (Fig. 2d) shows that the isolated hook region is monomeric, whereas the CD profile and NMR spectral dispersion indicate that the hook is disordered in solution (supplemental Fig. S1). To examine whether the hook region could induce oligomerization by interacting with β1a-SH3/GK, a titration experiment involving these two fragments was performed. Given that no labeled β1a-hook peaks shifted upon addition of β1a-SH3/GK (data not shown), it is reasonable to conclude that here is no evidence of any self-interaction between these two components in full-length β1a.
Table 1.
The oligomeric state of β1a constructs
The oligomeric state of β1a constructs in solution was calculated by dividing the observed weight-averaged molecular mass with that of the theoretical monomer. Molecular masses greater than 1 MDa eluted close to the void volume of the gel-filtration column and furthermore cannot be accurately determined by MALS; thus they have been designated as “aggregated” protein. Errors in the calculation of observed molecular mass are given. The average protein concentration (μm) corresponding to each peak at the detector is given.
| Protein | Molecular mass (kDa) |
Oligomeric state | [Protein] @ refractometer μm | |
|---|---|---|---|---|
| Observed | Theoretical monomer | |||
| β1a | >1000 | Aggregate | ||
| 57.1 ± 1.1 | 57 | 1.00 | 0.27 | |
| β1a-core | >1000 | Aggregate | ||
| 104 ± 1 | 2.42 | 0.35 | ||
| 67 ± 0.8 | 1.56 | 0.89 | ||
| 38 ± 0.1 | 43 | 0.88 | 0.86 | |
| 29 ± 0.1 | Contaminant | |||
| β1a-SH3/GK | 35.2 ± 0.2 | 37.2 | 0.95 | 9.7 |
| β1a-hook | 11.0 | 9.6 | 1.14 | |
| BSAa | 200 ± 1.6 | 2.99 | 0.38 | |
| 132.8 ± 0.5 | 1.98 | 1.2 | ||
| 67.1 ± 0.1 | 67 | 1.00 | 6.8 | |
a BSA was not monomeric.
Figure 2.

The solution molecular mass(es) of β1a constructs. Full-length β1a (a), β1a-core (b), β1a-SH3/GK (c), and β1a-hook (d) constructs were analyzed by SEC-MALS. Proteins (0.1 mg) were applied to an analytical Superdex 200 size exclusion column. They were eluted in 20 mm Tris-HCl at pH 8.0 and 150 mm potassium chloride at room temperature. Samples were reduced with 1 mm dithiothreitol prior to application. The elution profile was monitored by the change in refractive index (continuous blue line). The molecular masses (kDa; secondary axis) corresponding to peaks are shown as discrete points. Bovine serum albumin (non-monomeric) was analyzed as a standard.
Structure of the β1a subunit (β1a-SH3/GK)
Because of the monomeric nature of β1a-SH3/GK, this construct was selected for crystallographic analysis. Despite trialling numerous crystal screening conditions, β1a-SH3/GK could only be crystallized in the presence of the I-II loop (AID) peptide: a clear point of distinction from the β2a and β3 isoforms where structures have been solved with and without AID. We solved the structure of β1a-SH3/GK to a resolution of 1.83 Å by PHASER (15), using the structure of the rat β2a homolog in complex with an α subunit peptide (PDB code 1T0J). Theβ1a-SH3/GK structure bears a marked similarity with the structures of other isoforms (Fig. 3a) (5–7) displaying backbone root mean square deviations of 0.64, 0.74, and 0.89 Å with β isoforms 2a (PDB code 1T0H), 3 (PDB code 1VYT), and 4 (PDB code 1VYU) (over 265, 269, and 268 C-α atoms, respectively). Briefly, the structure is made up of well-conserved SH3 and GK domains sharing some of the features observed for the MAGUK family of proteins (Fig. 3a). The SH3 domain consists of five antiparallel β-strands with the truncated hook region separating the fourth and fifth β-strands. The SH3 domain and its RT-loop are sandwiched between an N-terminal helix (α1) and another helix that lies C-terminal to β4 (α2) through a network of hydrophobic interactions. The structure of all isoforms thus far examined show that the canonical SH3 polyproline-binding site is occluded by the RT-loop and the α2 helix, and this is also true for β1a (Fig. 3a). The GK domain consists of a five-stranded parallel β-sheet surrounded by six α-helices and forms an intramolecular interaction with the SH3 domain through a series of hydrogen bond and van der Waals contacts. The I-II loop AID peptide binds the GK domain in a hydrophobic groove that is situated on the opposite side of the SH3 domain. Similar to the other β isoforms, upon binding the DHPR I-II loop (AID) adopts an α-helical conformation that predominantly contacts the α3, α6, and α9 helices of β1a by means of several hydrophobic contacts and to a lesser extent hydrogen bond and ionic interactions as displayed in Fig. 3b.
Figure 3.

