

Abstract
The efficiency and type of pathway chosen to repair DNA double-strand breaks (DSBs) are critically influenced by the nucleosome packaging and the chromatin architecture surrounding the DSBs. The Swi/Snf (PBAF and BAF) chromatin-remodeling complexes contribute to DNA damage-induced nucleosome remodeling, but the mechanism by which it contributes to this function is poorly understood. Herein, we report how the Baf200 (Arid2) PBAF-defining subunit regulates DSB repair. We used cytological and biochemical approaches to show that Baf200 plays an important function by facilitating homologous recombination-dependent processes, such as recruitment of Rad51 (a key component of homologous recombination) to DSBs, homology-directed repair, and cell survival after DNA damage. Furthermore, we observed that Baf200 and Rad51 are present in the same complex and that this interaction is mediated by C-terminal sequences in both proteins. It has been recognized previously that the interplay between distinct forms of Swi/Snf has profound functional consequences, but we understand little about the composition of complexes formed by PBAF protein subunits. Our biochemical analyses reveal that Baf200 forms at least two distinct complexes. One is a canonical form of PBAF including the Swi/Snf-associated Brg1 catalytic subunit, and the other contains Baf180 but not Brg1. This distinction of PBAF complexes based on their unique composition provides the foundation for future studies on the specific contributions of the PBAF forms to the regulation of DNA repair.
Keywords: chromatin remodeling, DNA damage, DNA recombination, DNA repair, homologous recombination, Baf200, PBAF, Rad51
Introduction
The genome in eukaryotic cells is assembled into chromatin, a complex structure that influences fundamental processes of DNA metabolism, including replication, gene expression, and repair. In the process of repairing DNA, damage in the form of double-strand breaks (DSBs)2, nucleosome packaging, and the chromatin architecture surrounding the DSB can present an important problem for DNA damage signaling, as well as the efficiency and choice of the pathway utilized for repair of the damaged DNA (1). To counteract nucleosomes impeding the proper DNA damage response, the cell has evolved a number of systems involving chromatin-remodeling complexes (2–8) that alter the nature of the interaction between DNA and histones by inducing nucleosome sliding and disruption, and histone eviction or exchange, thus facilitating the repair of DSBs.
In mammals, Swi/Snf chromatin remodelers have been implicated in transcriptional regulation, development, differentiation, and failures in the proper function of these enzymes contribute to tumorigenesis (reviewed in Refs. 9–16). Swi/Snf complexes can be grouped into two subfamilies, Brg1/Brahma-associated factors (BAF) and PBAF (polybromo-associated BAF), with distinct subunits and functions. PBAF and BAF have considerable overlap in subunit composition, including the Brg1 ATPase; however, PBAF is defined by specific subunits such as Brd7, Baf180, and Baf200. Swi/Snf has been proposed to play a major role in preventing cancer via transcription-independent tumor suppressor activities (9, 17). Notably, genes encoding Swi/Snf components are among the most commonly mutated genes in human tumors, with an estimated 20% of all cancers bearing Swi/Snf mutations (17–19). Uncovering the functional interactions of Swi/Snf with DNA damage signaling and repair pathways is an important challenge in the field, and will provide a necessary foundation to understand its role in cancer.
Several lines of evidence suggest that PBAF components play critical roles in DNA damage repair. First, mediated by Brg1 interaction with acetylated H3 through its bromodomain, Brg1 and γH2AX cooperatively act in a feedback activation loop to facilitate DNA damage signaling at initial stages of the DNA damage response (20). Second, components of the PBAF complex, but not BAF, participate in repressing RNA PolII-mediated transcription at a start site located in cis to DNA DSBs (21). Third, Brg1 knockdown was found to result in defective Rad51 filament assembly with increased replication protein A (RPA) retention, suggesting that PBAF and/or BAF may act at specific steps of the homologous recombination pathway (22). The ability of Brg1 to promote Rad51 foci was not dependent on the ATPase activity of Brg1, but instead was modulated by an interaction between Brg1 and Rad52. In agreement with a role of PBAF in DNA damage signaling and/or in the DNA repair machinery function, germ cell-specific ablation of Brg1 resulted in impaired recombination during meiosis (23, 24), and Brd7−/− mice show impaired spermatogenesis because of apparent meiotic DNA repair defects (25). Although these results suggest that PBAF components are critical to the DNA repair process, they also leave several questions regarding the mechanisms that underlie PBAF function. For example, are there protein subunits of PBAF that participate in different steps of the DNA damage response? Are there biochemically/structurally distinct PBAF subcomplexes? What is the composition of these complexes? To answer these questions and elucidate PBAF mechanisms of action in DSB repair, we employed biochemical, cellular, and genetic approaches and focused on Baf200, an essential yet uncharacterized PBAF regulatory subunit.
Baf200 was first identified in biochemical experiments designed to purify Swi/Snf complexes. This work also demonstrated that Baf200, but not Baf180, is required for PBAF to mediate expression of an interferon-responsive gene, suggesting that Baf200 is an important targeting subunit of PBAF (26). Subsequent work showed a role for Baf200 in gene regulation: 1) Baf200 promotes osteoblast differentiation, which supports the idea that it is an important factor in preserving cellular identity and tissue-specific gene expression (27); 2) depletion of Baf200 in human lymphoid CEM cells impairs Tat-activated transcription of the HIV LTR (28); 3) Baf200 interacts with the serum response factor and activates the promoter of cardiac genes, suggesting that Baf200 may serve as a transcription co-activator (29). The latter is possibly the cause of the requirement of Baf200 for normal heart morphogenesis and coronary artery development (30). Evidence for distinct Swi/Snf complexes and their relevance in transcription has been recently obtained from genome-wide chromatin association studies on PBAF/BAF subunits including Baf200 (31). This observation prompted us to investigate the composition of complexes formed by PBAF subunit components, as physical evidence for structurally distinct forms of PBAF has not yet been obtained. In this work, we show that Baf200 and Baf180 can be found in biochemically/structurally different PBAF complexes.
Although it is abundantly clear that Baf200 is involved in regulation of gene expression, its roles in other important cellular processes, such as DNA repair, are poorly understood. Indeed, Baf200 action in facilitating DNA DSB repair may directly impact maintenance of genome stability and help explain the increasing number of recurrent inactivating mutations in Baf200 found in different types of cancer (Refs. 17, 32, and 33 and references within). In this work, we focus on understanding the requirements of Baf200 in DNA DSB repair. We unmask a dual mechanism by which Baf200 regulates the cellular DNA damage response. That is, Baf200 acts in concert with the DNA repair machinery (i.e. Rad51) to promote the repair of DNA DSBs. In light of our findings that Baf200 can form structurally distinct complexes with other subunits of PBAF, we discuss the importance of a previously unrecognized complexity to the PBAF-dependent epigenetic regulation of DNA repair.
Results
Baf200 expression is important for DNA repair
To characterize the role of Baf200 in DNA repair, we analyzed the sensitivity of Baf200-depleted cells to the DNA-damaging agent etoposide (Fig. 1, A–C) and ionizing radiation (Fig. 1D) inducing DNA DSBs. U2OS cells treated with any of three different RNAi designed against Baf200 (Fig. 1, C and D) displayed increased sensitivity compared with control cells. This sensitivity is comparable with that observed upon depletion of Brg1 (Fig. 1A) and is apparently not caused by changes in topoisomerase II or Rad51 expression (Fig. 1B). Although this is consistent with a model in which PBAF complexes containing Brg1 play a central role in DNA repair, the mechanism of function of the PBAF-specific regulatory subunits such as Baf200 remains unclear.
Figure 1.

Baf200 depletion sensitizes cells to DNA damage. A, U2OS transfected with the indicated siRNA and treated with etoposide was analyzed 10 days later to measure the number of surviving cells by flow cytometry. B, Western blot showing knockdown of Brg1 or Baf200. Shown is a comparison of three different siRNA targeting Baf200 in U2OS cells corresponding to experiments in C and D. Note that no changes in topoisomerase II or Rad51 are observed after depletion of Baf200. Molecular weights for each blot are indicated. C and D, U2OS cells transfected with the indicated siRNA against Baf200 were treated with etoposide (C) or ionizing radiation (D) and analyzed 10 days later to measure the number of surviving cells by flow cytometry. The mean ± S.D. is shown.
