

Abstract
In budding yeast (Saccharomyces cerevisiae) the multilayered spindle pole body (SPB) is embedded in the nuclear envelope (NE) at fusion sites of the inner and outer nuclear membrane. The SPB is built from 18 different proteins, including the three integral membrane proteins Mps3, Ndc1, and Mps2. These membrane proteins play an essential role in the insertion of the new SPB into the NE. How the huge core structure of the SPB is anchored in the NE has not been investigated thoroughly until now. The present model suggests that the NE protein Mps2 interacts via Bbp1 with Spc29, one of the coiled-coil proteins forming the central plaque of the SPB. To test this model, we purified and reconstituted the Mps2-Bbp1 complex from yeast and incorporated the complex into liposomes. We also demonstrated that Mps2-Bbp1 directly interacts with Mps3 and Ndc1. We then purified Spc29 and reconstituted the ternary Mps2-Bbp1-Spc29 complex, proving that Bbp1 can simultaneously interact with Mps2 and Spc29 and in this way link the central plaque of the SPB to the nuclear envelope. Interestingly, Bbp1 induced oligomerization of Spc29, which may represent an early step in SPB duplication. Together, this analysis provides important insights into the interaction network that inserts the new SPB into the NE and indicates that the Mps2-Bbp1 complex is the central unit of the SPB membrane anchor.
Keywords: cell biology, membrane protein, molecular cell biology, recombinant protein expression, Saccharomyces cerevisiae
Introduction
The GDa yeast spindle pole body (SPB),3 the functional equivalent of the mammalian centrosome, organizes nuclear and cytoplasmic microtubules and, therefore, is a major player in chromosome segregation and positioning of the nucleus (for review, see Ref. 1). Similar to the much smaller nuclear pore complex (NPC, 40–70 MDa), the SPB is embedded within the nuclear envelope at fusion sites between the inner (INM) and outer nuclear membrane (ONM, 2).
From the 18 different proteins forming the SPB, 10 proteins build up the cylindrical 0.5-GDa core unit of the SPB, which is organized in three vertical layers, the outer, central, and inner plaques (1, 3). Four proteins, Sfi1, Cdc31, the tail-anchored membrane protein Kar1, and the SUN domain containing membrane protein Mps3 (4), constitute the half-bridge/bridge being essential for the once per cell cycle duplication of the SPB (for review, see Refs. 5 and 6). A network of four proteins, Mps2, Bbp1, Ndc1, and Nbp1, collectively named the SPB insertion network (SPIN), is essential for insertion of the newly formed SPB into the nuclear envelope (for review, see Ref. 7). Mps2 is an integral membrane protein containing one transmembrane domain (8, 9) and interacts with Bbp1 (10) and the integral membrane protein Mps3 (4). Experimental data indicate the localization of the C-terminal domain of Mps2 in the perinuclear space (4, 10). Mps2 and Bbp1 each contain two predicted coiled-coil regions (CC), and based on the interaction of subfragments from both proteins, it was proposed that CC2 of Mps2 interacts with CC1 of Bbp1 (Ref. 11, supplemental Fig. S1A). The integral membrane protein Ndc1 contains six or seven transmembrane helices in the N-terminal half and has a large soluble C-terminal domain (12). Ndc1 is not only a SPB component but also associates with NPCs (13) where it forms together with the proteins Pom34 and Pom152 membrane rings that anchor the NPC to the pore membrane (14). Consistent with its localization, Ndc1 plays a role in NPC biogenesis (15–18).
The SPIN component Nbp1 is a monotopic inner nuclear membrane protein that contains an N-terminal amphipathic α-helix, which serves as an in-plane membrane anchor (19). Proteins containing amphipathic α-helices can either sense membrane curvature (20) or remodel membranes (21–23). Nbp1 interacts with both Ndc1 and the Mps2-Bbp1 complex (10, 24) and may be involved in fusion of nuclear membranes (19).
The central plaque of the SPB is formed by the interacting Spc42 and Spc29 proteins (25, 26). Spc42 forms a regular hexagonal lattice within the central plaque (27). Spc29, by interacting with Spc110, links the central plaque with the γ-tubulin complex of the inner plaque (26). The C terminus of Spc42 connects the central plaque of the SPB via the protein Cnm67 to the outer plaque (28).
How the huge core structure of the SPB is embedded in the nuclear envelope is poorly understood. However, Bbp1 (29, 30), which is present at the periphery of the central plaque, interacts with the nuclear envelope protein Mps2 and the central plaque protein Spc29 (10, 31), raising the possibility of its role as a linker between the SPB core and the nuclear envelope. Such a model predicts the existence of a stable ternary Mps2-Bbp1-Spc29 complex that may anchor additional SPIN components to the SPB.
Here, we address the important question of how a structure like the SPB is embedded in the nuclear envelope. We show purification and characterization of the Mps2-Bbp1 complex and the importance of coiled-coil interactions between Mps2 and Bbp1 for complex formation. Using purified Spc29 and the Mps2-Bbp1 complex, we reconstituted a high molecular weight ternary Mps2-Bbp1-Spc29 complex. Moreover, our experiments indicate that Bbp1 has the ability to oligomerize Spc29. The high molecular weight Mps2-Bbp1-Spc29 complex may resemble an early step in SPB duplication leading to the formation of the satellite, a SPB precursor. The Mps2-Bbp1 complex also interacts with the purified Mps3 and Ndc1 proteins, suggesting that Mps2-Bbp1 has a key role in recruiting SPIN proteins to the periphery of the central plaque. In summary, SPIN components show multiple biochemical interactions that are likely important for the insertion of the new SPB into the nuclear envelope.
Results
Mps2 and Bbp1 form a stable membrane-bound complex
Mps2 and Bbp1 are key mediators of the link between the nuclear envelope and the SPB because of their interactions with the integral membrane protein Mps3 and the central plaque component Spc29 (4, 31). To understand how Mps2 and Bbp1 fulfill their central role in membrane insertion of the SPB, we purified both proteins from Saccharomyces cerevisiae (32). The soluble His6-Mps2-(1–306) protein, comprising the cytoplasmic domain of Mps2, runs as a 304-kDa protein by gel filtration (supplemental Figs. S1, A and B). In contrast, Mps2-His10 that was purified in the presence of the detergent lauryl dimethylamine-N-oxide (LDAO) had an apparent molecular mass of ∼436 kDa (calculated molecular mass of the monomer, 46.8 kDa) (supplemental Fig. S1C). Next, we co-overexpressed MPS2-His10 together with BBP1 in S. cerevisiae. To correct for different expression rates of MPS2-His10 and BBP1 and to ensure that most of the purified Mps2-His10 protein was in complex with Bbp1, we transformed S. cerevisiae cells simultaneously with the plasmids p423-GAL1-BBP1, p424-GAL1-BBP1, p425-GAL1-BBP1, and p426-GAL1-MPS2-His10 and subsequently induced the GAL1 promoter by the addition of galactose (supplemental Fig. S2A). We purified the Mps2-His10-Bbp1 complex by Ni-NTA purification and gel filtration chromatography (Fig. 1, A–C). Mps2-His10 and Bbp1 co-eluted at 11.89–11.91 ml from the Superose 6 column in LDAO containing buffer (Fig. 1, A and E), whereas Mps2-His10 eluted at a larger volume (Fig. 1C; 13.39 ml). The elution volume of the Mps2-Bbp1 complex was unaffected when cells expressed an excess of MPS2 over BBP1 (supplemental Fig. S2A; GAL1-MPS2-His10 GAL1-BBP1 cells), suggesting a stable complex stoichiometry. Based on intensity measurements of the Coomassie Blue-stained bands, we estimated a molar ratio of 1.7 for Mps2 and Bbp1 in the complex (Fig. 1, A and B).
