

Abstract
Bacterial community structure can be combined with observations of ecophysiological data to build predictive models of microbial ecosystem function. These models are useful for understanding how function might change in response to a changing environment. Here we use five spring–summer seasons of bacterial community structure and flow cytometry data from a productive coastal site along the western Antarctic Peninsula to construct models of bacterial production (BP), an ecosystem function that heterotrophic bacteria provide. Through a novel application of emergent self-organizing maps we identified eight recurrent modes in the structure of the bacterial community. A model that combined bacterial abundance, mode and the fraction of cells belonging to the high nucleic acid population (fHNA; R2=0.730, P<0.001) best described BP. Abrupt transitions between modes during the 2013–2014 spring–summer season corresponded to rapid shifts in fHNA. We conclude that parameterizing community structure data via segmentation can yield useful insights into microbial ecosystem function and ecosystem processes.
Introduction
Ecosystem functions are physicochemical processes carried out by specific functional groups of organisms. Changes to a specific ecosystem function (perhaps caused by a change in the abundance of a functional group) can drive change to the overall biological or chemical characteristics of an ecosystem, thus predicting ecosystem function is a useful way to evaluate ecosystem processes. Incorporation of dissolved organic matter (DOM) into new biomass through bacterial production (BP) is a critical ecosystem function of heterotrophic marine bacteria (here meaning heterotrophic members of the domains Bacteria and Archaea). Relatively higher rates of BP indicate that the bacterial community is assimilating a larger amount of DOM. This repacking of DOM into microbial biomass facilitates the microbial loop, wherein organic matter is recycled to the higher trophic levels via bacterivory (Azam and Graf, 1983).
The western Antarctic Peninsula (WAP) is one of the most rapidly warming regions on the planet (Steig et al., 2009). Changing oceanographic conditions along the WAP have produced highly variable sea ice conditions (Stammerjohn et al., 2008), leading to shifts in primary production and phytoplankton community composition (Montes-Hugo et al., 2009). Because of this variability and long-term climatic forcing, the WAP is an ideal natural laboratory in which to observe changes to BP and other microbial ecosystem functions.
High rates of BP accompany intense seasonal phytoplankton blooms along the coastal marine environment of the WAP (Bowman et al., 2016; Kim and Ducklow, 2016). Whereas the ratio of BP to PP is low in the WAP relative to the global ocean (Bowman et al., 2016), the uptake of 3H-leucine in productive coastal waters can exceed 100 pmol l−1 h−1 (Kim and Ducklow, 2016), values typical for eutrophic temperate marine systems (Cottrell and Kirchman, 2003). BP is highly variable across the WAP coastal marine environment and between years, and can be strongly or weakly correlated to primary production (PP; Ducklow et al., 2012). Though recent work has shown a strong time-lagged correlation between chlorophyll a concentration and BP within a single season (Luria et al., 2016), in general the variability of this relationship makes it difficult to predict BP 'from the bottom–up' using PP or chlorophyll concentration as a proxy for labile DOM supply.
Although BP has been extensively studied along the WAP, there have been relatively few studies of bacterial community structure or composition. The lack of historical data on bacterial community structure partially reflects past methodological limitations. The first publication to use high-throughput sequencing of the 16S rRNA gene to characterize microbial community structure in the region emerged only in 2012 (Ghiglione and Murray, 2012) with data generated as part of the International Census of Marine Microbes (Amaral-Zettler et al., 2010). This was followed by a three-domain study of microbial community structure in onshore and offshore areas of the WAP that compared a single winter sample against a series of samples taken in the austral summer (Luria et al., 2014). An analysis of succession across the spring–summer season was not undertaken until 2013 (Luria et al., 2016). These studies identified distinct winter and summer bacterial and archaeal populations (Ghiglione and Murray, 2012), and a pronounced difference between summer surface water and water below the photic zone, with the structure of the microbial community observed at depth more similar to the winter microbial community (Luria et al., 2014; Bowman and Ducklow, 2015). Because the winter and deep samples share the common feature of reduced light and reduced primary production, a reasonable hypothesis is that the heterotrophic bacterial community responds to 'bottom–up' controls, namely the availability of labile organic carbon from phytoplankton (Ducklow et al., 2011; Bowman et al., 2016; Kim and Ducklow, 2016).
Bacteriophage and grazing also exert a 'top–down' control on bacterial community structure (Jürgens and Matz, 2002; Suttle, 2007). Although top–down structuring of bacterial communities has not been reported for coastal Antarctica, heterotrophic microbial eukaryotes are common (Moorthi et al., 2009; Garzio et al., 2013; Luria et al., 2014), and marine viruses are hypothesized to have increased significance at higher latitudes (Brum et al., 2015). Because mixotrophic cryptophytes and other putative bacterivores may be increasing in abundance along the WAP (Moline et al., 2004), the importance of top–down controls on bacterial community structure and function may be changing as well.