γH2AX accumulates rapidly after formation of a DSB, followed by reduction of the signal after DNA repair. To determine the effect of Baf200 and other PBAF components' depletion on DNA DSB repair, we tested whether RNAi-mediated depletion of Baf200, Baf180, and Brg1 in U2OS cells affected γH2AX foci number after DNA damage. The kinetics of γH2AX immunosignal was used to monitor foci formation and disappearance after inducing DNA damage using etoposide (Fig. 2, A and B) or ionizing radiation (Fig. 2C). A recent report has shown that siRNA depletion of Baf180 and Brg1 show higher γH2AX signals compared with control cells (21). Our experiments show that Baf200, Baf180, and Brg1 knockdown did not significantly induce γH2AX foci in nondamaged cells, as expected. Importantly, following etoposide or ionizing radiation exposure, cells transfected with siRNA control and cells depleted of PBAF components exhibited strong γH2AX, although Baf200-depleted cells (any of three different siRNAs targeting Baf200) showed reproducibly higher signals (Fig. 2, A–C). The number of γH2AX foci decreased in control cells at later time points (180 and 360 min), reflecting DNA repair. In contrast, there was a delay in the decrease of γH2AX foci number in cells depleted of Baf200, Baf180, and Brg1. Fig. 2, B and C, show the mean ± S.D. from three independent experiments initiated from a different set of cultured and treated cells. Statistical differences were examined using paired two-tailed Student's t test. For cells exposed to etoposide, comparison of control siRNA with all siRNA treatments for each time point, except siRNA Brg1 (360 min, p = 0.057) and siRNA Baf180 (10 min, p = 0.0002), resulted in p < 0.0001 (n = 150 cells; 95% confidence interval). For cells exposed to ionizing radiation, comparison of control siRNA with Baf200 siRNA treatments for each time point resulted in p < 0.0001 (n = 150 cells; 95% confidence interval). γH2AX kinetics analysis was performed with two additional siRNAs designed to target Baf200 (siRNA Baf200-2 and Baf200-3) (Fig. 2D). The results obtained were similar to those shown with siRNA Baf200-1 (Fig. 2B) in that siRNA Baf200 cell treatment results in increased γH2AX signal.
Figure 2.

Effect of Baf200 depletion on the DNA damage response. A, U2OS cells were treated with the indicated siRNA, followed by treatment with 10 μm etoposide for 20 min to induce damage, and allowed to repair DSBs for the indicated times. Representative images of cells immunostained with DAPI (nuclei) and γH2AX antibodies (DSBs) are shown. The scale bar represents 10 μm in all images. B, quantitative analysis of γH2AX foci formation and resolution after etoposide exposure from three independent experiments is shown. C, quantitative analysis of γH2AX foci formation and resolution after cells were treated with siRNA control and siRNA Baf200 and exposed to ionizing radiation from three independent experiments is shown. D, quantitative analysis of γH2AX foci formation and resolution after cells were treated with different siRNAs targeting Baf200 and exposed to etoposide to induce DNA damage. E, cell cycle distribution of U2OS cells before and after siRNA treatment. U2OS cells were transfected with the indicated siRNAs. After 72 h, cells were stained with propidium iodide, and the percentage of total U2OS cells at G2, S, and G1 cell cycle stages was measured by ArrayScan quantification. The data shown are from a single representative experiment out of three repeats; n = 10,000 cells analyzed from a single experiment. The mean ± S.D. is shown.
We found that depletion of Baf200 or Brg1 did not alter the cell cycle distribution (Fig. 2E), indicating that these effects on DNA repair kinetics are not caused by changes in the cell cycle phase.
In sum, we suggest a model in which Baf200 and Baf180 work together with Brg1 during the DNA damage response to stimulate DSB resolution.
Previous work has shown that depletion of Baf200 results in reduction of Baf180 (26), a result that we observed as well (Fig. 3A), but had no effect on the levels of other subunits that are in common to both the PBAF and the BAF complexes (data not shown). It is possible that the DNA repair phenotype arising from Baf200 depletion is, at least in part, a consequence of reduced Baf180 levels. To investigate this possibility, we expressed Baf180-GFP (21) in Baf180- or Baf200-depleted U2OS cells (Fig. 3B). We found that expression of Baf180-GFP restored γH2AX foci numbers after etoposide treatment in Baf180-depleted cells, but not in Baf200-depleted cells. Thus, Baf200 is required to stimulate DNA repair (Fig. 3C).
Figure 3.

Baf200 plays an important role in DNA DSB repair. A, Western blot showing Baf200 and Baf180 levels in whole U2OS lysates after treatment with the indicated siRNA. Molecular weights for each blot are indicated. B, Western blot showing expression of Baf180-GFP in U2OS cells treated with the indicated siRNAs. Molecular weights for each blot are indicated. C, U2OS cells treated with the indicated siRNAs were transfected with Baf180-GFP, treated with 10 μm etoposide for 20 min, and allowed to repair DSBs. Immunofluorescence analysis and quantitative analysis of γH2AX foci formation from three independent biological replicates (utilizing different sets of cultured and treated cells). n = 150 cells each; mean ± S.D. is shown.
Baf200 expression is important for homologous recombination repair of DSBs
Given the important role of Baf200 and Baf180 in the repair of DSBs (Fig. 2), we asked whether the homologous-directed repair (HDR) pathway is affected by loss of Baf200 or Baf180.
We used a U2OS reporter cell line containing an integrated split-GFP transgene reporter designed to measure the repair of a DSB by HDR (Fig. 4A) (34). The split-GFP transgene contains a restriction site for the I-SceI endonuclease, which is otherwise not found within the human genome. Unique DSBs were generated by expressing SceI (Fig. 4B). The control cells showed an increased number of GFP-positive cells upon expression of SceI, indicating the successful generation of functional GFP transgene by HDR. Previous studies had shown that depletion of Brg1 decreased the number of GFP-positive cells in assays monitoring HDR (22), and we observed this as well, similar to control cells depleted for Rad51 (35) (Fig. 4A). Depletion of Baf200 or Baf180, but not Baf250A, a BAF-specific subunit, significantly reduced the efficiency of HDR (p < 0.001). We conclude that Baf200 and Baf180 along with Brg1 regulate HDR of DSBs.
Figure 4.
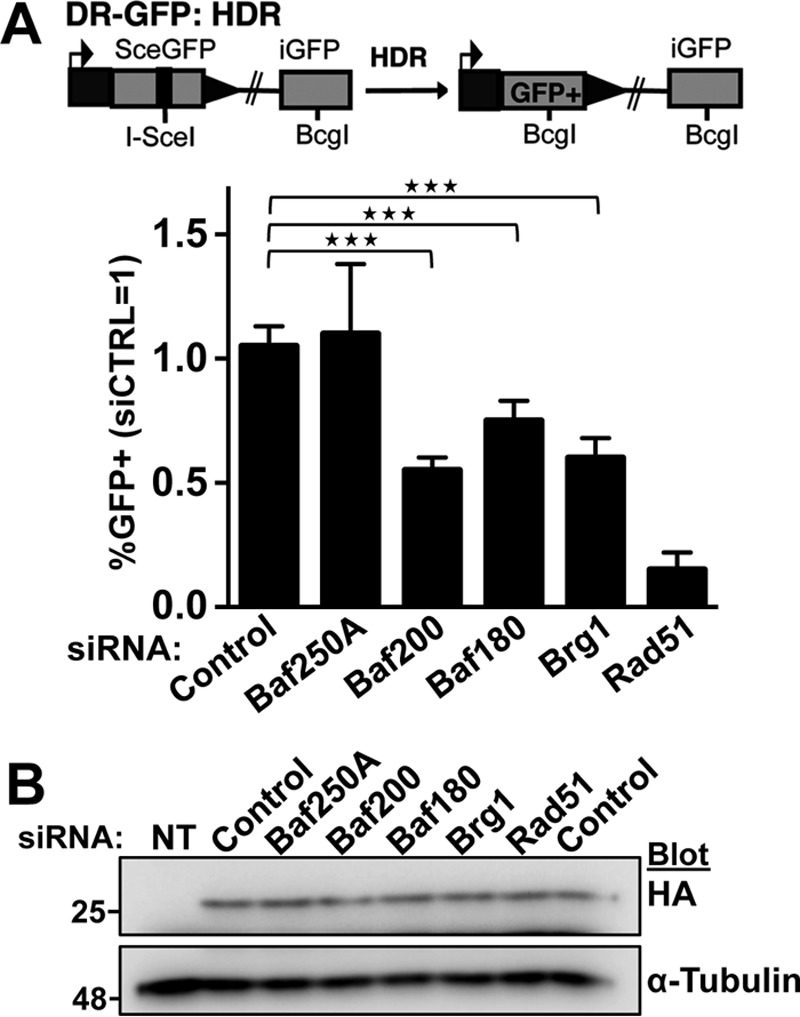
Baf200 and Baf180 expression is important for homologous recombination. A, a U2OS cell line with an integrated split-GFP transgene was used to report the efficiency of DSB repair through homologous recombination. The cell line was transfected with the indicated siRNA. After 24 h, cells were transfected with a plasmid expressing I-SceI to induce a DSB per cell, and 24 h later, GFP-positive cells indicative of DSB repair were quantified by FACS. Schematic of the GFP-HDR reporter used to monitor homologous recombination and quantitative analysis of GFP-positive cells is shown. For all experiments, the mean ± S.D. from three independent biological replicates (experiments that utilized independently cultured and treated sets of cells) is plotted. Shown are the frequencies of GFP-positive cells relative to parallel transfections with siControl. Statistical differences were examined using paired two-tailed Student's t test. Comparison of control siRNA treatment with Baf200, Baf180, Brg1, and Rad51 siRNA treatments resulted in p < 0.0001. Comparison of control siRNA treatment with Baf250A treatment resulted in a non-significant difference; ***, p < 0.001. B, a Western blot analyzing expression of I-Sce1-HA is shown. Molecular weights for each blot are indicated.