Figure 1.

Purification and reconstitution of the Mps2-Bbp1 membrane protein complex. A, purification of the Mps2-His10-Bbp1 protein complex from TKY384 yeast cells. Membrane proteins of galactose-induced TKY384 cells were solubilized with 1% LDAO, and Mps2-His10 was purified by IMAC and then analyzed by gel filtration on a Superose 6 column (upper panel) and subsequent SDS-PAGE and immunoblotting using anti-Mps2, anti-Bbp1, and anti-penta-His antibodies (lower panel). mAU, milliabsorbance units. B, the Mps2:Bbp1 ratio (as determined by quantifying the Coomassie Blue R250-stained gel shown in A with ImageJ; AU, absorbance units) was ∼1.7 and constant in the peak fractions of the gel filtration chromatography. C, comparison of Mps2 (blue line) and Mps2-Bbp1 (orange line) gel filtrations (upper panel). The peak fractions were analyzed by SDS-PAGE and Coomassie staining (lower panel). ΔMps2-His10 and ΔBbp1 are degradation products of Mps2-His10 and Bbp1, respectively. D, reconstitution of Mps2 and Mps2-Bbb1 complex into liposomes. Formation of proteoliposomes was analyzed by SDS-PAGE and subsequent Coomassie staining (upper panel) and immunoblotting (anti-Mps2/anti-Bbp1 antibodies; lower panel), respectively. Input, protein-lipid mixture diluted below critical micelle concentration (CMC) of the detergent and mixed with Nycodenz solution; Proteolip, floated proteoliposome fractions obtained after centrifugation. To visualize flotation of the formed proteoliposomes, the same volumes of inputs and floated proteoliposome fractions were analyzed. E, reconstitution of the Mps2-Bbp1 complex from purified Mps2-His10 and Bbp1-His10 proteins. Mps2-His10 and Bbp1-His10 were mixed in a molar ratio of ∼1:1, incubated for 15 min at room temperature, and then analyzed by gel filtration on a Superose 6 column (upper panel). In control experiments Mps2-His10, Bbp1-His10, and gel filtration-purified Mps2-His10-Bbp1 complex were treated in the same way. Proteins eluted from the Superose 6 column were analyzed by immunoblotting using an anti-Mps2 antibody (lower panel). The formed Mps2-His10-Bbp1-His10 complex eluted at ∼12.24 ml, and an additional “high molecular weight” Mps2-His10-Bbp1-His10 complex is marked with an asterisk. The reconstituted Mps2-His10-Bbp1-His10 complex could not be analyzed for the presence of Bbp1 because our Bbp1 antibody did not detect C-terminally tagged Bbp1. An anti-penta-His antibody did not allow discrimination of the similar-sized Mps2-His10 and Bbp1-His10 proteins.
Mass spectrometry of the Mps2-His10-Bbp1 complex showed that S232 of Mps2, which is located between the two coiled-coil domains, was phosphorylated (supplemental Fig. S2B; compare Ref. 33). However, phosphorylation of Mps2 and Bbp1 was not required for complex formation as the Mps2-Bbp1 complex also assembled in Escherichia coli (supplemental Fig. S2C).
The purified Mps2-His10-Bbp1 complex (and as a control, Mps2-His10) was reconstituted into liposomes by rapid dilution of the detergent (Fig. 1D). Mps2 and Bbp1 co-migrated with the fluorescently labeled liposomes in Nycodenz gradients. The formation of Mps2-Bbp1 proteoliposomes proved that Mps2-Bbp1 was indeed a stable membrane protein complex.
To test whether Mps2 and Bbp1 are capable of forming complexes without the help of additional factors, separately purified Bbp1-His10 (supplemental Fig. S2D) and Mps2-His10 proteins were used for complex reconstitution (Fig. 1E). Formation of the Mps2-His10-Bbp1-His10 complex was analyzed by gel filtration chromatography. Similar to the purified Mps2-His10-Bbp1 complex (Fig. 1E, bottom panel; 11.91 ml, corresponding to an apparent molecular mass of ∼1 MDa), in vitro assembled Mps2-His10-Bbp1-His10 complex was detected at an elution volume of 12.24 ml. In addition, a high molecular weight Mps2-His10-Bbp1-His10 complex (elution volume of 7.84 ml) eluted close to the void volume of the column (Fig. 1E, top, ∼7.5–7.7 ml). Thus, Bbp1 and Mps2 assemble in vitro into stable medium and high size molecular weight complexes.
Coiled-coil domain interactions are important for Mps2-Bbp1 complex stability
It has been shown that Bbp1 and Mps2 each contain two different coiled-coil domains: self-associating, homotypic ones for Bbp1-Bbp1 and Mps2-Mps2 homo-dimer formation and heterotypic coiled-coil interactions that allow assembly of Mps2-Bbp1 (Ref, 11; compare supplemental Fig. S1A). We asked if this hetero-association of coiled-coil domains is indeed the reason for Mps2-Bbp1 complex formation. The elution volume of Mps2 and Mps2-His10-Bbp1 in gel filtration experiments depended on the detergent type. If Fos-choline-12 (Fos12, n-dodecylphospho-choline) was used, the apparent molecular mass of Mps2-His10 was 215 kDa and not 419 kDa, as was the case in LDAO-containing buffer (Fig. 2A, top panel). This suggests dissociation of Mps2-Mps2 homo-assemblies by Fos12. In addition, although an Mps2-Bbp1 complex with an apparent molecular mass of more than 600 kDa was detectable in the presence of LDAO (Fig. 2B, bottom panel, M-B), this complex was absent when Fos12 was used (Fig. 2, A and B). The outcome of Fig. 2, A and B, led to the idea that Fos12 destabilizes certain types of coiled-coil interactions.
Figure 2.

The stability of the Mps2-Bbp1 complex is based on coiled-coil domain interactions. A, purification of the Mps2-Bbp1 complex failed using Fos12 as detergent. Mps2-His10 (upper panel) and Mps2-His10-Bbp1 (lower panel) were purified from galactose-induced yeast TKY376 and TKY377, cells respectively. The copy number of the BBP1 gene in TKY377 cells is lower than in TKY384 cells so that in this case not all purified Mps2 protein was in the Mps2-Bbp1 complex (compare Fig. 1 and supplemental Fig. S2A). Mps2 and Mps2-Bbp1 each were purified using the detergents Fos12 (left panel) and LDAO (right panel) and analyzed by gel filtration on a Superdex 200 column. Complexes are shown in a schematic way, because the exact number of monomers is not known. mAU, milliabsorbance units. B, immunoblot analysis of Mps2-Bbp1 purifications in the presence of Fos12 and LDAO, respectively. The fractions from the gel filtration experiments in A were analyzed by immunoblotting with anti-Mps2, anti-Bbp1, and anti-penta-His antibodies, respectively. The Mps2-Bbp1-complex (M-B) was only detectable when LDAO was used as detergent. The elution volume of Mps2 clearly depended on the used detergent und was significantly decreased in the Mps2-Bbp1 complex. C, the apparent molecular weight of the C-terminal Nbp1 domain was changed in the presence of Fos12 detergent. sfGFP-His6 (control protein), Nbp1-(174–319)-sfGFP-His6 (having a strong N-terminal coiled-coil (CC) domain from amino acids 174–219; 46.7 kDa), and Nbp1-(220–319)-sfGFP-His6 (41.2 kDa) were purified from E. coli, adjusted to 10 mm DTT, and incubated for 5 min at 4 °C with no detergent, 0.2% DDM, or 0.1% Fos12. Gel filtration on a Superdex 200 column using different buffer conditions (no detergent versus 0.2% DDM versus 0.1% Fos12 in 50 mm Tris/HCl, pH 8.0, 300 mm NaCl, 10% glycerol buffer) was followed by absorbance at 490 nm. The peak fractions (1–4) of the gel filtration runs were analyzed by SDS-PAGE to verify the correct sizes of monomeric proteins. D, the Mps2-Bbp1 complex purified using LDAO as detergent was disrupted by the addition of Fos12. Mps2-His10-Bbp1 was purified from galactose-induced yeast TKY377 cells using LDAO as detergent. Then, to one aliquot of the IMAC eluate, 1% Fos12 was added, and after 5 min of incubation at room temperature the reaction mixtures were analyzed by gel filtration on a Superose 6 column (with 0.1% LDAO in the running buffer). The eluted fractions were analyzed by immunoblotting with anti-Bbp1 antibodies.