The rapid rate of change in the marine environment—epitomized by the WAP—requires new methods to track changes in microbial community structure across time and space, and to link community structure with microbial ecosystem functions. Here we used five spring–summer seasons of 16S rRNA gene amplicon data from Arthur Harbor, a highly productive site on Anvers Island along the WAP, to identify patterns in the annual appearance of different marine bacterial assemblages. Through a novel application of emergent self-organizing maps (ESOMs; Kohonen, 2001), we reduced the dimensionality of the complex community structure data to a single 'mode' for each sample to which we attributed specific taxonomic and functional properties. Using mode in combination with flow cytometry data, indicative of bacterial abundance and physiological state, we were able to accurately predict both BP and cell-specific BP (per-cell BP), essential ecosystem functions of the heterotrophic bacterial community and individual bacterial cells, respectively. We were further able to use the flow cytometry data to identify distinct physiological transitions in the bacterial community that we interpret as shifts between bottom–up and top–down control states.
Materials and methods
16S rRNA gene sequence analysis
Samples were collected for the 2009–2010, 2010–2011, 2011–2012 and 2012–2013 spring–summer seasons from 10 m depth at Arthur Harbor Station B, located ~1 km from Palmer Station on Anvers Island, as described in the supplementary methods. Samples for the 2013–2014 spring–summer season were collected from the Palmer Station seawater intake (6 m depth) as described in the supplementary methods and Luria et al. (2016). In brief, DNA was extracted from 0.2 μm filters (with 3.0 μm pre-filtration) with the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) and the hypervariable V6 region of the 16S rRNA gene was sequenced on an Illumina Hi-Seq 1000. Quality-controlled 16S rRNA gene amplicon libraries were subsampled to 18 876 reads, the size of the smallest library, and community and metabolic structure was determined with the paprica v0.3.1 metabolic inference pipeline (Bowman and Ducklow, 2015). Paprica uses the pplacer (Matsen et al., 2010) phylogenetic placement software to place 16S rRNA gene reads on a reference tree created from 16S rRNA genes from all completed bacterial genomes. Here we use the term closest completed genome (CCG) to denote placement of a read to a terminal node on the paprica v0.3.1 reference tree, and closest estimated genome (CEG) to indicate placement to an internal node on the tree. CCGs are described by the complete taxonomic name of the strain corresponding to the terminal node while CEGs are described by the consensus taxonomy for all downstream nodes. We normalized the abundance of all CCGs and CEGs and their inferred enzymes and metabolic pathways according to their estimated 16S rRNA gene copy number. We took the mean of the relative abundances to average sample replicates.
The Kohonen package (Wehrens and Buydens, 2007) in R (R Core Team, 2014) was used to construct an ESOM based on the abundance of CCGs and CEGs. The ESOM represents sample similarity with topography; samples that are part of the same topographic feature are more similar. Our map space consisted of 5 × 5 circular nodes in a hexagonal, non-toroidal configuration. Each node is associated with a vector of sample properties called a codebook vector; here the sample properties were relative abundance. Clusters of nodes were identified using k-means clustering, with a reasonable value for k chosen through the evaluation of a scree plot of within-clusters sum of squares and the SIMPROF test in the R clustsig package (Whitaker and Christman, 2014). Hereafter, we refer to these clusters as 'modes' in community structure. To identify which CCG and CEG contributed the most to variance between ESOM nodes, likely to also be the top contributors to variance between modes, we applied a principal components analysis to the codebook vectors. Although the distance between nodes in a space defined by the first two principal components is not directly analogous to node topography in the ESOM, these interpretations of similarity are complimentary. An identical procedure was used to segment samples into functional modes according to the relative abundance of predicted metabolic pathways associated with all CCGs and CEGs. An exception was in the calculation of relative abundance; for each metabolic pathway relative abundance was calculated as a fraction of the most abundant pathway present in each sample.
A MIMARKS-compliant table of all 16S rRNA gene amplicon samples is provided as Supplementary Table S1. All 16S rRNA gene sequence data are available from the NCBI SRA at SRP091049 (2013–2014) and SUB2014638 (all other samples). The ESOMs and code for our analysis can be found at https://github.com/bowmanjeffs/palmer_timeseries.
Community succession and persistence
To determine whether some modes have a tendency to follow other modes (succession), and whether certain modes have a tendency to persist (persistence), we evaluated the frequency of transitions between modes. A simulation was used to assess the significance of the observed frequencies. In simulation the modes corresponding to the 16S rRNA gene samples were repeatedly randomized and the transitions tallied. Transitions in the randomized data set occurring more or less often than observed in the original data set were noted. Transitions in the 95th percentile after 100 iterations (that is that occurred more often in the original data than the random data in at least 95 iterations) were deemed statistically significant.