Baf200 recruits Rad51 to DSBs repaired by homologous recombination
Results in Fig. 4 showing that Baf200 and Baf180 are required for HDR suggest that Baf200/Baf180 and Rad51 act together to stimulate DSB repair. To test this, we examined whether Baf200, Baf180, and Brg1 interact with Rad51. Indeed, an antibody detecting Baf200 but not Brg1 could co-immunoprecipitate Rad51 from U2OS extracts (Fig. 5A). Furthermore, an antibody against Baf200 immunoprecipitated Rad51 and an antibody specific for Rad51 was able to pull down Baf200. These interactions are also observed in the presence of ethidum bromide, an agent commonly used to disrupt DNA-protein interaction, and after cells were treated with siBrg1 (Fig. 5B). In addition, we performed a yeast two-hybrid assay and found that Rad51 interacts with Baf200, but not with Brg1 or Baf180 or the chromatin remodeling factors Mrg15 or Smarcl1 (Fig. 5, C and D), again suggesting the specificity of Baf200 and Rad51 interaction. We conclude that Rad51 and Baf200 are part of the same complex.
Figure 5.

Baf200 interacts with Rad51. A–D, an interaction between Baf200 and Rad51 is supported by co-IP using U2OS cell total extract in the presence of DNA damage agent (etoposide) or ethidium bromide (A and B), and a direct yeast two-hybrid assay (C and D). For yeast two-hybrid assays, growth of control yeast was monitored on medium lacking tryptophan and leucine (TL). Positive interactions were monitored by growth on medium lacking tryptophan, leucine, histidine, and adenine (TLHA). C, Baf200, but not Baf180 or Brg1, shows a strong interaction with Rad51. D, the specificity of Rad51 interaction with Baf200 is also supported by the strong growth in TLHA plates of Baf200-Rad51 mating products, but not other chromatin remodeling factors. E, U2OS cells were transfected with control or Brg1 siRNA and treated with etoposide for 20 min. After removal of etoposide, cells were lysed at the indicated times, and the chromatin-associated fraction was evaluated by Western blotting with the indicated antibodies. ND represents a sample where cells were not exposed to etoposide (no DNA damage) and collected 30 min after DNA damage induction. Chromatin fractions were probed with the indicated antibodies. Laminin B was used as loading control, γH2AX was used to indicate an early stage of the DNA damage response, and the Rad51 protein was used as a marker for a later stage of the homologous recombination-directed DNA repair pathway. Blue stars mark strong events of Baf200 and Rad51 association with chromatin. The figure shows representative results obtained in one of three independent biological replicates (experiments that begin from a different set of cultured cells). F, AID-AsiSI-ER U2OS cells were used to analyze time course of protein binding to chromatin using Baf200-, Rad51-, and γH2AX-specific antibodies at a DSB specifically repaired by the HDR pathway (site A: Chr9 129732985). Auxin addition was considered time 0. ChIP signals were normalized to the maximum signal obtained for each antibody. Time course for each ChIP (antibody) at the indicated times (experiments initiated with a different set of cultured cells) was repeated three times. The mean ± S.D. is shown. G, schematic of Baf200 and Rad51 functional domains and truncation mutants created to map sites of Baf200-Rad51 interaction. The negative (−) and positive (+) symbols represent absence or presence of interaction. Four positive symbols represent maximum interaction strength.
If Baf200 and Rad51 work together to stimulate DNA repair, we expect that both proteins will show similar dynamics of accumulation on chromatin after induction of DSBs. To test this, we first determined the temporal relationship between distinct events of the DNA damage response and the loading of Baf200 on chromatin. We treated U2OS cells with 10 μm etoposide to induce DSBs, and subsequently monitored chromatin loading of Baf200 and markers of early (γH2AX) and late (Rad51) DNA repair (Fig. 5E, left panel). Furthermore, to determine whether Baf200 chromatin loading depended on Brg1, we performed the same experiment with cells depleted of Brg1 by RNAi (Fig. 5E, right panel). In cells treated with a control siRNA, we observed that the major peaks of Baf200 loading onto chromatin occur later in the DNA damage response (3–9 h), coinciding with Rad51 loading onto the chromatin. Notably, cells depleted of Brg1 show similar Baf200 and Rad51 recruitment (Fig. 5E, right panel). We also observed coincidental loading of Baf200 and Rad51 on chromatin after DNA damage in 293T cells depleted of Brg1 (results not shown). We then asked whether the coincidental temporal pattern of Baf200 and Rad51 association to chromatin may also be observed at individual DSBs. We used an AID-AsiSI-ER U2OS cell line (36) in which addition of tamoxifen (4OHT) induces AsiSI-mediated DSBs. After 4 h of 4OHT induction, auxin was added to the media culture to promote degradation of the AsiSI endonuclease, which allows kinetic studies of DSB repair at selected genomic sites by ChIP-qPCR. Our analysis revealed that auxin-induced degradation of AID-AsiSI-ER correlated well with repair of DSBs, revealed by decreasing γH2AX levels measured by ChIP signal at DSBs specifically repaired by homologous recombination (36) (Fig. 5F shows homologous recombination site A). As expected, Rad51 signal is stronger at later time points after auxin addition (maximum signal detected at 4 h). This is coincidental with the temporal pattern of chromatin loading corresponding to Baf200. In sum, we take these results as to suggest that Baf200 and Rad51 cooperate during DSB repair and that Rad51 and Baf200 loading to the chromatin do not depend on Brg1.
We further explored Baf200 and Rad51 interaction by using the direct yeast two-hybrid assay and C- and/or N-terminal Baf200 and Rad51 deletion mutants. We identified small regions located at or near the C terminus of each protein that were necessary and sufficient for the Baf200-Rad51 interaction (Fig. 5G and supplemental Fig. S1).
The suggested interaction of Baf200 and Rad51 led us to test the possibility that Baf200 may affect the recruitment of Rad51 to DSBs. To evaluate this, we first analyzed the recruitment of Baf200, Brg1, and Rad51 to DSBs by chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR) using a U2OS cell line in which 4OHT induces AsiSI-mediated DSBs (37) (Fig. 6, A and B). DSBs at certain genomic locations have been shown to be preferentially repaired by homologous recombination (aided by Rad51), whereas DSBs at other locations are preferentially repaired by non-homologous end joining (NHEJ) (36). We screened Baf200 and Brg1 localization at three sites shown to be preferentially repaired by homologous recombination and Rad51 (36). We found that Baf200, Brg1, and Rad51 are enriched at these three different DSBs within the genome (Fig. 6A). Our results obtained with Brg1 recruitment are consistent with results from a previous report (22). Importantly, Rad51 recruitment to two DSB sites repaired by homologous recombination is decreased (either delayed or depleted) following depletion of Baf200 or Brg1 (Fig. 6B). In contrast, in a site predominantly repaired by NHEJ, XRCC4 (a key component of the NHEJ DNA repair pathway) recruitment did not show significant decrease after depletion of Baf200 or Brg1. Consistent with our results above (Fig. 2), γH2AX recruitment was not reduced after depletion of Baf200, indicating that loss of Baf200 did not decrease AsiSI accessibility, the efficiency of DSB induction, or early signaling in response to DNA damage (Fig. 6C). In sum, Baf200 and Brg1 are required for efficient recruitment of Rad51 to a subset of DSBs repaired by homologous recombination.