To test this model, we purified the C-terminal soluble fragment of SPIN component Nbp1 Nbp1-(174–319) that homo-dimerizes via coiled-coil interactions (11). As a control, we used the Nbp1-(220–319) fragment lacking the coiled-coil region (Fig. 2C). In the absence of detergent, Nbp1-(220–319)-sfGFP-His6 eluted at an apparent molecular mass of ∼74 kDa, whereas the corresponding Nbp1 fragment with the coiled-coil region (Nbp1-(174–319)-sfGFP-His6) run at 188 kDa, confirming the homo-dimerization of Nbp1 by coiled-coil interactions. We next analyzed Nbp1 fragments in the presence of either n-dodecyl-β-d-maltoside (DDM) or Fos12. The apparent molecular mass of Nbp1-(174–319)-sfGFP-His6 changed from 257 kDa to 116 kDa when DDM was exchanged for Fos12, whereas sfGFP-His6 and Nbp1-(220–319)-sfGFP-His6 remained unaffected by this detergent change.
We concluded that Fos12 disrupts homotypic coiled-coil interactions of the Bbp1-Mps2 complex and, therefore, dissociates Bbp1-Bbp1 and Mps2-Mps2 binding. Purification of the Mps2-Bbp1 complex in the presence of LDAO with subsequent addition of Fos12 triggered partial disassembly of the Mps2-Bbp1 complex (Fig. 2D). It is, therefore, reasonable to assume that Fos12 also disrupts heterotypic coiled-coil interactions between Mps2 and Bbp1.
Reconstitution of the Mps3-Mps2-Bbp1 complex
The inner nuclear membrane protein Mps3 is a member of the SUN protein family (supplemental Fig. S3A; Ref. 4) and one of the components of the SPB half-bridge. Purified Mps3 from yeast was found in a complex with histone (34) and ribosomal proteins (supplemental Fig. S3B) and could be reconstituted into liposomes (supplemental Fig. S3C). We next confirmed Mps3-Mps2 complex formation by size-exclusion chromatography (Fig. 3, A–C). The same approach was used to reconstitute an Mps3-Mps2-Bbp1 complex (Fig. 3, D–G). Using DDM as detergent, Mps2-Bbp1 was eluted in two complexes from the Superose 6 column (Figs. 3, F and G, and supplemental Fig. S6; a “high molecular weight complex” eluting next to the void volume of the column and the “medium molecular weight” complex eluting as seen with LDAO at ∼12 ml). Adding Mps3 in molar excess to Mps2-Bbp1 led to a shift of the elution volume of both Mps2-Bbp1 complexes (Fig. 3, E and G). The formation of the Mps3-Mps2-Bbp1 complex became visible as an increase of the Mps2, Bbp1, and Mps3 blot signal intensities at an elution volume from ∼8 to 10 ml (Fig. 3, E and G). Thus, Mps3 forms a stable complex with Mps2-Bbp1.
Figure 3.

Interaction of the SUN-domain protein Mps3 with the Mps2-Bbp1 complex. A–C, reconstitution of a Mps3-Mps2 complex. A, Mps3-His10 and Mps2-His10 were separately purified by solubilization with DDM and subsequent IMAC. Mps3 (in molar excess) was mixed with Mps2 and analyzed by SDS-PAGE and Coomassie staining. B, gel filtration of Mps2-His10, Mps3-His10, and the Mps3-His10-Mps2-His10 mixture was performed on a Superose 6 column (red line, experimental data; blue line, calculated data if Mps2-His10 and Mps3-His10 do not interact). mAU, milliabsorbance units. Complex formation is visible as a shoulder in the chromatogram. C, proteins eluted from the Superose 6 column were analyzed by immunoblotting with anti-Mps2 antibodies. The formation of the Mps3-Mps2 complex was visible as an increase of the Mps2 blot signal intensity at an elution volume of ∼9 to 11 ml. D–G, reconstitution of the Mps3-Mps2-Bbp1 complex. Mps3-His10 and the Mps2-His10-Bbp1 complex were separately purified by solubilization with DDM and subsequent IMAC. Mps3 (in molar excess) was mixed with Mps2-Bbp1 (inputs were analyzed by SDS-PAGE and Coomassie staining: 1, Mps3-His10; 2, Mps3 + Mps2-His10-Bbp1) (D) and analyzed by gel filtration on a Superose 6 column (complex formation is visible as a shoulder in the chromatogram) (E). F, in control experiments the Single proteins Mps3-His10 and the Mps2-His10-Bbp1 complex were treated in the same way. Elution from the gel filtration column was followed by absorbance at 280 nm (F) and immunoblotting with anti-Bbp1, anti-Mps2 and anti-Mps3 antibodies, respectively (G).
Interaction of the integral membrane protein Ndc1 with the Mps2-Bbp1 complex
To understand the function of Ndc1 in anchoring SPBs and NPCs to the nuclear envelope in more detail, characterization of Ndc1 was of special importance. We solubilized Ndc1-His10 and Ndc1-sfGFP-His10 with detergent (Figs. 4A and supplemental Fig. S3D) and demonstrated that the purified proteins could be reconstituted into liposomes (Fig. 4B). We next focused on the interaction of Ndc1 with Bbp1. NDC1-His10 and untagged BBP1 were co-overexpressed in yeast (Fig. 4C). Surprisingly, Bbp1 co-eluted with Ndc1 from the Ni-NTA column, whereas in the absence of Ndc1 there was only an unspecific interaction of Bbp1 with the Ni-NTA beads (Fig. 4C, Bbp1 immunoblots). Interestingly, traces of Mps2 co-eluted with Ndc1 and Bbp1 (MPS2 was not overexpressed in this experiment), indicating the presence of Ndc1-Bbp1-Mps2 interactions. The interaction between Ndc1 and Bbp1 is probably weak as Bbp1 was copurified in the amounts substoichiometric to Ndc1. Our data support the idea that Ndc1, Mps2, and Bbp1 form a network of membrane-associated proteins that may be crucial for SPB membrane insertion.
Figure 4.