Identification of HNA and LNA bacteria
Flow cytometry samples were collected and processed as described in Luria et al. (2016). To identify clusters of high (HNA) and low nucleic acid (LNA) bacteria for samples stained with SYBR Green I (Invitrogen, Carlsbad, CA, USA), all flow cytometry events from the 2013–2014 austral spring–summer season with a fluorescence intensity >3500 relative fluorescence unit at 533/30 nm after excitation with a 488 nm laser were concatenated, and a training set of 50 000 events was selected at random. The training set was used to build an ESOM in a manner similar to that described for the community structure data, except that the map space consisted of 76 × 76 nodes. All available parameters were used to construct the map (forward scatter, sidescatter, FL1; 533/30 nm, FL2; 585/40 nm, FL3; >670 nm, FL4; 675/25 nm) after log transformation of signal height. K-means clustering was again used to identify clusters of nodes. The resulting model was used to classify the complete data set (2010–2014) into the identified clusters. The fraction of cells belonging to the HNA population was calculated by dividing the number of HNA cells by the total number of cells, excluding presumed non-cellular material. Raw Flow Cytometry Standard-format files are available on the Palmer LTER DataZoo (Palmer LTER Science Team).
Flow cytometry and BP data were not always taken on the same day as samples for 16S rRNA analysis. To allow a comparison of flow cytometry, BP and 16S rRNA gene data, the flow cytometry and BP data were extended by linear interpolation with the R package zoo (Zeileis and Grothendieck, 2005). For each season interpolation was limited to the period during which data were collected (no extrapolation was made before or after the sample period). For the 2013–2014 season, the abundance and fraction of cells belonging to the high nucleic acid population (fHNA) anomalies were calculated by dividing the difference between each time point (x) and the seasonal mean (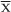 ) by the seasonal mean.
) by the seasonal mean.
The flow cytometry and community structure data were used to estimate the absolute abundance of CEG or CCG of interest by multiplying relative abundance by total bacterial abundance. Because our community structure data do not include members of the domain Archaea, this calculation does not represent the true absolute abundance. Nonetheless it provides an useful estimate, particularly during the summer months when archaeal abundance is expected to be low (Church et al., 2003; Ghiglione and Murray, 2012).
Linear models of BP
BP data for all seasons were obtained from the Palmer LTER DataZoo (Palmer LTER Science Team) BP was determined by the uptake of 3H-leucine; the data and methods are described in previous work (Ducklow et al., 2012; Kim and Ducklow, 2016; Luria et al., 2016). Stepwise linear regression was used to evaluate which combination of parameters best described the observed rate of BP. The available parameters were mode, functional mode, fHNA and bacterial abundance. Linear models were constructed using the lm command in R (R Core Team, 2014), and nested models were compared by analysis of variance and relative likelihood based on Akaike's information criterion according to Equation 2:
A relative likelihood below 0.05 (that is a model <0.05 times as likely to minimize the information loss as the best scoring model) was considered statistically significant. Because many of the parameters used in the model were correlated, which can lower the predictive power of linear models, variance-inflation factors were calculated with the vif command implemented in the R package car (Fox and Weisberg, 2011).
Results
Bacterial abundance and fHNA
We identified four clusters of flow cytometry events based on inspection of the within-clusters sum of squares scree plot (Supplementary Figure S1a). Three of the clusters were largely defined by their fluorescent signal at 533 nm (Supplementary Figure S1b–d) and interpreted as an HNA population, a middle nucleic acid population and a LNA population. A fourth population that varied widely in fluorescence at 533 nm was assumed to be non-cellular. Bacterial abundance (defined as all SYBR Green I-stained events above our intensity threshold) and fHNA varied widely across all time points. For the 2013–2014 season bacterial abundance ranged from 1.1 × 105 to 1.7 × 106 bacteria ml−1 (Figure 1a, mean=7.7 × 105 bacteria ml−1, s.d.=4.0 × 105 bacteria ml−1), whereas fHNA ranged from 0.04 to 0.75 (Figure 1b, mean=0.36, s.d.=0.16). For the 2013–2014 season the difference in abundance and fHNA anomalies was consistently negative prior to 23 December 2013 and largely positive after that date (Figure 1c). During the 2013–2014 season the absolute abundance of Candidatus Pelagibacter ubique HTCC1062, the most abundant CCG associated with the Candidatus Pelagibacter genus, increased during the spring bloom from 7.2 × 104 bacteria ml−1 on 26 October 2013 to 3.35 × 105 bacteria ml−1 on 18 March 2014 (Figure 1a).
Figure 1.