Figure 6.

Baf200 is enriched on the chromatin at DSB sites and is required for Rad51 recruitment to DSBs. A, DSBs were induced by tamoxifen (4OHT) treatment of AsiSI-U2OS cells containing estrogen receptor-tagged AsiSI. DSB sites, known to be preferentially repaired by homologous recombination, were evaluated for Rad51, Brg1, and Baf200 recruitment by ChIP followed by qPCR. HDR site A: Chr9 129732985, HDR site B: Chr22 37194040, and HDR site C: Chr22 19180307. B, AsiSI-expressing U2OS cells were transfected with the indicated siRNA, and DSBs were induced by 4OHT treatment. ChIP-qPCR was used to evaluate Rad51 and XRCC4 recruitment to the same two AsiSI sites preferentially repaired by homologous recombination (sites A and B) shown in A and one site preferentially repaired by NHEJ (site 1: Chr18 7556705). A and B, for all experiments, the mean ± S.D. from three independent biological replicates (experiments that utilized different sets of cultures and siRNA-treated cells) is plotted. Statistical differences noted in B were examined using paired two-tailed Student's t test. Comparison of control siRNA treatment with Baf200 siRNA and Brg1 siRNA treatments resulted in ***, p < 0.001. C, DSB-dependent phosphorylation of H2AX (γH2AX) and recruitment of Rad51 to HDR site A. D, quantitative analysis of RPA foci formation and resolution after cells were treated with control siRNA and siRNA targeting Baf200 and exposed to etoposide to induce DNA damage. The mean ± S.D. is shown.
In agreement with a model in which Baf200 promotes Rad51 loading at recombination sites, we observed that siRNA Baf200-treated cells show a significant increased number of RPA foci compared with control cells (Fig. 6D). Statistical differences were examined using paired two-tailed Student's t test. Comparison of control siRNA with Baf200 siRNA treatment for each time point resulted in p < 0.0001 (n = 150 cells; 95% confidence interval).
Baf200 and Baf180 participate in distinct PBAF subcomplexes
We next investigated whether Baf200 and other PBAF components can participate in complexes with different subunit compositions. Similar to that observed in Fig. 5E, both Baf200 and Baf180 associate with chromatin after DNA damage induction in U2OS cells, even upon RNAi-mediated depletion or CRISPR/Cas-mediated knock-out of Brg1 (Fig. 7, A and B). These data support the idea that Baf200 and Baf180 are able to form complexes that associate with chromatin independently of Brg1. Furthermore, similar to control cells, we observed that in cells treated with Brg1 siRNA or in a Brg1−/− cell line, Baf200 immunoprecipitated Rad51 and that Rad51 is able to pull down Baf200 (Fig. 7C). These results suggest that when incorporated into a Brg1-independent PBAF complex Baf200 is able to interact with Rad51.
Figure 7.

Biochemically distinct complexes composed by Baf200 and Baf180 coexist in U2OS cells. A, Western blot of chromatin fractions showing Baf200, Baf180, Brg1, and Lamin B levels after treatment of U2OS cells with the indicated siRNA and etoposide (10 μm for 20 min). B, Western blot showing Baf200 and Baf180 levels in etoposide-treated Brg1 knock-out U2OS cells generated using CRISPR/Cas. Total (T), soluble (S), and chromatin-bound (C) cell lysate fractions were analyzed with the indicated antibodies. C, co-IP was performed using cell total extract from U2OS cells transfected with siRNA control, siRNA Brg1, and a Brg1−/− U2OS cell line. Molecular weights for each blot are indicated. D, U2OS nuclear cell lysates were subjected to sucrose gradient fractionation (5–20%) and analyzed by Western blotting with the indicated antibodies. Only fractions 14–25 of a total of 30 fractions are shown. Stars mark the peaks of Baf200 and Brg1. One representative experiment of three independent replicates (utilizing independently cultured cells, processed and resolved in sucrose gradients) is shown. E, quantitative analysis of results in D. F, 293T nuclear cells lysates were subjected to sucrose gradient fractionation (5–20%) and analyzed by Western blotting with the indicated antibodies. Only fractions 14–31 of a total of 38 fractions are shown.
To determine whether a Brg1-independent Baf200/Baf180 complex occurs in cells that are not depleted of Brg1, we used sucrose gradient fractionation of nuclear extracts to separate distinct complexes, and detected different PBAF and BAF protein components within gradient fractions by immunoblotting (Fig. 7D). None of the components could be detected in eluted fractions predicted to contain free subunits (data not shown), suggesting that they all reside predominantly in large, multiprotein complexes. Several subunits of the PBAF and BAF complexes, including common subunits (Brg1, Baf170, Baf155, and Snf5) and the BAF-specific Baf250A, were present with maximum signal in fraction 17. The Baf200 and Baf180 sedimentation profiles are different from those of Brg1, suggesting that Baf200 and Baf180 participate in a complex or complexes distinct from the canonical PBAF complex containing Brg1. Furthermore, Baf200 shows a bimodal distribution with a minor peak in fraction 17, overlapping with the maximum signal observed for Brg1, and a major peak in fraction 20, overlapping with the maximum signal for Baf180 (Fig. 7, D and E). We also observed that Baf200 and Baf180 sedimentation profiles are different from those of Brg1 in 293T cells (Fig. 7F). These data suggest that Baf200 forms at least two biochemically/structurally distinct complexes. One of them is similar to that described for a canonical form of PBAF containing Brg1 and common PBAF/BAF components. The second complex contains the majority of the available pool of Baf200 and Baf180. Other known PBAF and BAF components were not abundant in fraction 20; thus, this complex may have minor participation of other PBAF/BAF core components, or there may be other complex components of unknown identity that co-sediment with Baf200 and Baf180.
To identify protein subunits participating in the distinct Brg1-containing and Brg1-independent Baf200/Baf180-complexes, we performed co-immunoprecipitation analyses with lysates of U2OS cells treated with siRNA control (Fig. 8A, lines 1–4) and U2OS cells treated with siRNA Brg1 (lines 5 and 6) or siRNA Baf200 (lines 7 and 8). As expected, when samples from cells treated with a control siRNA (line 1) were used for immunoprecipitation with unspecific IgG antibodies, no PBAF/BAF component was detected by Western blotting (line 2). We then used the resulting flow through to perform immunoprecipitation with Brg1 antibodies (see experiment design, bottom panel Fig. 8A). We observed that PBAF/BAF core components (Baf170, Baf155, and Snf5) and a BAF-specific subunit (Baf250A) co-immunoprecipitate with Brg1 (Fig. 8A, lane 3). Consistent with the results from our sucrose gradient experiments, only a small fraction of Baf200 and Baf180 co-immunoprecipitate with Brg1. We then used the resulting flow through from Brg1 immunoprecipitation experiments, which is now depleted of Brg1-containing PBAF complexes (Fig. 8B), for immunoprecipitation with Baf200-specific antibodies (Fig. 8A, line 4). In these samples, Baf180, and a minor fraction of Baf170 and Snf5, co-immunoprecipitated with Baf200. Similarly, we found that Baf200 interacts with Baf180, Baf170, and Snf5 in U2OS cells treated with siRNA Brg1 (Fig. 8A, lane 6). As expected, no PBAF/BAF component was detected after immunoprecipitation with Brg1 antibodies when we used nuclear extract from siRNA Brg1-treated cells (line 5). We conclude that Baf180 is a major partner of Baf200, and that at least some common PBAF/BAF core components can also interact with Baf200/Baf180 in complexes that do not include Brg1. In addition, we found that Brg1 forms a stable complex with most PBAF common subunits in Baf200-depleted cells (Fig. 8A,lane 7).
Figure 8.