Interaction of the nuclear envelope protein Ndc1 with the Mps2-Bbp1 complex. A, purification of Ndc1-sfGFP and Ndc1-sfGFP-His10. Upper panel, after disruption of induced yeast TKY321 cells with glass beads and centrifugation at 12,000 × g, a green layer is visible on top of undisrupted cells. Lower panel, IMAC-purified Ndc1-His10 eluted at 9.84 ml from the Superdex 200 column (used detergent, Fos12; for more details see supplemental Fig. S3D), whereas IMAC-purified Ndc1-sfGFP-His10 eluted at 9.47 ml. These elution volumes correspond to apparent molecular masses of 357 kDa (Ndc1-His10) and 423 kDa (Ndc1-sfGFP-His10). mAU, milliabsorbance units. B, reconstitution of Ndc1-His10 and Ndc1-sfGFP-His10 into liposomes. IMAC-purified Ndc1-His10 and Ndc1-sfGFP-His10 proteins were reconstituted into liposomes by rapid dilution of the detergent Fos12. A, Ndc1-sfGFP-His10 with non-labeled lipid mix; Ndc1-sfGFP-His10 (B) and Ndc1-His10 (C) with lipid mix containing rhodamine-DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanol-amine-N-(lissamine rhodamine B sulfonyl). The floated proteoliposomes obtained after density gradient centrifugation (upper panel of the figure; see green floating Ndc1-sfGFP-His10 proteoliposomes in A) were then analyzed by SDS-PAGE and subsequent Coomassie staining (lower panel; 1, 20-μl input (before centrifugation); 2, 10-μl floated proteoliposome fraction). C, copurification of (Mps2)-Bbp1 with Ndc1. Membrane proteins solubilized with LDAO were purified from galactose induced TKY375 (GAL1-NDC1-His10/GAL1-BBP1) and TKY383 (GAL1-BBP1) cells by IMAC. Expression and elution from the Ni-NTA column was followed by SDS-PAGE (MB, Mps2-His10-Bbp1 protein as control protein; PS, 100-, 70-, and 55-kDa standard proteins; T, total proteins = crude cell extract; S, soluble proteins; M, membrane fraction; SM, membrane proteins solubilized with 1% LDAO) and immunoblotting using anti-Bbp1 antibodies (blue blots) and anti-Mps2 antibodies (gray blots), respectively.
Reconstitution of Spc29-Bbp1 and Mps2-Bbp1-Spc29 complexes
The central plaque protein Spc29 contains a coiled-coil domain (residues 7–45) that does not homo-associate in isolation. In addition, Spc29 carries a second, strongly self-associating coiled-coil region (residues 129–174) that likely forms homo-trimers (35). Previously we have shown that Bbp1 interacts with Spc29 in pull down experiments (31) and an interaction between both proteins has been implicated based on yeast two-hybrid data (10). To gain more insights into the interaction of Spc29 with Bbp1 and with the Mps2-Bbp1 membrane protein complex, we purified full-length His-sfGFP-Spc29 from yeast (Fig. 5, A and B) in the presence of RNase A to remove RNA, which otherwise co-purified with Spc29 (supplemental Fig. S4, A and B). The reason for this co-purification of Spc29 with RNA is presently unknown and requires further investigation.
Figure 5.
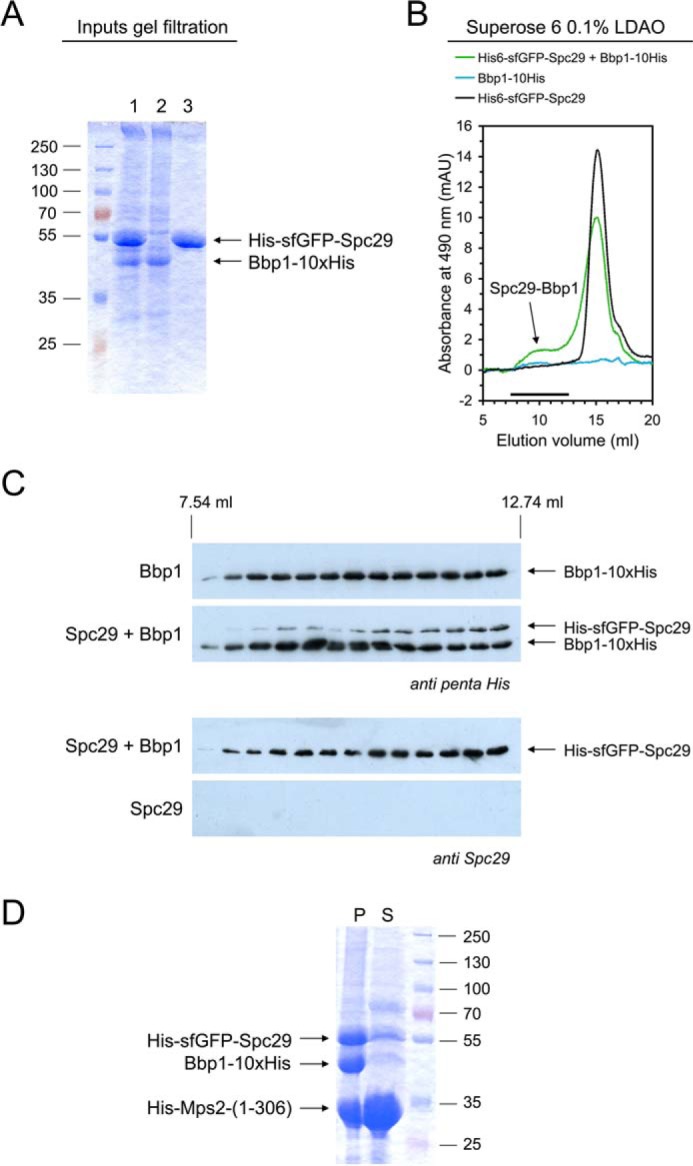
Reconstitution of Spc29-Bbp1 complexes. A and B, a molar excess of His-sfGFP-Spc29 was mixed with Bbp1-His10 in LDAO-containing buffer for 15 min at room temperature. The single Bbp1-His10 and His-sfGFP-Spc29 proteins were treated in the same way. Protein solutions were analyzed by SDS-PAGE and Coomassie staining (1, His-sfGFP-Spc29 + Bbp1-His10; 2, Bbp1-His10; 3, His-sfGFP-Spc29) (A) and by gel filtration on a Superose 6 column following the absorbance at 490 nm (absorbance maximum of GFP proteins) (B). mAU, milliabsorbance units. An arrow indicates the Spc29-Bbp1 complex. The shoulder in the Spc29 chromatogram in presence of Bbp1 indicates Spc29 oligomerization. C, proteins eluted from the Superose 6 column were subjected to immunoblotting using anti-penta-His and anti-Spc29 antibodies, respectively. D, coprecipitation of the soluble N-terminal domain of Mps2 together with Spc29 and Bbp1. His-Mps2-(1–306) (in molar excess), Bbp1-His, and His-sfGFP-Spc29 co-precipitated at a low LDAO concentration (∼0.03%). The assay was centrifuged for 5 min at 20,000 × g and separated into pellet (P) and supernatant (S) and analyzed by SDS-PAGE and Coomassie staining. The supernatant still contained soluble His-Mps2-(1–306) but only minor amounts of Spc29 and Bbp1.
Reconstitution of the Spc29-Bbp1 complex was achieved by incubating Bbp1–10His with a molar excess of His-sfGFP-Spc29. Formation of the complex was indicated by gel filtration (apparent molecular mass of ∼4 MDa; Fig. 5, B and C). We ensured in control experiments that the single proteins did not form higher homo-oligomers.
Our model suggests that interaction of Mps2-Bbp1 with Spc29 anchors the core structure of the SPB in the nuclear envelope. This predicts the existence of the ternary Mps2-Bbp1-Spc29 complex. In a first experiment we observed that at a low detergent concentration His-Mps2-(1–306) (a soluble protein lacking the transmembrane domain), which was purified from E. coli (supplemental Fig. S1B), Bbp1-His and His-sfGFP-Spc29 co-precipitated, indicating the formation of large oligomeric complexes (Fig. 5D). At 0.1% LDAO, soluble ternary Mps2-(1–306)-Bbp1-Spc29 complex assembled (supplemental Fig. S4, C–F). We next reconstituted the Mps2-Bbp1-Spc29 complex from purified Mps2-Bbp1 and Spc29 in DDM-containing buffer (Fig. 6, A–D). The medium molecular weight Mps2-Bbp1 complex (which is also in DDM-containing buffer the main species if the Mps2-Bbp1 complex concentration is low; Fig. 6, B and D) was incubated with a molar excess of Spc29. This promoted formation of a Mps2-Bbp1-Spc29 complex eluting at 7.5 ml within the void volume of the Superose 6 column (Fig. 6, B–D). Interestingly, incubation of Spc29 with Mps2-Bbp1 not only led to formation of the ternary Mps2-Bbp1-Spc29 complex but also to homo-oligomeric Spc29 complexes eluting between ∼9.0 to ∼14 ml (Fig. 6, B, middle chromatogram, and D), thus at a higher apparent molecular weight than only Spc29. In contrast, no Spc29 was eluted in the analyzed range between 7.14 and 12.74 ml in the absence of Mps2-Bbp1 (Fig. 6D, bottom panel). These data suggest that the Mps2-Bbp1 complex induces oligomerization of Spc29 without being part of the Spc29 oligomeres. Spc29 oligomerization was also observed in the presence of Bbp1 (Fig. 5, B and C) but not by Mps2 (Fig. 6B and supplemental Fig. S5), suggesting that the interaction with Bbp1 was responsible for the oligomerization of Spc29.