Bacterial abundance, the fraction of high nucleic acid bacteria (fHNA) and difference in anomalies (Δ anomaly) for these measures for the 2013–2014 spring/summer season. (a) Bacterial abundance, including the estimated absolute abundance of Candidatus Pelagibacter ubique HTCC1062, Dokdonia sp. MED134 and Rhodobacteraceae. (b) The fraction of cells belonging to the high nucleic acid population (fHNA). (c) The difference between the fHNA and bacterial abundance anomalies (Δ anomaly), with the black lines giving the measurement time points.
Mode and functional mode
Based on inspection of the within-cluster sum of squares plot, we identified eight modes based on the relative abundance of CCGs and CEGs (Supplementary Figure S2a) and eight modes based on inferred metabolic pathways (not shown). Although SIMPROF analysis of both ESOMs identified 11 significant hierarchical clusters, we selected the lower number for k-means clustering to be conservative, and to increase the number of samples contained within each cluster. In the principal components analysis of node codebook vectors, the nodes were largely segregated by mode in a space defined by PC1 and PC2 (Supplementary Figure S2b). The top seven eigenvectors (that is, CCG or CEG) by magnitude in PC1 and PC2 together accounted for 71.4% of PC1 and 69.3% of PC2. Further eigenvectors contributed comparatively little to PC1 or PC2. By order of magnitude in PC1 and PC2 the top eigenvectors corresponded to Candidatus P. ubique HTCC1062, Dokdonia MED134, Rhodobacteraceae, Candidatus Thioglobus singularis PS1, Owenweeksia hongkongensis DSM 17368, Planktomarina temperata RCA23 and Leisingera methylohalidivorans DSM 14336. It is important to note that these associations are to the matching CCG available in GenBank at the time the paprica v0.3.1 database was constructed (6 June 2016), or to the CEGs in the paprica v0.3.1 database, and should not be taken as a definitive classification at the strain level.
The relative abundances of the taxa associated with the top eigenvectors in the principal components analysis (Supplementary Figure S2b) largely defined the modes in community structure (Figure 2). Candidatus P. ubique HTCC1062 was most abundant in mode 6 and also abundant in mode 8 (Figure 2d). D. Dokdonia MED134 was most abundant in mode 7, and was often abundant in modes 3 and 5 (Figure 2e). Rhodobacteracea, the only CEG among the major contributors to variance, was most abundant in mode 5 but was also abundant in modes 2 and 3 (Figure 2f). Candidatus T. singularis PS1 was most abundant in mode 1, and was also abundant in modes 4, 7 and 8 (Figure 2g). O. hongkongensis DSM 17368 was largely restricted to mode 2 (Figure 2h). P. temperata RCA23 was most abundant in mode 4 and was also abundant in modes 4 and 6 (Figure 2i). L. methylohalidivorans was most abundant in mode 5 (not shown).
Figure 2.

Properties of the emergent self-organizing map (ESOM) nodes for community structure. (a) Nodes colored and numbered according to mode membership (colors correspond to the modes in Figures 6 and 7). Note that owing to the toroidal configuration of the map not all modes are contiguous. (b) Location of objects (samples) within the map. Samples located close to a node center have a relative abundance similar to that which defines the codebook vector for that node. Samples located further from the center have a relative abundance that is less similar. (c) Mean distance between sample relative abundances and the node codebook vector. Higher mean distance indicates that the samples contained in the node are less well defined by that node. (d–i) Relative abundance of the indicated closest completed genome (CCG) or closest estimated genome (CEG) in the node code vector. For all panels the thick black lines define the cluster (mode) boundaries. A full color version of this figure is available at the ISME journal online.
When classified by mode, samples showed distinct trends in estimated genomic character including mean genome length and 16S rRNA gene copy number (Figure 3). Mode 7 samples (n=7) had the largest mean genome size (mean=2.92 × 106, s.d.=2.22 × 105) and greatest mean number of 16S rRNA gene copies (mean=2.47, s.d.=0.43). Mode 8 samples (n=18) had the smallest mean genome size (mean=2.19 × 106, s.d.=1.28 × 105) and lowest mean 16S rRNA gene copy number (mean=1.56, s.d.=0.12).
Figure 3.

Predicted genomic characteristics for modes. (a) Mean 16S rRNA gene copy number for all samples assigned to each mode. (b) Mean genome length for all samples assigned to each mode. (c) 16S rRNA gene copy number as a function of genome length (R2=0.73, P<<1 × 10−5), points are shaded according to GC content. (d) Presence or absence of metabolic pathways and degradative metabolic pathways for select CEGs and CCGs.