X-ray crystal structure of DHPR β1a-SH3/GK complexed with AID peptide. a, cartoon representation of the DHPR β1a-SH3/GK complexed with AID. The split architecture of the SH3 domain is shown in gray with its labeled RT-loop highlighted. The RT-loop is sandwiched between the α1 (pink) and α2 helices (green), which are involved in the occlusion of the polyproline binding site. The GK domain is displayed in orange, and the DHPR I-II AID peptide binding ligand is in blue. The yellow shading denotes the putative polyproline binding site. b, close-up of the interaction between the AID peptide (blue) and β1a-SH3/GK (gray) highlighting contributing residues facilitating AID binding.
Thermal stability of β1a-SH3/GK versus β2a-SH3/GK constructs
The thermal stability of a protein is often a good indicator of its crystallization potential and is typically used to screen for crystallization conditions (16). Because it was possible to crystallize β2a-SH3/GK but not β1a-SH3/GK in the absence of AID, the temperature stability of both core constructs were examined using DSF. For all constructs (Fig. 4a), the transition melting curves showed only one inflection point (Fig. 4b), suggesting that either the SH3 and GK domains unfolded independently at similar temperatures or, more likely, because of their ability to interact, a synergic unfolding event had taken place. The Tm values of all βSH3/GK constructs are summarized in Fig. 4c.β1a-SH3/GK was found to be ∼3 °C less stable than β2a-SH3/GK, whereas the addition of AID increased the Tm of β2a-SH3/GK by ∼6.5 °C and β1a-SH3/GK by ∼8 °C, respectively, indicating the extent that AID stabilizes these constructs. These results may explain the difficulties surrounding the crystallizationβ1a-SH3/GK and why the addition of AID facilitates this process. As part of our preliminary studies, DSF studies were also performed with the full hook region (β1a-core). This experiment revealed a ∼3° reduction in Tm compared with truncated core (β1a-SH3/GK). The addition of AID did stabilize β1a-core to similar level as seen for β1a-SH3/GK, but neither the stand-alone protein nor its AID complex could be crystallized despite using a variety of screening conditions.
Figure 4.

Thermal denaturation measurements of βSH3/GK constructs in the absence and presence of AID peptide. a, amino acid composition of βSH3/GK constructs (black and red numbers denote β1a and β2a, respectively). b, temperature denaturation curves for selected βSH3/GK constructs with and without AID. c, summary of transition melting temperatures for the constructs β1a-SH3/GK, β2a-SH3/GK, β2a-SH3(β1aRT)GK, and β1aSH3/β2aGK. Each data set represents two independent sets of quadruplicate measurements. Paired Student's t test analyses for ± AID were performed relative to β1a-SH3/GK. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine which regions of the β subunits are responsible for their differences in stability, we have analyzed the sequences of all four β isoforms. Fig. 5a shows that the β1a isoform most resembles β2a, with a sequence identity of 60%. This value increased to 78% upon removal of the N- and C-terminal domains and hook region (Fig. 5b). Further deletion of the GK domain only resulted in a modest 2% increase in homology (Fig. 5c), whereas a 13% increase was observed by deletion of the SH3 domain (Fig. 5d), indicating that the SH3 domain is primarily responsible for the sequence diversity between core region of β1a and β2a. Based on this analysis, it is evident that the SH3 domain displays a level of sequence diversity among the β isoforms that warrants further investigation; therefore two β-chimeras were constructed and subjected to thermal denaturation. Substitution of the β1a SH3 domain onto a β2a background (β1a-SH3/β2aGK) resulted in a reduced Tm compared with the β2a-SH3/GK (Fig. 4, b and c). A reduction in Tm was also evident upon substitution of the β1a SH3 RT-loop onto a β2a background (β2a-SH3(β1aRT)/GK), indicating that unique sequence elements in the SH3 domain of β1a increase the temperature instability for the β1a-SH3/GK. This suggests that from a thermal stability viewpoint, the SH3 domain of the two isoforms β1a and β2a are not interchangeable.
Figure 5.

DHPR-β isoforms have high sequence identity and structural similarity. The primary sequences of the mouse protein cores, which matched the primary sequence of solved crystal structures (β1a, A2A454558–454; β2, Q8CC2668–485; β3, P5428516–375; and β4, Q8R0S448–398), were aligned pairwise using ClustalO. The boundaries of the core were defined by Simple Molecular Architecture Research Tool (SMART) and encompassed the VGCC domain through to the guanylate kinase domain. a, β; b, βSH3/GK; c, β SH3 domain; d, β GK domain. The sequence identity is shown as a percentage beside the arrows linking each isoform.
Affinity measurements of βSH3/GK constructs and the DHPR AID peptide
Previous binding values between β subunits and AID peptides have been measured to range between 5 and 50 nm (8); however, the affinity between β1a and its corresponding AID peptide has not been measured. The affinities of various βSH3/GK constructs for the DHPR AID peptide were measured using isothermal calorimetry (ITC) and showed stoichiometric binding of 1:1 (Fig. 6 and Table 2). The affinity of β1a-SH3/GK for skeletal AID was measured to be 4.9 ± 2.1 nm, whereas a value of 17.1 ± 3.3 nm was observed for β2a-SH3/GK. Substitution of the β1a RT-loop onto β2a (β2a-SH3(β1aRT)/GK) had a negligible effect on binding (16.5 ± 4.2 nm), but replacing the entire SH3 domain (β1a-SH3/β2aGK) increased the affinity (7.0 ± 2.8 nm) to values comparable with that observed for β1a.