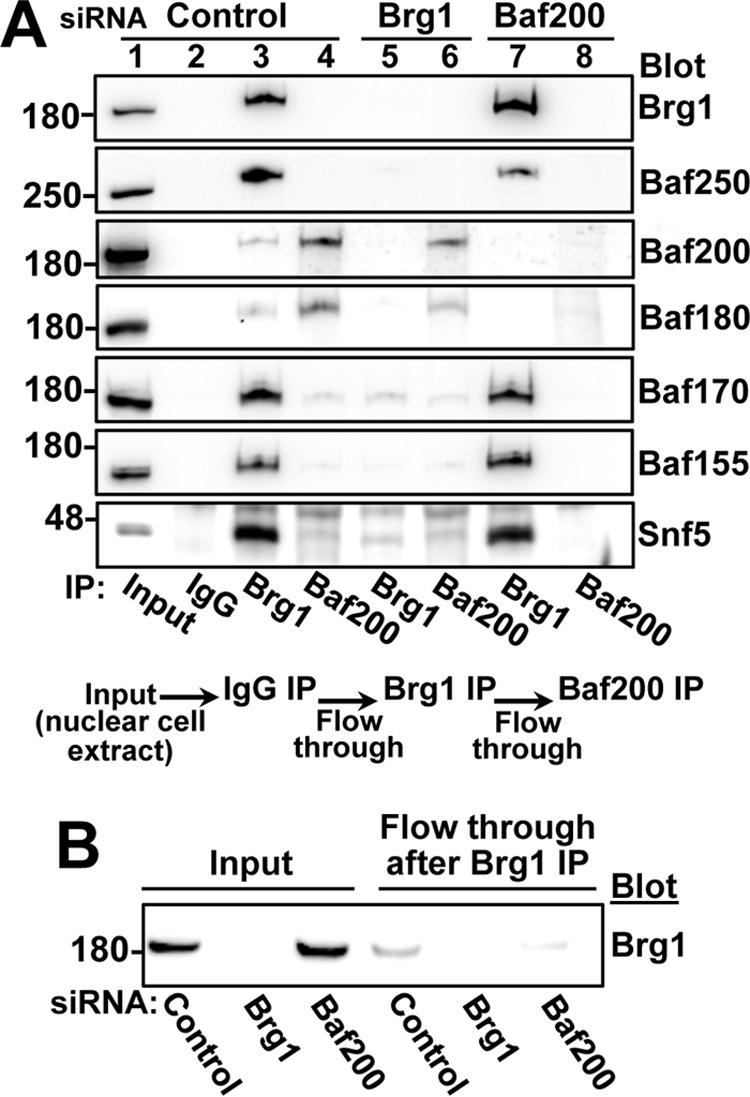
Distinct complexes composed by Baf200 and Baf180 coexist in 293T and U2OS cells. A, immunoprecipitation followed by Western blotting analysis of U2OS cells transfected with the indicated siRNAs. The schematic at the bottom of the panel summarizes the procedure: for each siRNA-treated condition, cell lysates were sequentially immunoprecipitated with unspecific IgG, followed by anti-Brg1 and then anti-Baf200 antibodies. Molecular weights for each blot are indicated. B, Western blot showing residual Brg1 protein after U2OS nuclear extract were immunoprecipitated (immunodepleted) with Brg1-specific antibodies.
Collectively, our results show that Baf200 forms at least two distinct complexes. One complex is a canonical form of PBAF, includes Brg1 and known PBAF components; the other complex contains Baf180, and possibly Smarca5, Snf5, Baf170, and Baf155, but not Brg1.
Discussion
Approximately 20% of cancers have been estimated to have mutations in Swi/Snf components, which would make this family of genes among the most commonly mutated in cancer (Ref. 17 and references within). Although this underscores the importance of PBAF in human cancer, much remains to be determined regarding the mechanisms by which it prevents tumorigenesis. Our work demonstrates that PBAF components can be found in biochemically/structurally distinct complexes, and that Baf200 is required for normal DNA damage response. These findings reveal new aspects important to understanding the roles of PBAF subunits in maintaining genome stability and as tumor suppressors.
The role of Baf200 in the DNA damage response
Although Baf200 is a recognized specific subunit of the PBAF chromatin-remodeling complex and has been implicated in gene regulation, its role in DNA repair is poorly understood. Our results suggest that Baf200 regulates the DNA damage response by promoting recombinational DNA repair dependent on Rad51 (Fig. 9).
Figure 9.
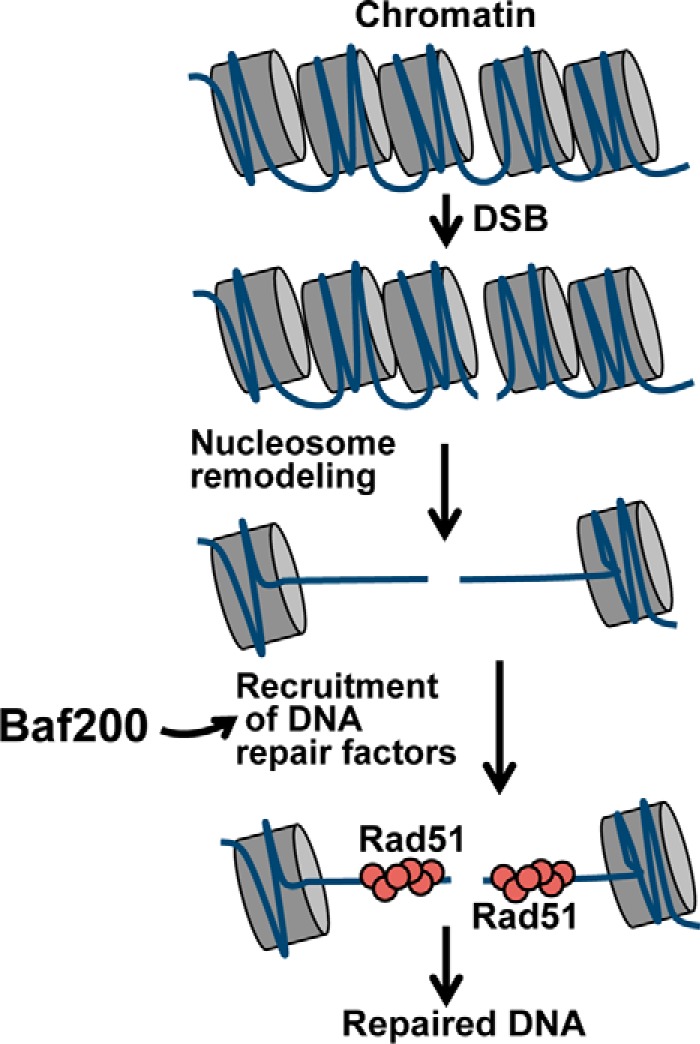
A model for the role of Baf200 in regulating DNA DSB repair. We propose that Baf200 acts at a critical step of the DNA repair progress by recruiting DNA repair factors to promote homologous recombination-dependent repair of DSBs.
PBAF subunits participate in biochemically/structurally distinct complexes
Until recently, PBAF has been considered a functional entity with a relatively fixed composition of subunits, in which Brg1 is the catalytic core and Baf200 and Baf180 are PBAF-specific subunits with regulatory functions. Our results show a more extensive diversification of PBAF components than previously thought. We demonstrate the coexistence of at least two biochemically distinct complexes with different subunit compositions; one of them is similar to that described for the canonical form of PBAF and the second is a previously unrecognized form that contains Baf200, Baf180, and possibly other core components, but not Brg1. Our results provide physical evidence for structurally distinct complexes composed by a particular combination of PBAF subunits.
What might be the functions of these distinct Baf200-containing complexes? The existence of functionally diverse Swi/Snf or PBAF subcomplexes working in transcription regulation and development has been suggested previously (31, 38, 39). Furthermore, a recent report suggests that most genes regulated by Swi/Snf are controlled by biochemically distinct forms of the complex, and the overall expression of a gene is the product of the interaction between these different complexes (31). In light of these results, we speculate that the distinct forms of PBAF we identified may have important functions in regulating distinct aspects of DSB repair. For example, it is possible that Brg1/Baf200/Baf180-containing complexes act at early stages of the DNA damage response (i.e. in signaling the DNA damage) and Brg1-independent complexes may act later in DNA repair by coordinating functions with Rad51 to promote homology-directed repair of DSBs. Although hypothetical, in view of recent studies suggesting that PBAF-mutant cancers depend on residual complexes for their aberrant growth (31, 40), it is possible that the complexes we identified contribute to specialized essential cell functions that sustain cancer cell viability and resistance to therapy. Future studies should investigate these possibilities.