Model predictions of BP
Of the 75 16S V6 rRNA gene datasets available to define the modes in community structure and function, 43 were taken during the time period covered by the seasonal interpolation of BP and flow cytometry data (Figure 4). Overall, mode accounted for 46.3% of the variance in BP and 70.9% of the variance in cell-specific BP (Table 1, Figures 5a and b). Functional mode was not correlated with BP, but accounted for 53.5% of the variance in cell-specific BP. The categorical predictors of mode and functional mode were themselves highly correlated according to the χ2-test (χ2=85.27, P=0.0010). The stepwise addition of other predictors to the mode models for BP and cell-specific BP generally improved both models, with bacterial abundance, mode and fHNA defining the best scoring model for both (accounting for 73.0% of the variance in BP and 76.4% of the variance in cell-specific BP), however, for cell-specific BP this was not a significant improvement in model fit over abundance and mode alone (Table 1). Substituting functional mode for mode accounted for a slightly lesser degree of variance in BP models (62.5% and 62.1%, respectively).
Figure 4.

Concurrence of flow cytometry (FCM) and 16S rRNA gene sample data with bacterial production (BP). Each point indicates a single 16S rRNA gene sample, early and late season samples typically did not have flow cytometry data available (white points).
Table 1. Results for stepwise regression of BP and cell-specific BP.
| Model | R2 BP | Relative likelihood based on AIC | R2 cell-specific BP | Relative likelihood based on AIC | VIF |
|---|---|---|---|---|---|
| Functional mode | 0.155 | 4.6 × 10−11 | 0.535*** | 1.0 × 10−6 | NA |
| Mode | 0.463*** | 8.1 × 10−7 | 0.709*** | 0.024 | NA |
| fHNA | 0.360* | 2.5 × 10−7 | −0.008 | 8.2 × 10−13 | NA |
| Mode+fHNA | 0.508*** | 3.6 × 10−6 | 0.725*** | 0.054 | 1.98 |
| Abundance | 0.506*** | 6.5 × 10−5 | 0.032 | 1.9 × 10−12 | NA |
| Abundance+mode | 0.670*** | 0.019 | 0.748*** | 0.38 | 1.46 |
| Abundance+mode+fHNA | 0.730*** | 1.0 | 0.764*** | 1.0 | 1.54, 2.58, 2.09 |
Abbreviations: AIC, Akaike's information criterion; BP, bacterial production; fHNA, fraction of cells belonging to the high nucleic acid population; VIF, variance-inflation factors. ***P<0.001, **P<0.01, *P⩽0.05.
Figure 5.

Visualization of models for bacterial production (BP) and cell-specific BP. (a) BP as a function of mode. (b) Cell-specific BP as a function of mode. (c) BP as a function of the fraction of cells belonging to the high nucleic acid population (fHNA). (d) Cell-specific BP as a function of fHNA. (e) BP as a function of bacterial abundance. (f) Cell-specific BP as a function of bacterial abundance.
Mode succession and persistence
Only the transition from mode 1 to mode 8 met our criteria for a statistically significant succession (Figure 6b). Although this transition occurred only four times in our data set (Figure 6a), this number was exceeded in only 1 of 100 random rearrangements of the modes. Low BP characterized modes 1 and 8 (Figure 5a), and the transition between these modes always occurred during either the onset or collapse of the seasonal peak in BP (Figure 4). Mode 1 had a relatively high abundance of Candidatus T. singularis (Figure 2g), whereas mode 8 had high proportions of Candidatus P. ubique HTCC1062 (Figure 2b). Consistent with the low BP and timing of modes 1 and 8, both modes have low 16S rRNA gene copy number (Figure 3a) and small, low-GC content genomes (Figures 3b and c). Except for mode 1, all of the modes exhibited significant persistence, with mode 8 being the most common and also the most frequently recurring mode (Figure 6).
Figure 6.