Figure 6.

ITC curves for βSH3/GK constructs titrated with AID. ITC isotherms and curves for the constructs β1a-SH3/GK (a), β2a-SH3/GK (b), β2a-SH3(β1aRT)GK (c), and β1aSH3/β2aGK (d). Binding and thermodynamic parameters are displayed in Table 2.
Table 2.
Collated ITC data for β-SH3/GK constructs titrated with AID
| n | KD | t test p value (to β1a-SH3/GK) | N | ΔH | ΔS | ΔG | |
|---|---|---|---|---|---|---|---|
| nm | kCal/mol | Cal/mol·K | kJ/mol | ||||
| β1a-SH3/GK | 3 | 4.9 ± 2.1 | NAa | 0.96 | −27.1 | −52.6 | −45.7 |
| β2a-SH3/GK | 3 | 17.1 ± 3.3 | 0.013 | 0.80 | −29.33 | −62.7 | −42.6 |
| β2a-SH3(β1aRT)/GK | 3 | 16.5 ± 4.2 | 0.002 | 0.72 | −26.8 | −54.2 | −44.8 |
| β1a-SH3/β2aGK | 3 | 7.0 ± 2.8 | 0.26 | 1.01 | −23.6 | −41.8 | −46.5 |
a NA, not applicable.
Discussion
In contrast to other DHPR β subunit isoforms, β1a is expressed only in skeletal muscle and is an exclusive partner of DHPR α1s. β1a is also essential in the functional assembly of skeletal muscle triads and is required to form DHPR tetrads (17). Given the unique functional features of β1a, it has been important to characterize its structural and biophysical properties. Isolated, full-length β1a exists primarily in large, multimeric assemblies. Removal of the instrinsically disordered N- and C-terminal regions shifted its oligomerization profile, resulting in a series of multi-sized aggregates, whereas further excision of the hook region gave rise to a well-behaved monomeric species. This indicates that the hook region is intimately involved in the oligomerization process. CD, SEC-MALS, and NMR confirmed that the hook region is monomeric and intrinsically disordered but, based on NMR titration experiments, is unlikely to promote oligomerization by interacting with the β1a core SH3 and GK domains. Although the exact role of the hook region in oligomerization of β1a remains undefined, a possible role may be to enable domain swapping between the SH3 and GK domains, a hypothesis first proposed for another MAGUK protein, PSD-95 (18). The functional role of the hook region from various β isoforms has been examined previously and identified as an important element in regulating DHPR channel inactivation (19). In the case of β1a, removal of the hook region has been reported to reduce intracellular calcium release (12). What is unknown and of future interest is whether the hook region that plays a role in the aggregation of β1a in vitro is implicated in DHPR tetrad formation?
The crystal structure of the monomeric β1a-SH3/GK complexed with the AID peptide has been solved to a resolution of 1.86 Å, making it the most highly resolved β-structure so far determined. As shown in Fig. 7A, the structure shows a high degree overlap with structural elements from other β-structures with the only real point of difference visible in loop regions in GK domain between strand β7 and helix α5, as well as the residual hook region. Although the majority of hook region has been removed, the remaining hook residues display a well-defined loop structure. As previously discussed, the β1a SH3 domain exhibits the greatest sequence variation between the β isoforms; therefore it was of interest to focus more closely on this region. Despite a clear difference in the composition of the β1a SH3 RT-loop between various isoforms, this region reveals a remarkable degree of overlap with other β-structures (Fig. 7B) with the RT-loop stabilized by a series of hydrophobic interactions between the two α-helical segments effectively occluding the canonical SH3 binding site. A clear point of difference in this structure, however, is the absence of a salt bridge between an acidic RT-loop and a conserved basic residue in α2, which is present in other β isoform structures (Fig. 7C). It is likely that this interaction may help in stabilizing the position of the RT-loop.
Figure 7.

Structural comparisons between β1a isoforms. A, backbone superposition of β1a with structures that have been crystallized with and without AID peptide. The color key corresponds to β isoforms. B, overlay of β X-ray crystal structures showing the SH3 domain, the α1 and α2 helices, and the RT-loop. The β1a structure is depicted in red. C, the X-ray crystal structure of the SH3 domain of rabbit β2a (PDB code 1t3l). The α2 helix and the RT-loop are highlighted in magenta and green, respectively, and interact through a salt bridge involving the side chains of Glu76 and Lys124 (shown). These structural elements occlude the polyproline binding site, which is displayed as a pale orange line. The sequence alignment of the α2 helix and the RT-loop is displayed for all β-subunit isoforms with the arrows denoting charged residues involved in a salt bridge that is absent in the β1a RT-loop.