Figure 6.

Reconstitution of the Mps2-Bbp1-Spc29 membrane protein complex anchoring the spindle pole body in the nuclear envelope. A and B, molar excess of His-sfGFP-Spc29 was mixed either with purified Mps2-His10-Bbp1 complex or with purified Mps2 (control) in DDM containing buffer for 15 min at room temperature. The single proteins His-sfGFP-Spc29, Mps2-Bbp1-His10 complex, and Mps2-His10 were treated in the same way. All protein purifications were performed in the presence of RNase A. Protein solutions were analyzed by SDS-PAGE and Coomassie staining (Inputs gel filtration: 1, Spc29 + Mps2-Bbp1; 2, Spc29) (A) and by gel filtration on a Superose 6 column following the absorbance at 280 nm (left chromatogram) and 490 nm (middle chromatogram) (B). mAU, milliabsorbance units. The right chromatogram shows the control experiment with Mps2 (extended analysis in supplemental Fig. S5). C, the elution of Mps2-Bbp1-Spc29 and Spc29 from the Superose 6 column was compared with the elution of standard proteins. D, proteins eluted from the Superose 6 column were subjected to immunoblotting using anti-Spc29 antibodies and a mixture of anti-Bbp1 and anti-Mps2 antibodies, respectively. The assembled Mps2-Bbp1-Spc29 complex was eluted within the void volume of the Superose 6 column at 7.50 ml. Interaction with Mps2-Bbp1 also led to the formation of higher molecular weight Spc29 complexes by oligomerization (h.-o. Spc29/homo-oligomeric Spc29; in the absence of Mps2-Bbp1, no Spc29 was eluted from the Superose 6 column within the analyzed volume range as shown in the bottom blot). E, model for the in vitro formation of the Mps2-Bbp1-Spc29 complex. We assume that in presence of the membrane protein complex Mps2-Bbp1, the central plaque protein Spc29 is oligomerized and that in a second step a high molecular weight Mps2-Bbp1-Spc29 complex is formed that may resemble an early satellite structure or the central plaque of the SPB. For simplicity reasons, detergent micelles are not shown. F, different views on a simplified central plaque structure of the SPB (Spc42 and the other plaques of the SPB are not shown). The membrane protein complex Mps2-Bbp1 anchors the Spc29 layer of the SPB to the nuclear envelope at fusion sites of the INM and ONM. Due to additional interactions of Spc29 with Spc42 and Spc110, respectively, the entire SPB is stably inserted into the nuclear envelope.
Additional studies also showed that the high molecular weight Mps2-Bbp1 complex (eluting at 7.83 ml) formed a complex with Spc29 that eluted at 7.73 ml (within the void volume of the column; supplemental Fig. S6, A and B). In a mixture of Mps2-Bbp1 and Mps2, Spc29 specifically interacted with Mps2-Bbp1 and not with Mps2 (supplemental Fig. S6, C–E and the control experiment in Fig. 6B). Thus, Bbp1 in the Mps2-Bbp1 complex interacts with Spc29 to oligomerize Spc29 and to form an Mps2-Bbp1-Spc29 complex.
Discussion
SPBs and NPCs both are inserted into fusion sites of the outer and inner nuclear membranes (discussed and reviewed in Refs. 36–38). Genetic and initial biochemical studies indicate that complexes containing Mps2, Mps3, Nbp1, and Ndc1 could function as membrane anchor for the SPB (for review, see Ref. 7). However, in vitro studies of SPB membrane insertion and anchoring are hampered by the difficulty of purifying coiled-coil and integral membrane proteins. The coiled-coil domain is a common feature of SPB proteins that is used for the oligomerization of plaque proteins (35). It has been estimated that a diploid SPB contains ∼1000 copies each of Spc42 and Spc110 (30), indicating that there might be a similar number of Spc29 molecules in the central plaque.
Here, we focused on the biochemical characterization of membrane anchor proteins of the SPB and of the central plaque protein Spc29 with a focus on the role of the Mps2-Bbp1 complex in membrane tethering of the SPB. We were able to purify a stable Mps2-Bbp1 complex and incorporated this complex into liposomes resembling the lipid composition of yeast endoplasmic reticulum. This proves the stability of the Mps2-Bbp1 complex in a native lipid environment. Based on binding of isolated coiled-coil regions, it was suggested that the Mps2-Bbp1 complex contains both subunits in a 4:1 ratio (11). However, our measurements indicate that the Mps2:Bbp1 ratio is closer to ∼1.7. At present, the total number of Mps2 and Bbp1 molecules in the complex is unknown. Because of very similar molecular masses of Mps2-His10 (46.8 kDa) and Bbp1 (45.4 kDa), light scattering (error in molecular mass determination of membrane protein complexes is ∼5%) cannot be used to determine unambiguously the stoichiometry of the Mps2-His10-Bbp1 complex (39).
Interestingly in the presence of the detergent Fos12, the apparent molecular weight of purified Mps2 was only half that with LDAO, although Fos12 and LDAO form similar sized micelles of ∼20 kDa (see Anatrace product information). LDAO and Fos12 both are zwitterionic detergents with a dodecyl chain; however, in Fos12 this dodecyl chain is linked to a negatively charged phosphate group, whereas in LDAO the dodecyl chain is linked to a positively charged nitrogen atom. Our analysis suggests that Fos12 can disrupt coiled-coil interactions. The underlying mechanism is not known but likely Fos12 destabilizes hydrophobic interactions within coiled coils. We propose that coiled-coil interactions are also essential for Mps2-Bbp1 complex formation in vivo and thus in membrane insertion and anchoring of the SPB.
Ndc1 is a shared component of NPCs and SPBs (13) and from biochemical and genetic studies it was already known that Ndc1 plays a role in NPC (14) and SPB (12) membrane insertion and anchoring. A recent structured illumination microscopy (SIM) study showed that a ring of Bbp1-mTourqoise2 co-localizes with Ndc1-YFP at the SPB (40). Our co-purification experiments of Ndc1 and Bbp1 (Fig. 4C) support an interaction between Ndc1 and Bbp1-Mps2. Whether this interaction is direct has to be addressed in further studies. We also showed that Mps3 not only interacts with Mps2 (4) but also with the Mps2-Bbp1 complex. The Mps3-Mps2-Bbp1 complex may play a role early in SPB-duplication, as the inability of mps2–381 to form a bridge or satellite at the restrictive temperature is related to decreased binding affinity of the Mps2 C terminus to the SUN-domain of Mps3 (4). In addition, the SUN protein Mps3 interacts with Ndc1, and it has been proposed that this interaction controls the distribution of Ndc1 between the SPB and the NPCs (41). Taken these data together, it is reasonable to assume the existence of a quaternary Mps3-Mps2-Bbp1-Ndc1 complex. Biochemical data suggest that in this complex Ndc1 and Mps3 interact independently with Mps2-Bbp1. It is, however, possible that Mps3 assists the binding of Ndc1 to the Mps2-Bbp1 complex.