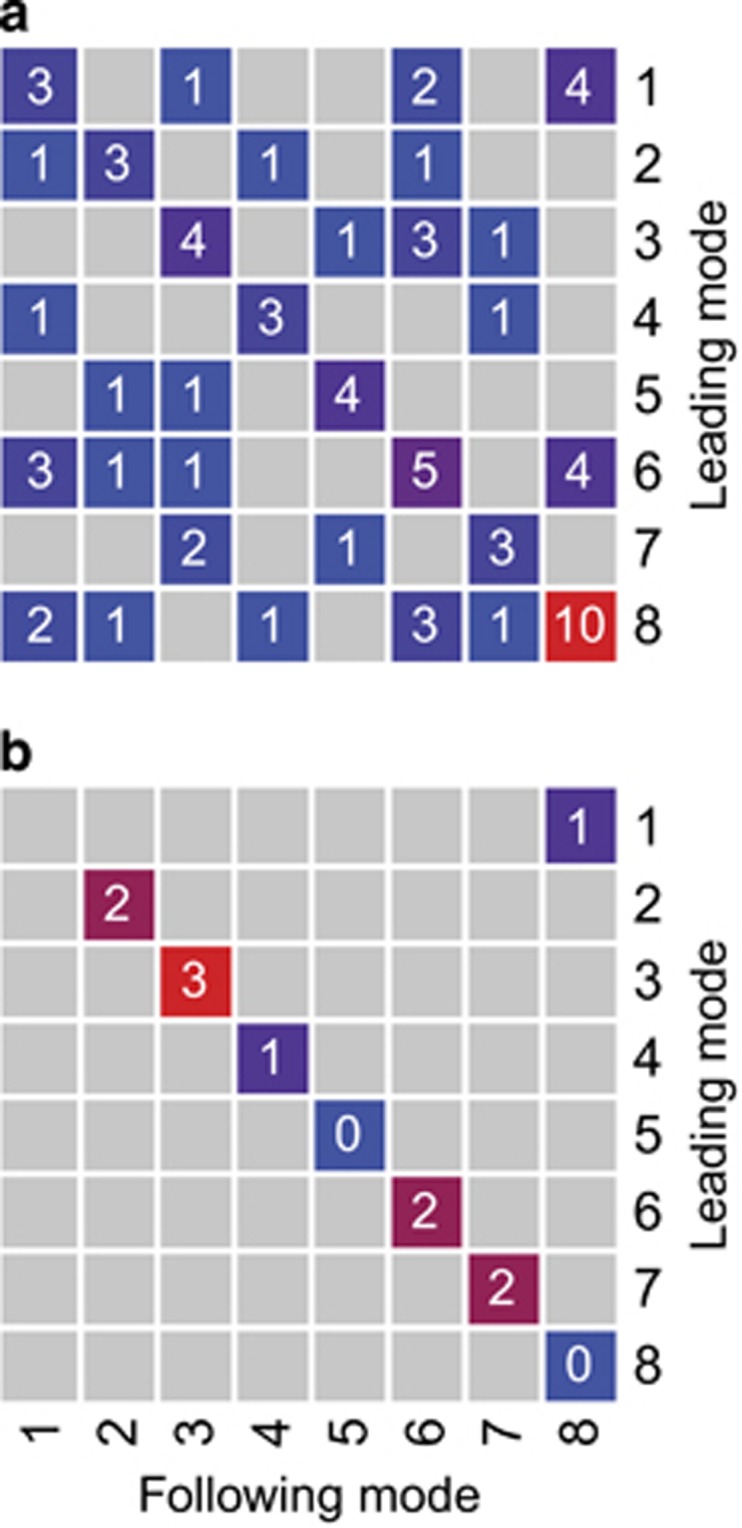
Mode persistence and succession. (a) The number of occurrences of each possible transition. Gray squares without a number indicate that the transition was not observed. Squares are colored according to the value noted inside each square. (b) Statistically significant transitions. The value inside each square gives the number of times the number of transitions given in Figure 6a was exceeded in 100 random rearrangements of the modes. Only values below five are shown, squares are colored according to value. A full color version of this figure is available at the ISME journal online.
Discussion
In this study BP was best described by a model of fHNA, bacterial abundance and mode. The improvement in model fit resulting from the addition of fHNA is consistent with previous studies in temperate regions (Table 1). In a temperate estuary in Waquoit Bay, MA, USA, Morán et al. (2011) observed a correlation between HNA and LNA abundance and BP (R2=0.41 and 0.49, respectively). In a complimentary study in seasonally oligotrophic waters off the Iberian Peninsula, Morán et al. (2007) also found a significant correlation between HNA cell abundance and BP, albeit with a modest amount of variance explained (R2=0.16). For our coastal setting along the WAP, and using strictly objective criteria for fHNA, we observed a correlation between fHNA and BP close to that observed for Waquoit Bay (R2=0.36, Table 1). Because the chlorophyll a concentrations and BP values during the spring bloom were much higher for our study area than for the oligotrophic study area of Morán et al. (2007), the higher correlation between BP and fHNA may reflect proportionally greater carbon utilization by the HNA population. Thus, although HNA abundance is a poor predictor of BP in the oligotrophic ocean where LNA cells are major contributors to BP, it may be a better predictor of BP in high-biomass coastal marine settings where HNA cells are the major contributors.