This argument is strengthened by the Tm measurements performed for the various β constructs. Comparison of coreβ1a-SH3/GK and β2a-SH3/GK consistently showed a marked difference in their Tm values of ∼3° with β2a-SH3/GK showing greater stability. Substitution of the β1a SH3 domain or the SH3 β1a RT-loop onto a β2a background served to lower the Tm values with respect to β2a, suggesting that the presence of specific amino acid residues within the β2a SH3 domain (in particular the RT-loop) stabilized the protein. The addition of the AID peptide resulted in a striking Tm increase for all constructs ranging from a maximum ∼8° C increase in β1a-SH3/GK to a minimum of ∼6.5° C in β2a-SH3/GK. This increase in Tm suggests that β1a exhibits a less stable, more dynamic structure that is markedly stabilized by the addition of AID, thereby facilitating crystallization. Even though there is an increase in the stability of β2a-SH3/GK upon the addition of AID, its presence is not a prerequisite for the crystallization of β2a (6).
The ITC results performed in this study indicate that the binding affinity of the AID peptide to β1a-SH3/GK is approximately three times tighter than that of β2a-SH3/GK, a result that is consistent with the trend observed in the thermal stability experiments (see above). However, of particular note was the Kd obtained for the β1a-SH3/β2aGK chimera, which was similar to that observed for β1a-SH3/GK. This result suggests that despite the AID-binding site being located on the GK domain, changes in the SH3 domain influenced binding on the other side of the molecule. Such an observation can be rationalized by considering the intramolecular interaction between the two domains as seen in all β X-ray crystal structures. In the case where the X-ray structures have been solved for both the apo/β and AID/β complexes (β2 and β3 isoforms), there are subtle but clear differences at the GK/SH3 domain interface as observed by changes in salt-bridge and hydrogen-bond connectivities (supplemental Fig. S2) and by overall changes in the buried surface area between the two domains (6). Prior to the structural determination of the AID-binding site, it was assumed that AID interacted with what was described as the β-interaction domain, which spans the SH3, hook, and GK domains. This assumption was based directly on studies where several β-interaction-domain point mutations directly impacted AID binding (20), a finding that highlights the intricate interaction network that exists throughout the molecule extending from the AID-binding site in the GK domain though to the SH3 domain. In this context, it is understandable that swapping the SH3 domain would influence AID binding to the GK domain as observed for the β1a-SH3/β2aGK chimera.
The intramolecular interaction between the SH3 and GK domains may provide a framework that enables allosteric changes to propagate throughout the molecule. A conformational change in β could conceivably be triggered by any number of biological events including depolarization of the surface plasma membrane. Several studies have suggested that the interaction between the β subunit and AID are reversible (9, 21). Interestingly, based on a 3.6 Å cryo-electron microscopy model of DHPR (CaV1.1), it was predicted that conformational changes in the DHPR α1 S6 transmembrane helix can be translated through to the β1a subunit via AID, giving rise to the displacement of β1a (4). Based on the findings derived from our study, it is clear that the biophysical properties of β subunits are highly sensitive to AID binding and may be a determining factor that controls its conformation and binding of other partner molecules. Furthermore, the temperature stability and AID-binding profiles observed for β1a-SH3/GK and β2a-SH3/GK are sufficiently different to suggest that the sequence variability between the two isoforms represents another layer of complexity that may give rise to isoform-specific binding partners.
In the past, the role of the β subunit has been limited to enhancing localization of the α subunit to the plasma membrane, as well as regulating calcium channel gating; however, we are now discovering newly assigned functions independent of these roles, including gene regulation (22). A number of studies have openly raised the possibility that the SH3 domain of DHPR β subunits may engage with partner proteins possessing a SH3 recognition motif (PXXP) (12, 23); however, the crystal structure of all β subunits show that the traditional SH3 binding site is occluded. The information derived from the present study indicates that the SH3 domain in β1a exhibits a degree of structural plasticity that under certain conditions may allow other proteins possessing SH3 recognition motifs to engage the β1a subunit.
Experimental procedures
Production of recombinant proteins and peptide synthesis
All proteins were expressed recombinantly in Escherichia coli (BL21 DE3) in 2× YT supplemented with yeast nitrogen base without amino acids, iron chloride, and the appropriate antibiotic. The DHPR α1s AID peptide (357QQLEEDLRGYMSWITQGE374) was synthesized by the Biomolecular Resource Facility of the John Curtin School of Medical Research (Australian National University, Canberra, Australia) using an Applied Biosystems 430A peptide synthesizer and purified by reverse-phase HPLC on a Jupiter 300 C4 column. Peptides were eluted using a linear gradient from buffer A (deionized water and 0.1% TFA) and buffer B (acetonitrile and 0.1% TFA). Purified peptide fractions were identified by mass spectroscopy using an AB MDS Sciex 4800 MALDI-TOF-TOF mass analyzer.
DHPR β1a
The gene sequence encoding full-length mouse β1a subunit (NCBI code NM_031173) was amplified by PCR. The PCR product was cloned into pHUE (24), which encodes an N-terminal polyhistidine-ubiquitin tag. Transformed bacteria were cultured at 37 °C until A600 nm = ∼0.6. Protein expression was induced by adding isopropyl β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mm. The cells were cultured for a further 3 h. The protein was purified by nickel-agarose chromatography. Ubiquitin was removed by digestion with UBP41 (produced in-house), a polyhistidine-tagged ubiquitin-dependent protease (24). The protein was further purified by preparative electrophoresis using a Bio-Rad model 491 prep cell. The protein was refolded by dialysis into 50 mm sodium phosphate buffered at pH 8 and 300 mm sodium chloride. The protein was concentrated and stored at −80 °C.