Experimental procedures
Antibodies
Antibodies against Baf200 were purchased from Abcam (ab510190), Bethyl (A302-230A), and Sigma (SAB2702507). We also generated polyclonal antibodies against human Baf200 using peptides directed against the N-terminal (RERRPSQPHTQSGGT) and C-terminal (PREEGKSKNNRPLRTSQC) sequences of the protein. The Baf180 (A301-591A) antibody was from Bethyl. Antibodies against α-tubulin (66031-1) and the Brg1 (21634-1) were from Proteintech Group. Baf155 (D7F8S), Baf170 (D8O9V), Baf250 (D2A8U), and Snf5 (D9C2) antibodies were from Cell Signaling. Anti-Rad51 (H-92) and anti-H2B (FL-126) were from Santa Cruz Biotechnology. Anti-RPA (Ab-3) was from Oncogene. The antibody against phospho-histone H2A.X (Ser-139) clone JBW301 was from Millipore (05-636) or from Cell Signaling (2577S). Lamin B1 antibody was from Abcam (ab16048). The HA antibody was from BioLegend (16B12). Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories: HRP-conjugated goat anti-rabbit IgG Fc fragment specific (111-035-008), HRP-conjugated mouse anti-rabbit IgG light chain specific (211-032-171), HRP-conjugated goat anti-mouse IgG Fcγ fragment specific (115-035-071), ChromPure rabbit IgG whole molecule (011-000-003), rhodamine-conjugated goat anti-mouse IgG, F(ab′)2 (115-026-072), and FITC-conjugated goat anti-rabbit IgG, F(ab′)2 (115-096-047).
Plasmids and siRNAs
pEGFP-C3 BAF180 plasmid (21) was obtained from Dr. Jessica Downs (University of Sussex, Brighton, UK). I-SceI endonuclease expression vector pCBASceI was purchased from Addgene. For yeast two-hybrid assays, full-length proteins or truncations were cloned into pGBKT7 (bait) or pGADT7-AD (prey) as indicated. Baf200 truncations (1–250, 200–750, and 1200–1835) were cloned into the pcDNA3.1 plasmid.
All siRNAs were purchased from Bioneer Inc. The efficiency of knocking down was analyzed in each experiment by Western blotting. The control siRNA (SN-1003) does not recognize any human mRNA. The sense sequences of siRNA duplex were the following: siBaf200 (1007749) CAGUUUCACAGGGUCAACA(dTdT), siBaf180 (1112705) GAUUUGUACCUUCGAACAA(dTdT), siBrg1 (1141405) CUCUCUCAACGCUGUCCAA(dTdT), siRad51 (1126153) CCAGCUCCUUUAUCAAGCA(dTTd), siBaf250A (1007732) GUAUAUCCAGUGUCUCUAU(dTdT).
Cell culture, transfections, and induction of DSBs
All cells were grown in a 5% CO2 humidified incubator at 37 °C. Cells were cultured as monolayers in Dulbecco's Modified Eagle Medium (Invitrogen) supplemented with 10% heat-inactivated FBS and glutamine. AsiSI-ER-U2OS cells were selected using 1 mg/ml of puromycin. Brg1 knock-out cell line was generated by co-transfecting U2OS cells with pRP[CRISPR]-hCas9-U6>hSMARCA4_10_66984_20nt vector carrying the following BRG1 targeting sequence: GAAGATTACTTTGCGTATCG (VectorBuilder, Inc) and pGFP-Puro plasmids at a ratio of 10:1. Transfected cells were selected with puromycin (3 μg/ml) for 2 weeks and clones were analyzed according to Brg1 expression.
For siRNA transfection, 2 × 105 cells were treated with Lipofectamine RNAiMAX (Thermo Fisher Scientific) and 60 pmol of siRNA according to the manufacturer's instructions. For all other experiments, 4 × 105 cells were transfected using Lipofectamine 2000 following manufacturer's instructions.
DSBs were generated by incubating cells with 10 μm etoposide for 20 min at 37 °C or exposing cells to ionizing radiation. After that, cells were incubated with fresh medium to allow DNA damage repair.
Cell survival curve
U2OS cells were treated with the indicated siRNAs and 24 h later seeded in low density (4,000 cells/well of 6-well plate). DNA damage was induced 48 h after siRNA treatment by incubating these cells with indicated concentrations of etoposide for 20 min or by exposure to ionizing radiation. Cells were placed in the cell incubator for 10 days, and cells were collected and the number was measured by FACS. All samples were measured for a period of 1 min in BD FACSCalibur. Number of cells treated with the corresponding siRNA and no DNA damage was computed as 100%. Typically, 8,000 cells were counted for the condition in which cells were treated with control siRNA and no etoposide.
ChIP experiments using AsiSI-ER-U2OS cells
AsiSI-ER-U2OS cell lines were kindly provided by Gaelle Legube. Cells were cultivated as described above and DSBs were induced by addition of 4-OHT (300 nm) (Sigma I5148) for 3 h. The ChIP assay was performed as described previously (36, 37), with slight modifications. Briefly, the samples were cross-linked by adding paraformaldehyde to the cell culture media to a final concentration of 1% and incubated at room temperature for 10 min. The cross-linking reaction was halted by adding 1:10 volume of 0.125 m glycine and incubating for 5 min at room temperature. Then, cells were washed with cold 1× PBS and harvested. Cell pellets were suspended in buffer: 5 mm PIPES, pH 8.0, 85 mm KCl, and 0.5% IGEPAL supplemented with protease inhibitors (Thermo Fisher Scientific), and homogenized using a Dounce homogenizer with tight pestle B. Nuclei were pelleted by centrifugation at 2,000 × g during 10 min. Nuclei pellets were suspended in lysis buffer (50 mm Tris-HCl, pH 8.1, 10 mm EDTA, 1% SDS) and incubated on ice during 30 min before sonication. Samples were diluted in ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, 167 mm NaCl) and precleared with protein A/G beads (Sigma-Aldrich). Samples were then incubated with the indicated antibodies overnight at 4 °C. Antibody-protein complexes were recovered by incubating the samples with agarose beads coated with protein A/G. Beads were then washed once with low-salt washing buffer (0.2% sarcosyl, 2 mm EDTA, 50 mm Tris-HCl, pH 8.0) and four times with washing buffer (500 mm LiCl, 1% IGEPAL, 1% sodium deoxycolate, 100 mm Tris-HCl, pH 8.8). Protein-DNA complexes were eluted twice with elution buffer (1% SDS, 100 mm NaHCO3) for 15 min at 65 °C. Cross-linking was reversed by adding 5 m NaCl and RNase A to the samples and incubating overnight at 62 °C. Then, proteins were digested by incubation with proteinase K for 2 h at 45 °C. ChIP and input DNA were purified with phenol/chloroform, precipitated, and analyzed in quadruplicate by real-time qPCR (StepOnePlus Real-Time PCR System, Applied Biosystems) using the SYBR Green qPCR SuperMix (Invitrogen) according to the manufacturer's instructions.
We used distal sites for qPCR amplification of selected AsiSI sites (36). All samples were calibrated to amplification from input DNA. Three biological replicates were performed. The primer sequences were as follows.
Site A, Chr9 129732985_800-bp form AsiSI site_FW (5′-TATGGGACCAAGCGAGTAGG-3′); site A, Chr9 129732985_800-bp form AsiSI site_REV (5′-TGCCTCACACACACACCCATA-3′); site B, Chr9 129732985_800-bp form AsiSI site_FW (5′-GGGTATGGAGCTGCCTCTAA-3′); site B, Chr9 129732985_800-bp form AsiSI site_REV (5′-GACAAAGATGGCTGGAGGAG-3′); site C, 19180307_dist_FW (5′-CCCATCTCAACCTCCACACT-3′); site C, 19180307_dist_REV (5′-CTTGTCCAGATTCGCTGTGA-3′); and site 1, Chr18 7556705_80-bp form AsiSI site_FW (5′-TCCCCTGTTTCTCAGCACTT-3′); site 1, Chr18 7556705_80-bp form AsiSI site_REV (5′-CTTCTGCTGTTCTGCGTCCT-3′).