We were not able to purify a stable Mps2-Bbp1-Spc29 complex from yeast cells overexpressing the three genes, probably because the interaction between Spc29 and Bbp1 is not as strong as the interaction between Mps2 and Bbp1. However, reconstitution of the ternary Mps2-Bbp1-Spc29 complex from purified Mps2-Bbp1 and Spc29 proteins was successful. The Mps2-Bbp1-Spc29 complex eluted within the void volume of the Superose 6 gel filtration column. That the exclusion limit is 40 MDa indicates the formation of large Mps2-Bbp1-Spc29 oligomers. This oligomerization reaction may be of importance for the assembly of the SPB satellite in early G1 phase of the cell cycle.
In the presence of Mps2-Bbp1 (but not Mps2; Fig. 6, B–D, and supplemental Fig. S5) a portion of Spc29 eluted at a higher molecular weight than the single protein Spc29 without obvious complex formation with Mps2-Bbp1 (Fig. 6D). We propose that Mps2-Bbp1 can change the structure of Spc29 leading to the self-oligomerization of Spc29. This property raises the possibility that formation of the high molecular weight Mps2-Bbp1-Spc29 complex occurs in vitro in two steps. First, the Mps2-Bbp1 complex oligomerizes the central plaque protein Spc29. Second, the high molecular weight Mps2-Bbp1-Spc29 complex forms by the binding of Mps2-Bbp1 (and low molecular weight Mps2-Bbp1-Spc29) to the Spc29 platform (Fig. 6E).
The known localization of Mps2-Bbp1 at the periphery of the central plaque together with its binding to Mps3, Ndc1, and Spc29 supports the model that in vivo the Mps2-Bbp1 complex anchors the SPB to fusion sites of the INM and ONM (Fig. 6F). Spc29 has a central role in this process: first, Spc29 together with Spc42 forms the central plaque. Second, at the central plaque periphery Spc29 gains the ability to cross-link the central Spc42-Spc29 layer with the integral membrane proteins Ndc1 and Mps3 via the interaction with the Mps2-Bbp1 complex. Third, Spc29 together with Spc42 anchors the γ-TuSC receptor Spc110 to the inner side of the SPB (26). Finally, as mentioned above, the direct interaction of Spc29 with the membrane-bound Mps2-Bbp1 complex anchors the central plaque into the nuclear envelope.
Experimental procedures
Yeast strains, growth conditions, and preparation of popcorn cells
Plasmids and yeast strains are listed in supplemental Tables S1 and S2, respectively. Yeast strain ESM356–1 was a derivative of S288C. Synthetic complete (SC) SC-Ura or SC-Ura-Leu or SC-Ura-Leu-His-Trp media were used for growth of the yeast cells for protein purifications. Glucose was added in precultures to repress expression under the control of the GAL promoter. Main cultures used for protein purifications contained 3% raffinose, and gene expression was then induced at an A600 of 1–2 by adding 2% galactose. After induction with galactose, yeast cells were further incubated for 6–8 h at 30 °C and 200 rpm. Cells were harvested by centrifugation and washed with TN buffer (50 mm Tris/HCl, pH 7.4, at room temperature, 150 mm NaCl). The washed cell pellets were then resuspended in TN buffer containing 2 mm PMSF (1 ml of buffer/1 g of cell pellet (wet weight)) and dripped into liquid nitrogen. Frozen popcorn cells were stored at −80 °C.
Plasmid construction
Cloning of the SPB genes was performed using standard techniques. The DNA sequence of all cloned genes was confirmed.
Purification of HisTag proteins from E. coli
For purification of HisTag proteins, 2 liters of E. coli BL21-CodonPlus-(DE3)-RIL cells transformed with the appropriate plasmid were grown at 30 °C, induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of 0.4 to 0.5, and shaken for an additional 1.5–2 h at 30 °C. Cells were then harvested and disrupted by sonication in 40 ml of 50 mm Tris-HCl (either pH 7.4 or pH 8.0 at room temperature), 300 mm NaCl buffer supplemented with protease inhibitors (and in some experiments with RNase A). A cleared lysate obtained by centrifugation was adjusted to 20 mm imidazole and then applied to immobilized metal ion affinity chromatography (IMAC). After washing the Ni-NTA-column with TN buffer supplemented with 20 mm imidazole (reducing the NaCl concentration from 300 to 150 mm NaCl), HisTag proteins were eluted with TN buffer containing 500 mm imidazole and 10% glycerol. If the protein was not used for interaction studies, washing and eluting steps were performed with 300 mm NaCl. Ni-NTA eluates were adjusted to 10 mm DTT and concentrated with Vivaspin 500 ultrafiltration spin columns (Sartorius) if necessary, snap-frozen in liquid nitrogen, and stored at −80 °C.
Purification of integral membrane proteins from yeast cells
Expression and purification of yeast integral membrane proteins was carried out according to Hays et al. (32) yielding milligram quantities of protein (as estimated from Coomassie-stained SDS gels) if the best suited detergent was used. Yield was reduced for the Mps2-Bbp1 complex in cases DDM (for reconstitution experiments) was used instead of LDAO.
All purification steps were performed at 4 °C using precooled buffers. 50 g of galactose-induced yeast popcorn cells were disrupted using glass beads (BioSpec, 0.5 mm diameter) and a French Pulverisette. Glass beads were then separated using 50-ml syringes, and the crude cell extracts were centrifuged for 30 min at 12,000 × g at 4 °C. The membrane, cell debris, and organelle-containing layer on top of the pellet of unlysed cells was resuspended and washed with TN buffer containing 1 mm PMSF. After centrifugation (30 min at 12,000 × g at 4 °C) the pellets were resuspended in a total volume of 20 ml of TN buffer containing 1 mm PMSF and 1% detergent (Fos12 or DDM (both obtained from Anatrace) or LDAO (obtained from Sigma) and transferred to a 50-ml Falcon tube. Membrane proteins were solubilized for 1–2 h using a tube roller mixer. To the supernatant obtained after centrifugation (30 min at 40,000 × g) 2 ml of Ni-NTA-agarose (Qiagen), 20 mm imidazole (for His6Tag) or 40 mm imidazole (for His10Tag) and TN buffer up to a total volume of 50 ml were added (decreasing the detergent concentration from 1.0% to 0.4%). Binding of HisTag proteins to the Ni-NTA beads was achieved by mixing the suspension for 30 min on a roller mixer. IMAC was then performed as gravity flow chromatography. The Ni-NTA beads were washed with 6–8 ml of TN buffer containing 0.1% of the used detergent (0.2% for DDM) and either 20 or 40 mm imidazole; HisTag proteins were subsequently eluted with 500 mm imidazole in TN buffer containing 0.1% detergent (0.2% for DDM) and 10% glycerol and then adjusted to 10 mm DTT. Proteins were concentrated using Vivaspin 500 ultrafiltration spin columns, snap-frozen in liquid nitrogen, and then stored at −80 °C.
Purification of Bbp1 and Spc29 from yeast cells
Bbp1-His10 was purified as described for the integral membrane proteins but using 2.5% LDAO for solubilization of the protein. His-sfGFP-Spc29 was solubilized using 1% Fos12. Washing and eluting the Ni-NTA beads with buffers containing no detergent (alternatively IMAC was also used to exchange the detergent Fos12 to LDAO) removed the detergent. Purification of His-sfGFP-Spc29 from yeast was also possible using LDAO as detergent, but protein yields were lower in this case. RNase A was added during cell disruption and solubilization.