DHPR β1a-core
The β1a·core construct was also expressed with a hexahistidine-ubiquitin tag. Bacteria transformed with pHUE-β1a·core were cultured at 37 °C to an A600 nm level of ∼0.6. Protein expression was induced with 0.1 mm IPTG and cultured for 3 h. The bacteria were resuspended in 20 mm Tris-HCl buffered at pH 8.0, 500 mm sodium chloride, and 30 mm imidazole (Buffer O) containing 10% glycerol and EDTA-free protease inhibitor mixture (Roche). The cells were lysed by the addition of lysozyme (1 μg·ml), DNase I (1 μg·ml), and RNase H (1 μg·ml). The suspension was also passed through a French press (3 × 1500 p.s.i.). The cleared lysate was applied to HisTrap column and washed with buffer O and then buffer O containing 1.5 m sodium chloride. The protein was eluted using a gradient of up to 500 mm imidazole in buffer O. The protein was de-ubiquitinylated using UBP41 and simultaneously dialyzed into buffer O containing 5 mm 2-mercaptoethanol. The tag, uncleaved protein, and de-ubiquitinylase were removed by passing the protein solution over HisTrap column. The flowthrough was dialyzed into 20 mm Tris-HCl buffered at pH 8.0 and 50 mm sodium chloride (buffer Q) for 2 h at 4 °C. The protein solution was applied to anion-exchange resin and eluted with buffer Q with a gradient of up to 1 m sodium chloride. The eluate was applied to a size-exclusion column. Peaks containing β1a·core were pooled, concentrated to 1 mg·ml−1, snap frozen, and stored at −80 °C.
DHPR βSH3/GK constructs (β1a-SH3/GK, β2a-SH3/GK, β1a-SH3/β2aGK, and β2a-SH3(β1aRT)/GK)
Genes encoding hexahistidine-tagged β constructs were synthesized and cloned into a custom vector pJ411KanR with a high copy number origin of replication (DNA 2.0). Truncated and chimeric protein constructs were constructed to include the following residues: βSH3/GK (68–192 ∼ 252–462), β2a-SH3/GK (24–144 ∼ 202–422), β1a-SH3/β2aGK (68–192 ∼ 252–272 ∼ 225–422), and β2a-SH3(β1aRT)/GK (24–66 ∼ 108–132 ∼ 90–144 ∼ 202–422), where bold denotes β1a and non-bold denotes β2a numbering. Bacteria transformed with the plasmids were cultured at 37 °C in 2× YT and kanamycin (50 μg·ml−1) to an A600 nm level of ∼0.3. They were further cultured at 16 °C for 1 h. Protein expression was induced with IPTG (0.4 mm), and the cells were cultured overnight. The bacterial pellet was resuspended in 50 mm phosphate buffer at pH 7.0, 500 mm sodium chloride, and 30 mm imidazole (buffer A) containing an EDTA-free protease inhibitor mixture (Roche), lysozyme (1 μg·ml−1), DNase I (1 μg·ml−1), and RNase H (μg·ml−1). The cells were lysed using a French press (3 × 1500 p.s.i.). The cleared lysate was applied to a HisTrap column. The resin was washed with buffer A and then buffer A containing 1.5 m sodium chloride. The protein was eluted using a gradient of imidazole up to 500 mm. The hexahistidine tag was cleaved with HRV 3C protease (produced in-house) and simultaneously dialyzed overnight in buffer A containing 5 mm 2-mercaptoethanol at 4 °C. The protein solution was applied to nickel-nitrilotriacetic acid resin to remove the hexahistidine tag-containing entities. The flowthrough was further purified by size-exclusion chromatography.
β1a-hook
The DNA sequence encoding hexahistidine-tagged HOOK was synthesized and cloned into a custom vector pJ411KanR with a high copy number origin of replication (DNA 2.0). Bacteria transformed with pHis3CkanR-HOOK were cultured at 37 °C in 2× YT and kanamycin (50 μg·ml−1). Protein expression was induced at an A600 nm level of ∼0.8 by the addition of IPTG (1 mm) and cultured for a further 4 h. The bacteria were resuspended in 20 mm Tris-HCl buffered at pH 8.0, 500 mm NaCl, and 30 mm imidazole (buffer H) containing 6 m guanidine-HCl. The cells were lysed using the French press (3 × 1500 p.s.i.). The cleared lysate was applied to nickel-nitrilotriacetic acid resin and incubated for 30 min at 4 °C. The resin was washed with buffer H containing 6 m urea and then with buffer H. The peptide was eluted with buffer H containing 300 mm imidazole. The acidified (pH < 5.0) eluate was applied to preparative C18 reversed-phase HPLC column. The peptide was eluted with a gradient of acetonitrile over a background of 0.1% trifluoroacetic acid in MilliQ water. The appropriate peaks were lyophilized and stored at −70 °C.