ChIP assays using AID-AsiSI-ER-U2OS
2–3 × 108 AID-AsiSI cells were treated with 300 nm 4OHT (Sigma, H7904) for 4 h. Cells were washed three times in prewarmed 1× PBS and incubated with 500 μg/ml of auxin for the indicated time periods. Cells were fixed in 0.3% paraformaldehyde for 30 min at 4 °C and then quenched with 0.125 m glycine for 5 min at room temperature. Cells were washed three times with cold 1× PBS and scrapped in 1× PBS. Pellets of cells were frozen in liquid nitrogen and stored at −80 °C. For each antibody used in ChIP, 50–75 × 106 cells were suspended in 2.5 ml of swelling buffer (25 mm HEPES, 1.5 mm MgCl2, 10 mm KCl, 0.1% IGEPAL) with 1 mm PMSF and incubated 10 min on ice. Cells were passed through a Dounce homogenizer (20 strokes with a B pestle), divided into two tubes, and centrifuged at 2,000 rpm for 7 min at 4 °C in a swinging bucket centrifuge. Cell pellets were suspended in 5 ml of sucrose buffer A (0.32 m sucrose, 15 mm HEPES, pH 7.9, 50 mm KCl, 2 mm EDTA, 0.5 mm EGTA, 0.5 mm PMSF), layered gently over 5 ml of sucrose buffer B (30% sucrose, 15 mm HEPES, pH 7.9, 50 mm KCl, 2 mm EDTA, 0.5 mm EGTA, 0.5 mm PMSF) in a 15-ml tube, and centrifuged 10 min at 3,000 rpm in a swinging bucket GH-3.8 rotor at 4 °C. Nuclei were washed once in 10 ml of wash buffer (0.32 m sucrose, 15 mm HEPES, pH 7.9, 50 mm KCl, 2 mm EDTA, 0.5 mm EGTA, 0.5 mm PMSF) and pelleted for 10 min at 2,000 rpm. Pellets were then suspended in 1.25 ml of immunoprecipitation (IP) buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 0.5 mm EGTA, 0.1% sodium deoxycolate, 0.5% N-lauroylsarcosine) with 1:200 protease inhibitor (Cocktail Set III, Calbiochem). The lysed nuclei samples were sonicated (QSonica S-4000 with a cup horn sonication system) using ice-cold polypropylene glycol as a transmitter of acoustic energy (set up: 14 pulses of amplitude 50, 10–30 s on-off). After sonication, Triton X-100 was added to a final concentration of 1% and samples were cleared by centrifugation at 13,000 rpm for 15 min at 4 °C. The supernatants were combined and 10% was reserved as DNA input. Chromatin content was estimated by NanoDrop and 100 μg of chromatin were immunoprecipitated with 0.2 μg of antibodies. The antibodies were incubated with the chromatin samples rotating overnight at 4 °C. 50 μl of ChIP-grade protein A/G magnetic beads (Life Technologies) were washed three times with IP buffer, blocked with IP buffer containing 0.5% BSA by rotating 1 h at 4 °C. Samples were incubated with the beads rotating during 3 h at 4 °C. Beads were washed three times in IP washing buffer (50 mm HEPES-KOH, pH 7.6, 250 mm LiCl, 1 mm EDTA, 1% IGEPAL, 0.7% sodium deoxycholate), once in TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA) supplemented with 50 mm NaCl, and once with TE buffer before elution in 200 μl 1% SDS, 100 mm NaHCO3 for 17 min at 65 °C with agitation. Supernatants were supplemented with 8 μl of 5 m NaCl and incubated overnight at 65 °C. 4 μl of 0.5 m EDTA, 8 μl of 1 m Tris-HCl, pH 6.5, and 3 μl of RNase A (10 mg/ml) were added to reactions and incubated for 30 min at 37 °C, followed by 5 μl proteinase K (20 mg/ml) at 56 °C for 1 h. Reactions were purified using MiniElute kit (Qiagen) and concentration was measured using Qubit fluorometric quantitation (Life Technologies) before analysis by qPCR.
Immunofluorescence and foci quantitation
The cells were prepermeabilized in ice-cold buffer (20 mm HEPES, pH 7.9, 50 mm NaCl, 300 mm sucrose, 3 mm MgCl2 and using 0.5% Triton X-100) for 5 min prior to fixation. Then, cells were fixed with 3.7% PFA/2% sucrose in 1× PBS for 15 min at room temperature and subsequently permeabilized with 0.2% Triton X-100 in 1× PBS for 10 min. Cells were incubated overnight at 10 °C with primary antibody diluted in blocking solution (0.2% pork skin gelatin in 1× PBS), washed in 1× PBS, and incubated with secondary antibodies 1:300 diluted in blocking solution. Coverslips were mounted using Fluoromount-G (EM Sciences). Images were acquired using a Zeiss AxioPlan 2ie microscope fitted with a 63 × 1.4 NA objective, Roper CoolSnap camera, and custom acquisition software. Quantification of foci and immunofluorescence intensity was done on ∼150–200 cells per slide. Briefly, using ImageJ software (National Institutes of Health) 10 cells were randomly picked per picture using DAPI channel. Next, we used home-developed macros in ImageJ to determine cell area and total fluorescence intensity of each cell. Finally, the number of foci per cell was determined by the function Find Maxima (with output type count) with specific noise tolerance and threshold calculated by a calibration curve performed for each experiment.
Immunoblotting and immunoprecipitation
Cells were collected in 1× PBS and fractioned in chromatin extraction buffer (0.1% Nonidet P-40, 50 mm Tris-HCl, pH 7.9, 150 mm NaCl, 3 mm MgCl2, 3 mm EDTA, 10% glycerol, 1 mm DTT and protease inhibitors). For immunoblotting experiments, pellets containing chromatin-bound proteins were washed in chromatin extraction buffer and proteins were solubilized with sample buffer (4% SDS, 160 mm Tris-HCl, pH 6.8, 20% glycerol, 100 mm DTT, and 0.005% bromphenol blue). For immunoprecipitation experiments, cells were lysated using IP buffer (0.5% Triton X-100, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 3 mm MgCl2, 10% glycerol, 1 mm DTT, EDTA-free protease inhibitors and Benzonase). Proteins from the soluble fraction were immunoprecipitated with antibodies prebound with protein A ultralink resin beads (Thermo Fisher Scientific). After rotation at 4 °C for 6 h, the beads were washed four times with ice-cold IP buffer, and bound proteins were eluted by boiling for 5 min with SDS-PAGE sample buffer. Proteins were separated by 4–15% gradient SDS-PAGE under reducing conditions and transferred to nitrocellulose membranes. The blots were probed with individual primary antibodies as indicated, and then incubated with HRP-conjugated donkey anti-mouse or rabbit antibodies as required. In all blots, proteins were visualized by enhanced chemiluminescence.
GFP-DNA repair pathway reporters
U2OS cells containing chromosome-integrated expression cassette reporters (34) were used to evaluate DSB repair induced by I-SceI nuclease. Once DNA damage was repaired, it reconstituted GFP expression. 24 h after treatment with indicated siRNAs, a U2OS cell line DR-GFP (homologous recombination directed) was transfected with a plasmid containing the I-SceI endonuclease. 48 h after transfection, reconstitution GFP expression was analyzed by flow cytometry (BD FACSCalibur).
Sucrose density gradient centrifugation
Protein extracts were obtained from U2OS cells using a buffer containing 5% sucrose, 0.5% Triton X-100, 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 3 mm MgCl2, 1 mm DTT, EDTA-free protease inhibitors and Benzonase). Protein extracts were placed on top of a 12-ml sucrose gradient bed (5–20% in lysis buffer). Samples were centrifuged using a Beckman SW Ti 40 rotor (Beckman Coulter; Fullerton, CA) at 175,000 × g for 20 h at 4 °C. Finally, fractions were collected from the top of the tube and protein fractions were analyzed by Western blotting as described.
Yeast two-hybrid assay
Full-length mouse Baf200 and Rad51 were cloned into pGADT7-AD (Clontech) to produce fusions to the Gal4 DNA binding and activation domains. Plasmids containing full-length and fragments of Rad51 (amino acids 1–340, 1–100, 1–270, 100–230, 100–230, 100–270, 100–340, and 250–340) and Baf200 (amino acids 1–455, 1–1164, 1–1391, 1383–1835, and 1616–1835) were constructed by cloning the appropriate PCR products in pGBKT7 (Clontech). All fusions were confirmed by sequencing. Two-hybrid assays were conducted in the AH109 strain background. After mating, colonies containing both plasmids were selected using media lacking tryptophan and leucine. Interactions between partners were assayed by growth on synthetic media lacking tryptophan, leucine, adenine, and histidine. Transformations were carried out according to the matchmaker kit manual (BD Biosciences).
Statistical reporting
Statistical analysis methods are described in the text or figure legends. GraphPad Prism (version 6.0f) package was used for generation of graphs and all statistical analysis.
Author contributions
R. O. C. conceived the idea for the project, designed and conducted most experiments, analyzed results, and wrote the paper. L. P., V. G., A. F., M. F. G., and A. F. conducted some experiments, analyzed results, and edited the paper. R. J. P. conceived the idea for the project, designed most experiments and conducted experiments, analyzed results, and wrote the paper.