HNA and LNA populations are thought to be largely a function of taxonomy as opposed to activity (Sherr et al., 2006; Vila-Costa et al., 2012; Morán et al., 2015), although this is based on a limited number of studies largely from temperate regions. In our study mode was highly correlated with fHNA (R2=0.52, P=1.63 × 10−5) and some fHNA minima or maxima corresponded to shifts in community structure (Figure 7), suggesting a link between taxonomy and flow cytometric population. Despite this a considerable fraction of variance in fHNA could not be accounted for by mode. Because the addition of fHNA to our model for BP significantly improved the model fit (Table 1), we suggest that for the coastal WAP, fHNA to some extent reflects community physiology. This idea does not necessarily contradict the findings of Vila-Costa et al. (2012), who noted that some Gammaproteobacteria were found in both the HNA and LNA populations. The putative links between fHNA and community physiology may be in part due to our ability to distinguish between HNA and middle nucleic acid bacteria, a distinction not taken into account by previous environmental BP studies. Under different growth conditions (that is, carbon limited and carbon replete) a community of a fixed composition should express a different fluorescent signal. Thus, although the LNA population might consist of bacteria with small genomes, the HNA and middle nucleic acid populations might consist of bacteria with comparatively larger genomes in more or less active states, respectively.
Figure 7.

Bacterial community dynamics during the 2013–2014 spring–summer season. (a) The fraction of cells belonging to the high nucleic acid population (fHNA). (b) Flow-cytogram of green fluorescence (533 nm) and sidescatter for 10 January 2014. (c) Flow-cytogram of green fluorescence (533 nm) and sidescatter for 23 January 2014. (d) Flow-cytogram of green fluorescence (533 nm) and sidescatter for 18 February 2014. (e) Relative abundance of the 30 most abundant closest completed genomes (CCGs) and closest estimated genomes (CEGs). The mode assigned to each sample is given by the color bar at the top of the heatmap. A full color version of this figure is available at the ISME journal online.
Interestingly, taxonomic mode was a better predictor of BP and cell-specific BP than functional mode (Table 1). Although metabolic structure is well correlated with community structure for WAP marine bacterial communities, potential function is much better conserved across communities than taxonomy (Bowman and Ducklow, 2015). We suggest that the high correlation between mode and BP observed in this study results from the more resolved view of structure provided by taxonomy; ecologically meaningful shifts in community structure may be more readily observed by changes in taxonomy than by changes in functional composition (Bowman and Ducklow, 2015). There are several possible explanations for this, including the greater conservation of potential function over taxonomy between communities, disparity between potential function and gene expression patterns (expressed function) for functionally similar but taxonomically distinct communities, and our inability to identify key rare enzymes and/or assign them to taxa during metabolic inference.
Grazing by bacterivorous eukaryotes is thought to preferentially impact the HNA population (Gonzalez et al., 1990; del Giorgio et al., 1996; Jürgens and Matz, 2002). This process, confirmed for the WAP in limited studies (Garzio et al., 2013), impacts BP directly by removing the fastest growing members of the bacterial community, and can correspond to a permanent shift in bacterial community structure (Figure 7). Similarly, through the 'kill the winner' model, marine viruses should disproportionately impact the fastest growing bacteria (Fuhrman and Suttle, 1993). In contrast to these top–down controls on BP, phytoplankton biomass serves as a bottom–up control (Bowman et al., 2016; Kim and Ducklow, 2016). To assess time periods when one mode of control might dominate over the other, we looked at the difference in the seasonal anomalies of fHNA and bacterial abundance (Δanomaly: abundance anomaly−fHNA anomaly) across the 2013–2014 season (Figure 1c). Despite the low abundance of HNA bacteria early in the season, the fHNA anomaly is higher than the abundance anomaly prior to 23 December 2013 (Figure 1c). After 23 December the fHNA anomaly is generally lower than the abundance anomaly, except for distinct time points where abundance decreases without a concurrent decrease in fHNA. We hypothesize that these negative and positive Δ anomaly phases indicate periods of bottom–up and top–down control, respectively.
For heterotrophic marine bacteria bottom–up and top–down controls result from the availability of DOM and predator and phage abundance, respectively. Consistent with the dominance of mode 8 by Candidatus Pelagibacter and Candidatus T. singularis PS1 (Figure 2), early in the season carbon is limiting and there may be insufficient grazers present to impose top–down control on HNA bacteria. At this time most phage are expected to be in a lysogenic phase (Brum et al., 2015). Following the onset of the spring phytoplankton bloom the availability of labile DOM becomes less important as a structuring mechanism, whereas grazing or viral events preferentially reduce fHNA. After each putative mortality event we anticipate a brief release from grazing pressure resulting from rapid prey evolution (Yoshida et al., 2007; Hiltunen and Becks, 2014) or phage or predator mortality, and a brief return to bottom–up control.