Gel filtration and determination of apparent molecular weights
Gel filtration of single proteins and protein complexes, respectively, was carried out at 4 °C using an ÄKTA Purifier System from GE Healthcare and prepacked columns for high resolution size exclusion chromatography (either a Superdex 200 10/300 GL or a Superose 6 10/300 GL column). Protein samples (purified by IMAC and concentrated with Vivaspin 500 ultrafiltration spin columns) were slowly thawed on ice and centrifuged for 5 min at 20,000 × g at 4 °C (to remove precipitated protein), and then 100 μl of protein aliquots of the supernatant were subjected to the used gel filtration column equilibrated in TN buffer containing 10% glycerol, 2 mm DTT, and 0.1% of the appropriate detergent (in case of DDM 0.2%) at a flow rate of 0.4 ml/min. The elution was followed by absorbance at 280 nm (and additional wavelengths indicated in the figures), and 0.4-ml fractions were collected. SDS-PAGE analysis with subsequent Coomassie staining of the gel filtration runs was carried out with 11.25–15-μl aliquots of each of the analyzed fractions. Apparent molecular weights were obtained using globular standard proteins ovalbumin (44 kDa), albumin (75 kDa), aldolase (158 kDA), ferritin (440 kDa), and thyroglobulin (669 kDa) for calibration of the gel filtration columns. Because the proteins analyzed in this study were not globular but coiled-coil domain proteins and because membrane proteins bind detergent, the calculated apparent molecular weights do not reflect the exact molecular weight/size of the analyzed proteins.
Reconstitution of protein complexes
The various proteins purified by IMAC (salt concentration 150 mm NaCl, pH 7.4) and concentrated with Vivaspin 500 ultrafiltration spin columns were mixed (normally using one of the interacting proteins in molar excess; total volume of the interaction assays was ∼130 to 200 μl), incubated for 15 min at room temperature, snap-frozen in liquid nitrogen, and stored at −80 °C. For gel filtration analysis 100-μl aliquots of interaction assays were subjected to a Superose 6 10/300 GL column (as described above); 3.75–15-μl aliquots of the samples were analyzed by SDS-PAGE and subsequent Coomassie staining (input control, visualizing the used ratio of the interacting proteins). In control experiments, single proteins (in some cases mixed with the used buffer) were treated in the same way. Interaction was followed by absorbance and immunoblot analysis of the eluted proteins. Complex formation can be seen by comparing the gel filtration chromatogram of a protein complex with the theoretical chromatogram obtained by adding the chromatograms of the single proteins (=chromatogram for non- interacting proteins in a mixture). Reconstitution of the complexes was tried out with the detergents LDAO and DDM; the successful detergent is mentioned in the respective figure.
Reconstitution of integral membrane proteins into liposomes
All lipids were purchased from Avanti Polar Lipids; sodium cholate was from Sigma. For the reconstitution experiments membrane proteins and lipids were used in a molar ratio of ∼1:1000. The lipid mixture used to reconstitute Ndc1, Mps2, Mps2-Bbp1, and Mps3, respectively, resembles the lipid composition of the yeast endoplasmic reticulum (but using cholesterol instead of ergosterol; Ref. 42) and was composed of 42 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 6 mol % 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), 20 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phos-phoethanolamine (POPE), 22 mol % l-α-phosphatidylinositol (liver and bovine), 10 mol % cholesterol (and optional 2 mol % of 1,2-dipalmitoyl-sn-glycero-3-phosphoethanol-amine-N-(lissamine rhodamine B sulfonyl) (rhodamine-DPPE)). 2.5 μmol of the dried lipid mixture was solved in 1 ml of protein solution (250 μl of Ni-NTA-purified protein in detergent, 500 μl of TN buffer, and 250 μl of 10% sodium cholate in TN buffer). Liposomes were formed by diluting the used detergent below the critical micelle concentration (with TN buffer containing 1 mm DTT) followed by desalting using PD10 spin columns (GE Healthcare) and flotation of the reconstituted liposomes in a Nycodenz gradient as described previously (43).
Antibodies
Anti-penta-His (Qiagen), affinity-purified anti-Bbp1 antibodies (10), affinity-purified anti-Mps2 antibodies (Schiebel laboratory), affinity-purified anti-Spc29 antibodies (Schiebel laboratory), HRP-conjugated goat anti-mouse (Jackson), and HRP-conjugated goat anti-rabbit (Jackson ImmunoResearch Laboratories) antibodies were used for protein detection by immunoblotting.
Author contributions
T. K. designed, performed, and evaluated all the experiments described in Figs. 1–6 and in the supplemental figures and prepared all the figures. Reconstitution of proteoliposomes was designed and performed by J. M. and T. K. T. K. and E. S. wrote the paper. All authors discussed the results and commented on the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Ruppert and his group for mass spectrometry performed at the ZMBH Core facility for mass spectrometry and proteomics and Diana Rüthnick for providing plasmids pDR009 and pDR012 as well as Wanlu Zhang for providing plasmid pZW003. Diana Rüthnick and Wanlu Zhang performed the first experiments to purify Mps2-Bbp1 from E. coli. We thank Thomas Söllner for helpful discussions and Ursula Jäckle and Stefanie Heinze for excellent technical assistance.
This was supported by a grant from the Deutsche Forschungsgemeinschaft (Collaborative Research Centre SFB638; to E. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1 and S2 and Figs. S1–S6.
- SPB
- spindle pole body
- DDM
- n-dodecyl-β-d-maltoside
- Fos12
- n-dodecylphosphocholine
- IMAC
- immobilized metal ion affinity chromatography
- INM
- inner nuclear membrane
- LDAO
- lauryl dimethylamine-N-oxide
- NPC
- nuclear pore complex
- ONM
- outer nuclear membrane
- SPIN
- SPB insertion network
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Jaspersen S. L., and Winey M. (2004) The budding yeast spindle pole body: structure, duplication, and function. Annu. Rev. Cell Dev. Biol. 20, 1–28 [DOI] [PubMed] [Google Scholar]
- 2. Byers B., and Goetsch L. (1975) Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. J. Bacteriol. 124, 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Toole E. T., Winey M., and McIntosh J. R. (1999) High-voltage electron tomography of spindle pole bodies and early mitotic spindles in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 10, 2017–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaspersen S. L., Martin A. E., Glazko G., Giddings T. H. Jr, Morgan G., Mushegian A., and Winey M. (2006) The Sad1-UNC-84 homology domain in Mps3 interacts with Mps2 to connect the spindle pole body with the nuclear envelope. J. Cell Biol. 174, 665–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rüthnick D., and Schiebel E. (2016) Duplication of the yeast spindle pole body once per cell cycle. Mol. Cell. Biol. 36, 1324–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seybold C., and Schiebel E. (2013) Spindle pole bodies. Curr. Biol. 23, R858–R860 [DOI] [PubMed] [Google Scholar]
- 7. Jaspersen S. L., and Ghosh S. (2012) Nuclear envelope insertion of spindle pole bodies and nuclear pore complexes. Nucleus 3, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muñoz-Centeno M. C., McBratney S., Monterrosa A., Byers B., Mann C., and Winey M. (1999) Saccharomyces cerevisiae MPS2 encodes a membrane protein localized at the spindle pole body and the nuclear envelope. Mol. Biol. Cell 10, 2393–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winey M., Goetsch L., Baum P., and Byers B. (1991) MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J. Cell Biol. 114, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schramm C., Elliott S., Shevchenko A., and Schiebel E. (2000) The Bbp1p-Mps2p complex connects the SPB to the nuclear envelope and is essential for SPB duplication. EMBO J. 19, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zizlsperger N., and Keating A. E. (2010) Specific coiled-coil interactions contribute to a global model of the structure of the spindle pole body. J. Struct. Biol. 170, 246–256 [DOI] [PubMed] [Google Scholar]