Supplementary Material
Acknowledgments
We thank Dean Dawson and Linda Thompson for invaluable encouragement, advice, and discussion.
This work was supported in whole or part by the National Institutes of Health Grant GM103636 and March of Dimes Grant FY14–256 (to R. J. P.). The authors declare that they have no conflicts of interest with the content of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. S1.
- DSB
- double-stand break
- BAF
- Brg1/Brahma-associated factors
- PBAF
- polybromo-associated BAF
- HDR
- homologous-directed repair
- qPCR
- quantitative PCR
- CRISPR
- clustered regularly interspaced short palindromic repeats
- Cas
- CRISPR-associated system
- RPA
- replication protein A
- IP
- immunoprecipitation.
References
- 1. Price B. D., and D'Andrea A. D. (2013) Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jeggo P. A., and Downs J. A. (2014) Roles of chromatin remodellers in DNA double strand break repair. Exp. Cell Res. 329, 69–77 [DOI] [PubMed] [Google Scholar]
- 3. Seeber A., Hauer M., and Gasser S. M. (2013) Nucleosome remodelers in double-strand break repair. Curr. Opin. Genet. Dev. 23, 174–184 [DOI] [PubMed] [Google Scholar]
- 4. Papamichos-Chronakis M., and Peterson C. L. (2013) Chromatin and the genome integrity network. Nat. Rev. Genet. 14, 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goodarzi A. A., Jeggo P., and Lobrich M. (2010) The influence of heterochromatin on DNA double strand break repair: getting the strong, silent type to relax. DNA Repair 9, 1273–1282 [DOI] [PubMed] [Google Scholar]
- 6. Vignali M., Hassan A. H., Neely K. E., and Workman J. L. (2000) ATP-dependent chromatin-remodeling complexes. Mol. Cell Biol. 20, 1899–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gerhold C. B., Hauer M. H., and Gasser S. M. (2015) INO80-C and SWR-C: guardians of the genome. J. Mol. Biol. 427, 637–651 [DOI] [PubMed] [Google Scholar]
- 8. House N. C., Koch M. R., and Freudenreich C. H. (2014) Chromatin modifications and DNA repair: beyond double-strand breaks. Front. Genet. 5, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brownlee P. M., Meisenberg C., and Downs J. A. (2015) The SWI/SNF chromatin remodelling complex: its role in maintaining genome stability and preventing tumourigenesis. DNA Repair 32, 127–133 [DOI] [PubMed] [Google Scholar]
- 10. Mohrmann L., and Verrijzer C. P. (2005) Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim. Biophys. Acta 1681, 59–73 [DOI] [PubMed] [Google Scholar]
- 11. Wang W. (2003) The SWI/SNF family of ATP-dependent chromatin remodelers: similar mechanisms for diverse functions. Curr. Top. Microbiol. Immunol. 274, 143–169 [DOI] [PubMed] [Google Scholar]
- 12. Choi K. Y., Yoo M., and Han J. H. (2015) Toward understanding the role of the neuron-specific BAF chromatin remodeling complex in memory formation. Exp. Mol. Med. 47, e155. [DOI] [PubMed] [Google Scholar]
- 13. Hargreaves D. C., and Crabtree G. R. (2011) ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kosho T., Miyake N., and Carey J. C. (2014) Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am. J. Med. Genet. C Semin. Med. Genet. 166C, 241–251 [DOI] [PubMed] [Google Scholar]
- 15. Narayanan R., and Tuoc T. C. (2014) Roles of chromatin remodeling BAF complex in neural differentiation and reprogramming. Cell Tissue Res. 356, 575–584 [DOI] [PubMed] [Google Scholar]
- 16. Reisman D., Glaros S., and Thompson E. A. (2009) The SWI/SNF complex and cancer. Oncogene 28, 1653–1668 [DOI] [PubMed] [Google Scholar]
- 17. Hodges C., Kirkland J. G., and Crabtree G. R. (2016) The many roles of BAF (mSWI/SNF) and PBAF complexes in cancer. Cold Spring Harb. Perspect. Med. 6, a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kadoch C., and Crabtree G. R. (2013) Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shain A. H., and Pollack J. R. (2013) The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 8, e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee H. S., Park J. H., Kim S. J., Kwon S. J., and Kwon J. (2010) A cooperative activation loop among SWI/SNF, γ-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 29, 1434–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kakarougkas A., Ismail A., Chambers A. L., Riballo E., Herbert A. D., Künzel J., Löbrich M., Jeggo P. A., and Downs J. A. (2014) Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 55, 723–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qi W., Wang R., Chen H., Wang X., Xiao T., Boldogh I., Ba X., Han L., and Zeng X. (2015) BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J. Cell Sci. 128, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Gu H., Lin H., and Chi T. (2012) Essential roles of the chromatin remodeling factor BRG1 in spermatogenesis in mice. Biol. Reprod. 86, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim Y., Fedoriw A. M., and Magnuson T. (2012) An essential role for a mammalian SWI/SNF chromatin-remodeling complex during male meiosis. Development 139, 1133–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H., Zhao R., Guo C., Jiang S., Yang J., Xu Y., Liu Y., Fan L., Xiong W., Ma J., Peng S., Zeng Z., Zhou Y., Li X., Li Z., Li X., Schmitt D. C., Tan M., Li G., and Zhou M. (2016) Knockout of BRD7 results in impaired spermatogenesis and male infertility. Sci. Rep. 6, 21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yan Z., Cui K., Murray D. M., Ling C., Xue Y., Gerstein A., Parsons R., Zhao K., and Wang W. (2005) PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 19, 1662–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu F., Flowers S., and Moran E. (2012) Essential role of ARID2 protein-containing SWI/SNF complex in tissue-specific gene expression. J. Biol. Chem. 287, 5033–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Easley R., Carpio L., Dannenberg L., Choi S., Alani D., Van Duyne R., Guendel I., Klase Z., Agbottah E., Kehn-Hall K., and Kashanchi F. (2010) Transcription through the HIV-1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology 405, 322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X., Azhar G., Zhong Y., and Wei J. Y. (2006) Zipzap/p200 is a novel zinc finger protein contributing to cardiac gene regulation. Biochem. Biophys. Res. Commun. 346, 794–801 [DOI] [PubMed] [Google Scholar]
- 30. He L., Tian X., Zhang H., Hu T., Huang X., Zhang L., Wang Z., and Zhou B. (2014) BAF200 is required for heart morphogenesis and coronary artery development. PLoS ONE 9, e109493, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Raab J. R., Resnick S., and Magnuson T. (2015) Genome-wide transcriptional regulation mediated by biochemically distinct SWI/SNF complexes. PLoS Genet. 11, e1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao H., Wang J., Han Y., Huang Z., Ying J., Bi X., Zhao J., Fang Y., Zhou H., Zhou J., Li Z., Zhang Y., Yang X., Yan T., Wang L., Torbenson M. S., and Cai J. (2011) ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget 2, 886–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zucman-Rossi J., Villanueva A., Nault J. C., and Llovet J. M. (2015) Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149, 1226–1239 e4 [DOI] [PubMed] [Google Scholar]
- 34. Gunn A., and Stark J. M. (2012) I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 920, 379–391 [DOI] [PubMed] [Google Scholar]
- 35. Courilleau C., Chailleux C., Jauneau A., Grimal F., Briois S., Boutet-Robinet E., Boudsocq F., Trouche D., and Canitrot Y. (2012) The chromatin remodeler p400 ATPase facilitates Rad51-mediated repair of DNA double-strand breaks. J. Cell Biol. 199, 1067–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aymard F., Bugler B., Schmidt C. K., Guillou E., Caron P., Briois S., Iacovoni J. S., Daburon V., Miller K. M., Jackson S. P., and Legube G. (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iacovoni J. S., Caron P., Lassadi I., Nicolas E., Massip L., Trouche D., and Legube G. (2010) High-resolution profiling of γH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 29, 1446–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brechalov A. V., Georgieva S. G., and Soshnikova N. V. (2014) Mammalian cells contain two functionally distinct PBAF complexes incorporating different isoforms of PHF10 signature subunit. Cell Cycle 13, 1970–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ryme J., Asp P., Böhm S., Cavellán E., and Farrants A. K. (2009) Variations in the composition of mammalian SWI/SNF chromatin remodelling complexes. J. Cell Biochem. 108, 565–576 [DOI] [PubMed] [Google Scholar]
- 40. Hohmann A. F., and Vakoc C. R. (2014) A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 30, 356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.