MALS of β1a constructs
Protein (0.1 μg), reduced with 1 mm DTT, was applied to a Superdex 200 Increase analytical column attached to an in-line refractometer and light scatterer (Wyatt). The protein was eluted using 20 mm Tris buffer at pH 8.0 and 150 mm potassium chloride at a flow rate of 0.5 ml·min−1 (Waters instrument).
Crystallization of β1a-SH3/GK
Initial crystallization trials were conducted at the Collaborative Crystallization Centre (http://www.csiro.au/c3) using the vapor diffusion method. Crystallization conditions were identified using the JCSG+ and PACT screens (25). Protein solution (150 nl) and screening solution (150 nl) were dispensed into the reservoirs of Innovadyne crystallization plates. The drops imaged by Rigaku Minstrel systems at 8 or 20 °C. Crystals were observed to grow in conditions G7 (20% PEG 3350, 0.1 m Bis-Tris propane, pH 7.5, 0.2 m sodium acetate) were selected for optimization. Optimized crystals for X-ray data collection were grown using the hanging-drop vapor diffusion methods using VDXTM plates (Hampton Research). 2-μl drops containing protein were mixed with equal volumes of precipitant (20% PEG 3350, 0.1 m Bis-Tris propane buffered at pH 8.0 and 0.2 m sodium acetate) on siliconized coverslips (Hampton Research), which were suspended over drops containing precipitant. The trays were equilibrated at 5 °C. Crystals appeared in 5 days and reached maximum size (bipyramids with maximum dimension of ∼200 μm) in 10 days. MiTeGen loops were used to manipulate crystals. For X-ray data collection, a crystal was transferred to artificial mother liquor containing 25% PEG 3350 for 2 min prior to transfer to flash cooling to 100 K using an Oxford Cryo-stream. CuKα X-rays were produced by a Rigaku 007HF rotating anode generator with Varimax optics. X-ray data were collected using a Mar345 desktop beamline. Diffraction data were integrated, merged, and scaled with the HKL2000 package (26). The structure was solved by PHASER (15), using the structure of the rat β2a homolog in complex with an α subunit peptide (PDB code 1T0J). Iterative cycles of model building and refinement were performed in COOT (27) and REFMAC5 (28). The X-ray data and model quality are given in Table 3.
Table 3.
X-ray data collection and refinement statistics for β1a-SH3/GK
| X-ray data | |
| Space group | P212121 |
| Unit cell parameters | a = 46.2, b = 69.4, c = 131.1 Å; α = β = γ = 90° |
| Resolution range (Å)a | 50–1.86 (1.93–1.86) |
| Total no. of observations | 249,097 |
| No. of unique reflections | 36,235 |
| I/σI | 35.45 (4.95) |
| Rmerge (%)b | 5.3 (36.4) |
| Rmeas (%) | 5.3 (36.4) |
| CC½ (%) | 98.8 (93.2) |
| Completeness | 99.8 (99.1) |
| Multiplicity | 6.9 (5.7) |
| Refinement statistics | |
| Resolution range (Å) | 22.8–1.86 (1.908–1.860) |
| No. of reflections (Rwork set) | 32,572 (2447) |
| No. of reflections (Rfree set) | 1801 (108) |
| Rwork (%)c | 16.87 (22.8) |
| Rfree (%) | 20.70 (29.6) |
| No. of atoms | 3,010 |
| áBñ of structure (Å2) | 15.49 |
| Root mean square deviation from ideal geometry | |
| Bond lengths (Å) | 0.019 |
| Bond angles (°) | 1.897 |
| Chiral centers (Å3) | 0.121 |
| General planes (Å) | 0.010 |
a The numbers in parentheses refer to the highest resolution bin.
b Rmerge = ΣhΣi|Ii − áIñ|/ΣhΣiIi.
c Rfactor = Σh|Fobs − Fcalc|/Σh|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively. Rfree was calculated from 5% of the diffraction data not used in refinement.
SYPRO orange thermal denaturation assays of DHPR βSH3/GK constructs
The proteins were either dialyzed overnight or gel-filtered (Superdex 200 Increase) in 20 mm HEPES buffered at pH 7.5, 150 mm sodium chloride, and 1 mm DTT (buffer T). Dialyzed proteins were centrifuged for 10 min at (20,000 × g) to remove aggregated species. The protein (0.1 mg·ml−1) solutions were dispensed in a 384-well plate in quadruplicate (EpMotion). SYPRO orange fluorescence (F) was monitored as the plate was heated from 25–90 °C at a rate of 1°/min (Q-PCR 7900). All constructs were analyzed in the presence or absence of 5-fold excess AID. A sigmoid curve (below) with variables corresponding to the gradient (m) and the point of inflection (i) was fitted using either MATLAB or Microsoft Excel. The average of the inflection points was taken as the melting temperature (Tm) for each construct.
| (Eq. 1) |
Each Tm represents the mean of two sets of quadruplicate measurements with the error range of the Tm represented by error bars.