- 12. Winey M., Hoyt M. A., Chan C., Goetsch L., Botstein D., and Byers B. (1993) NDC1: a nuclear periphery component required for yeast spindle pole body duplication. J. Cell Biol. 122, 743–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chial H. J., Rout M. P., Giddings T. H., and Winey M. (1998) Saccharomyces cerevisiae Ndc1p is a shared component of nuclear pore complexes and spindle pole bodies. J. Cell Biol. 143, 1789–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alber F., Dokudovskaya S., Veenhoff L. M., Zhang W., Kipper J., Devos D., Suprapto A., Karni-Schmidt O., Williams R., Chait B. T., Sali A., and Rout M. P. (2007) The molecular architecture of the nuclear pore complex. Nature 450, 695–701 [DOI] [PubMed] [Google Scholar]
- 15. Lau C. K., Giddings T. H. Jr, and Winey M. (2004) A novel allele of Saccharomyces cerevisiae NDC1 reveals a potential role for the spindle pole body component Ndc1p in nuclear pore assembly. Eukaryot. Cell 3, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Madrid A. S., Mancuso J., Cande W. Z., and Weis K. (2006) The role of the integral membrane nucleoporins Ndc1p and Pom152p in nuclear pore complex assembly and function. J. Cell Biol. 173, 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mansfeld J., Güttinger S., Hawryluk-Gara L. A., Panté N., Mall M., Galy V., Haselmann U., Mühlhäusser P., Wozniak R. W., Mattaj I. W., Kutay U., and Antonin W. (2006) The conserved transmembrane nucleoporin NDC1 is required for nuclear pore complex assembly in vertebrate cells. Mol. Cell 22, 93–103 [DOI] [PubMed] [Google Scholar]
- 18. Onischenko E., Stanton L. H., Madrid A. S., Kieselbach T., and Weis K. (2009) Role of the Ndc1 interaction network in yeast nuclear pore complex assembly and maintenance. J. Cell Biol. 185, 475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kupke T., Di Cecco L., Müller H. M., Neuner A., Adolf F., Wieland F., Nickel W., and Schiebel E. (2011) Targeting of Nbp1 to the inner nuclear membrane is essential for spindle pole body duplication. EMBO J. 30, 3337–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drin G., and Antonny B. (2010) Amphipathic helices and membrane curvature. FEBS Lett. 584, 1840–1847 [DOI] [PubMed] [Google Scholar]
- 21. Beck R., Sun Z., Adolf F., Rutz C., Bassler J., Wild K., Sinning I., Hurt E., Brügger B., Béthune J., and Wieland F. (2008) Membrane curvature induced by Arf1-GTP is essential for vesicle formation. Proc. Natl. Acad. Sci. U.S.A. 105, 11731–11736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ford M. G., Mills I. G., Peter B. J., Vallis Y., Praefcke G. J., Evans P. R., and McMahon H. T. (2002) Curvature of clathrin-coated pits driven by epsin. Nature 419, 361–366 [DOI] [PubMed] [Google Scholar]
- 23. Lee M. C., Orci L., Hamamoto S., Futai E., Ravazzola M., and Schekman R. (2005) Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122, 605–617 [DOI] [PubMed] [Google Scholar]
- 24. Araki Y., Lau C. K., Maekawa H., Jaspersen S. L., Giddings T. H. Jr, Schiebel E., and Winey M. (2006) The Saccharomyces cerevisiae spindle pole body (SPB) component Nbp1p is required for SPB membrane insertion and interacts with the integral membrane proteins Ndc1p and Mps2p. Mol. Biol. Cell 17, 1959–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elliott S., Knop M., Schlenstedt G., and Schiebel E. (1999) Spc29p is a component of the Spc110p-subcomplex and is essential for spindle pole body duplication. Proc. Natl. Acad. Sci. U.S.A. 96, 6205–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Knop M., and Schiebel E. (1997) Spc98p and Spc97p of the yeast γ-tubulin complex mediate binding to the spindle pole body via their interaction with Spc110p. EMBO J. 16, 6985–6995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bullitt E., Rout M. P., Kilmartin J. V., and Akey C. W. (1997) The yeast spindle pole body is assembled around a central crystal of Spc42p. Cell 89, 1077–1086 [DOI] [PubMed] [Google Scholar]
- 28. Schaerer F., Morgan G., Winey M., and Philippsen P. (2001) Cnm67p is a spacer protein of the Saccharomyces cerevisiae spindle pole body outer plaque. Mol. Biol. Cell 12, 2519–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park C. J., Song S., Giddings T. H. Jr, Ro H. S., Sakchaisri K., Park J. E., Seong Y. S., Winey M., and Lee K. S. (2004) Requirement for Bbp1p in the proper mitotic functions of Cdc5p in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 1711–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wigge P. A., Jensen O. N., Holmes S., Souès S., Mann M., and Kilmartin J. V. (1998) Analysis of the Saccharomyces spindle pole by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry. J. Cell Biol. 141, 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Araki Y., Gombos L., Migueleti S. P., Sivashanmugam L., Antony C., and Schiebel E. (2010) N-terminal regions of Mps1 kinase determine functional bifurcation. J. Cell Biol. 189, 41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hays F. A., Roe-Zurz Z., and Stroud R. M. (2010) Overexpression and purification of integral membrane proteins in yeast. Methods Enzymol. 470, 695–707 [DOI] [PubMed] [Google Scholar]
- 33. Keck J. M., Jones M. H., Wong C. C., Binkley J., Chen D., Jaspersen S. L., Holinger E. P., Xu T., Niepel M., Rout M. P., Vogel J., Sidow A., Yates J. R. 3rd, Winey M. (2011) A cell cycle phosphoproteome of the yeast centrosome. Science 332, 1557–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gardner J. M., Smoyer C. J., Stensrud E. S., Alexander R., Gogol M., Wiegraebe W., and Jaspersen S. L. (2011) Targeting of the SUN protein Mps3 to the inner nuclear membrane by the histone variant H2A.Z. J. Cell Biol. 193, 489–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zizlsperger N., Malashkevich V. N., Pillay S., and Keating A. E. (2008) Analysis of coiled-coil interactions between core proteins of the spindle pole body. Biochemistry 47, 11858–11868 [DOI] [PubMed] [Google Scholar]
- 36. Antonin W. (2009) Nuclear envelope: membrane bending for pore formation? Curr. Biol. 19, R410–R412 [DOI] [PubMed] [Google Scholar]
- 37. Antonin W., and Mattaj I. W. (2005) Nuclear pore complexes: round the bend? Nat. Cell Biol. 7, 10–12 [DOI] [PubMed] [Google Scholar]
- 38. Meseroll R. A., and Cohen-Fix O. (2016) The malleable nature of the budding yeast nuclear envelope: flares, fusion, and fenestrations. J. Cell. Physiol. 231, 2353–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Slotboom D. J., Duurkens R. H., Olieman K., and Erkens G. B. (2008) Static light scattering to characterize membrane proteins in detergent solution. Methods 46, 73–82 [DOI] [PubMed] [Google Scholar]
- 40. Burns S., Avena J. S., Unruh J. R., Yu Z., Smith S. E., Slaughter B. D., Winey M., and Jaspersen S. L. (2015) Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife, 0.7554/eLife.08586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen J., Smoyer C. J., Slaughter B. D., Unruh J. R., and Jaspersen S. L. (2014) The SUN protein Mps3 controls Ndc1 distribution and function on the nuclear membrane. J. Cell Biol. 204, 523–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weber T., Zemelman B. V., McNew J. A., Westermann B., Gmachl M., Parlati F., Söllner T. H., and Rothman J. E. (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.