ITC of DHPR βSH3/GK constructs with AID
DHPR β constructs were exchanged into 10 mm K3PO4 buffered at pH 7.0 and 150 mm KCl (buffer C) either using overnight dialysis or size-exclusion chromatography. The AID peptide was dissolved in buffer C, and the pH was adjusted. ITC was conducted in 10 mm potassium phosphate buffered at pH 7.0 and 150 mm potassium chloride at 25 °C using a VP-ITC (MicroCal). The reference power was set to 17 μCal·s−1, and the cell contents were stirred continuously at 300 rpm. We aimed for 50 μm AID in the titration syringe and 5 μm β constructs in the cell, although the true concentration of protein varied slightly between experiments. A small volume (3 μl) was initially injected. The next 10 and final 11 injections were of ∼350 pmol to obtain an accurate enthalpy change (ΔH). Smaller injections of ∼175 pmol were used to accurately determine gradient of the transition and the point of inflection, which correlate to the affinity (KA) and stoichiometry (N) of the interaction, respectively. We allowed 5-min delays between injections, which was extended to 6 min when necessary. Heat was corrected for the heat of dilution by titrating AID peptide into buffer using the same titration protocol. A binding isotherm was generated by plotting the heat change for each injection over the total delay interval against the molar ratio of AID to the β construct. The binding isotherm was modeled for a single site using non-linear least squares analysis (Origin 7.0 embedded in MicroCal software). All parameters, which include the stoichiometry (N), binding constant (KA), and the enthalpy change (ΔH), were allowed to vary during the fitting cycles.
The concentration of titrant (AID) and titrand (β constructs) were determined using a conventional UV-visible range spectrophotometer (A280 nm = ϵcl), where ϵAID @ 280 nm = 6.99 m−1·cm−1. The corrected extinction coefficients for the β constructs were determined using a conventional UV-visible range spectrophotometer and the theoretical extinction coefficient calculated (29) by ProtParam.
Statistical significance
The KD values and melting temperatures derived from ITC and DSF experiments, respectively, were analyzed for statistical significance using a paired t test. The criteria for a significant t test is considered to be a p value of <0.05 (30).
Author contributions
N. C. N., S. J., S. A., Y. K., and A. J. O. conducted the experiments, and N. C. N., S. J., A. J. O., P. G. B., A. F. D., and M. G. C. designed the experiments and wrote the paper.
Supplementary Material
Acknowledgments
We thank Dr. Damien Hall for help fitting curves using MATLAB and Dr. Hideki Onagi for advice on ITC experiments. We also thank the technical staff of the Bio21-C3 Center for help with crystallization.
The work was supported by grants from Australian National Health and Medical Research Council Grants APP1020589 and APP1002589 (to A. F. D., P. G. B., A. J. O., and M. G. C.) The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4ZW2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Figs. S1 and S2.
- DHPR
- dihydropyridine receptor
- ITC
- isothermal calorimetry
- AID
- α-interaction domain
- GK
- guanylate kinase
- PDB
- Protein Data Bank
- SH
- Src homology
- MAGUK
- membrane-associated guanylate kinase
- SEC-MALS
- size-exclusion chromatography-multi-angle light scattering
- DSF
- differential scanning fluorimetry
- IPTG
- isopropyl β-d-thiogalactopyranoside
- EC
- excitation-contraction
- RT
- arginine and threonine.
References
- 1. Obermair G. J., Tuluc P., and Flucher B. E. (2008) Auxiliary Ca2+ channel subunits: lessons learned from muscle. Curr. Opin. Pharmacol. 8, 311–318 [DOI] [PubMed] [Google Scholar]
- 2. Coronado R., Ahern C. A., Sheridan D. C., Cheng W., Carbonneau L., and Bhattacharya D. (2004) Functional equivalence of dihydropyridine receptor α1S and β1a subunits in triggering excitation-contraction coupling in skeletal muscle. Biol. Res. 37, 565–575 [DOI] [PubMed] [Google Scholar]
- 3. Wu J., Yan Z., Li Z., Yan C., Lu S., Dong M., and Yan N. (2015) Structure of the voltage-gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]
- 4. Wu J., Yan Z., Li Z., Qian X., Lu S., Dong M., Zhou Q., and Yan N. (2016) Structure of the voltage-gated calcium channel Cav1.1 at 3.6 A resolution. Nature 537, 191–196 [DOI] [PubMed] [Google Scholar]
- 5. Chen Y. H., Li M. H., Zhang Y., He L. L., Yamada Y., Fitzmaurice A., Shen Y., Zhang H., Tong L., and Yang J. (2004) Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature 429, 675–680 [DOI] [PubMed] [Google Scholar]
- 6. Opatowsky Y., Chen C. C., Campbell K. P., and Hirsch J. A. (2004) Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron 42, 387–399 [DOI] [PubMed] [Google Scholar]
- 7. Van Petegem F., Clark K. A., Chatelain F. C., and Minor D. L. Jr. (2004) Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richards M. W., Butcher A. J., and Dolphin A. C. (2004) Ca2+ channel β-subunits: structural insights AID our understanding. Trends Pharmacol. Sci. 25, 626–632 [DOI] [PubMed] [Google Scholar]
- 9. Campiglio M., Di Biase V., Tuluc P., and Flucher B. E. (2013) Stable incorporation versus dynamic exchange of β subunits in a native Ca2+ channel complex. J. Cell Sci. 126, 2092–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karunasekara Y., Rebbeck R. T., Weaver L. M., Board P. G., Dulhunty A. F., and Casarotto M. G. (2012) An α-helical C-terminal tail segment of the skeletal L-type Ca2+ channel β1a subunit activates ryanodine receptor type 1 via a hydrophobic surface. FASEB J. 26, 5049–5059 [DOI] [PubMed] [Google Scholar]
- 11. Eltit J. M., Franzini-Armstrong C., and Perez C. F. (2014) Amino acid residues 489–503 of dihydropyridine receptor (DHPR) β1a subunit are critical for structural communication between the skeletal muscle DHPR complex and type 1 ryanodine receptor. J. Biol. Chem. 289, 36116–36124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dayal A., Bhat V., Franzini-Armstrong C., and Grabner M. (2013) Domain cooperativity in the β1a subunit is essential for dihydropyridine receptor voltage sensing in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 110, 7488–7493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lao Q. Z., Kobrinsky E., Liu Z., and Soldatov N. M. (2010) Oligomerization of Cavβ subunits is an essential correlate of Ca2+ channel activity. FASEB J. 24, 5013–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dolphin A. C. (2003) β subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 35, 599–620 [DOI] [PubMed] [Google Scholar]
- 15. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ristic M., Rosa N., Seabrook S. A., and Newman J. (2015) Formulation screening by differential scanning fluorimetry: how often does it work? Acta Crystallogr. F Struct. Biol. Commun. 71, 1359–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schredelseker J., Dayal A., Schwerte T., Franzini-Armstrong C., and Grabner M. (2009) Proper restoration of excitation-contraction coupling in the dihydropyridine receptor β1-null zebrafish relaxed is an exclusive function of the β1a subunit. J. Biol. Chem. 284, 1242–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGee A. W., Dakoji S. R., Olsen O., Bredt D. S., Lim W. A., and Prehoda K. E. (2001) Structure of the SH3-guanylate kinase module from PSD-95 suggests a mechanism for regulated assembly of MAGUK scaffolding proteins. Mol. Cell 8, 1291–1301 [DOI] [PubMed] [Google Scholar]
- 19. Richards M. W., Leroy J., Pratt W. S., and Dolphin A. C. (2007) The HOOK-domain between the SH3 and the GK domains of Cavβ subunits contains key determinants controlling calcium channel inactivation. Channels 1, 92–101 [DOI] [PubMed] [Google Scholar]
- 20. De Waard M., Scott V. E., Pragnell M., and Campbell K. P. (1996) Identification of critical amino acids involved in α1-β interaction in voltage-dependent Ca2+ channels. FEBS Lett. 380, 272–276 [DOI] [PubMed] [Google Scholar]
- 21. Hidalgo P., Gonzalez-Gutierrez G., Garcia-Olivares J., and Neely A. (2006) The α1-β-subunit interaction that modulates calcium channel activity is reversible and requires a competent α-interaction domain. J. Biol. Chem. 281, 24104–24110 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y., Yamada Y., Fan M., Bangaru S. D., Lin B., and Yang J. (2010) The β subunit of voltage-gated Ca2+ channels interacts with and regulates the activity of a novel isoform of Pax6. J. Biol. Chem. 285, 2527–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miranda-Laferte E., Gonzalez-Gutierrez G., Schmidt S., Zeug A., Ponimaskin E. G., Neely A., and Hidalgo P. (2011) Homodimerization of the Src homology 3 domain of the calcium channel β-subunit drives dynamin-dependent endocytosis. J. Biol. Chem. 286, 22203–22210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Catanzariti A. M., Soboleva T. A., Jans D. A., Board P. G., and Baker R. T. (2004) An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 13, 1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newman J., Egan D., Walter T. S., Meged R., Berry I., Ben Jelloul M., Sussman J. L., Stuart D. I., and Perrakis A. (2005) Towards rationalization of crystallization screening for small- to medium-sized academic laboratories: the PACT/JCSG+ strategy. Acta Crystallogr. D Biol. Crystallogr. 61, 1426–1431 [DOI] [PubMed] [Google Scholar]
- 26. Otwinowski Z., and Minor W. (1997) Macromolecular Crystallography, part A, Academic Press, New York [Google Scholar]
- 27. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Skubák P., Murshudov G. N., and Pannu N. S. (2004) Direct incorporation of experimental phase information in model refinement. Acta Crystallogr. D Biol. Crystallogr. 60, 2196–2201 [DOI] [PubMed] [Google Scholar]
- 29. Edelhoch H. (1967) Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 30. Bonita R., Beaglehole R., and Kjellstrom T. (2006) Basic Epidemiology, 2nd Ed., pp. 73–74, World Health Organization, Geneva, Switzerland [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.