Several ecologically important points are implicit in this hypothesis. First, to achieve a positive Δ anomaly during the phytoplankton bloom the LNA population, which may be composed primarily of the small-genomed, low 16S rRNA gene copy number taxa associated with modes 1 and 8, must be increasing in abundance. This idea is consistent with the correlation-based network analysis of Luria et al. (2014) who found a strong association between SAR11 and a diatom operational taxonomic unit in summer samples from the coastal WAP, and Delmont et al. (2014) who observed a high abundance of Candidatus Pelagibacter (the predominant genus of the SAR11 clade) in association with a bloom of Phaeocystis in the Ross Sea. Although Candidatus Pelagibacter is often thought of as an oligotrophic specialist owing to its high relative abundance in the oligotrophic ocean, it is more accurately considered as a planktonic specialist that nonetheless prefers bloom conditions. Its dominance during non-bloom periods arises from its ability to persist under relatively low concentrations of bulk DOC, and potentially its lack of appeal to eukaryotic grazers and viruses owing to its small size and slow growth rate. Its absolute abundance decreases during bloom conditions as a result of the faster growth rate of motile, copiotrophic bacteria. Because those bacteria occupy a different ecological niche however, there is no competitive exclusion.
The ecological compatibility between Candidatus Pelagibacter and the phytoplankton bloom can be seen in the estimated absolute abundance of Candidatus P. ubique HTCC1062 during the 2013–2014 spring–summer season. The abundance of this CCG peaked on 6 January 2014 at 3.72 × 105 bacteria ml−1 at a time when the concentration of chlorophyll a was rapidly increasing and shortly after the Δ anomaly had transitioned to a positive phase (Figure 1c). Because Candidatus P. ubique HTCC1062 (and other non-motile heterotrophic marine bacteria) utilizes dissolved photosynthate as substrate for growth (Howard et al., 2006; Tripp et al., 2008; Sun et al., 2011), we do not find this result surprising.
Second, because several important shifts in bacterial community structure during the 2013–2014 spring–summer season corresponded to a strong positive Δ anomaly, top–down controls may be the major driver of bacterial community succession once the phytoplankton bloom is established. This idea follows concepts of community succession well established for phytoplankton (Sommer, 1989), and is supported by the inconsistent pattern of succession we observed between seasons (Figures 4 and 6). Rather than a specific functional progression driven by the availability of distinct substrates, this suggests an opportunistic 'takeover' by those bacterial taxa best able to fill the open niche. Although previous in situ work on bacterial community succession has largely focused on bottom–up controls (for example, Teeling et al., 2012; Luria et al., 2016), those findings do not contradict our assertion that top–down control can initiate community transitions. As grazing (or phage propagation) decreases the most rapidly growing members of the bacterial community, it opens new niches defined by substrate availability.
Our 5-year time series of bacterial community structure during the spring–summer season is the first multi-year study of bacterial community structure and dynamics for coastal Antarctica. Although the currently available data are sparse, with only 75 samples collected across 5 years, this study emphasizes the value of repeated, long-term data collection on microbial community structure and function. By using ESOMs to segment the microbial community into temporally discrete units we can more easily compare data sets that are discontinuous in time and space. Using the existing ESOM, future data can be classified as they become available or new, potentially more comprehensive maps can be constructed and applied retroactively to past data to create a consistent segmentation. In this way we can observe changes to the timing of critical transitions in microbial community structure, their possible mechanisms and functional implications.
Acknowledgments
We thank Sean O'Neill, Monica Stegman, Madie Willis and Sharon Grimm for help with sample collection during the 2013–2014 field season at Palmer Station. Naomi Shelton, Jamie Collins and Sebastian Vivancos assisted in the collection and analysis of samples for BP and flow cytometry. Annaliese Jones, Leslie Murphy and Beth Slikas assisted with genomic DNA extractions, library construction and next-generation sequencing. We would also like to thank the staff of Palmer Station and the crew of the ARSV Laurence M. Gould for their assistance. LAZ was funded under NSF OPP-1142114. JR was funded by NSF OPP-1141993. HWD was funded by NSF PLR-1440435. JB was funded by a Lamont-Doherty postdoctoral fellowship, a grant by the World Surf League PURE Ocean Health Initiative to HWD and NSF PLR-1641019. CL received funding as part of NSF OPP-1141993 to JR, as well as a Brown University Nicole Rosenthal Hartnett '91 Fellowship and Brown University-MBL Joint Graduate Program Tisch Funding.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
The authors declare no conflict of interest.
Supplementary Material
References
- Amaral-Zettler L, Artigas LF, Baross J, Bharathi L, Boetius A, Chandramohan D et al. (2010) A global census of marine microbes. In: Life in the World's Oceans: Diversity, Distribution and Abundance. Blackwell Publishing Ltd.: Oxford, pp 223–245. [Google Scholar]
- Azam F, Graf JS. (1983). The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 10: 257–263. [Google Scholar]
- Bowman JS, Ducklow HW. (2015). Microbial communities can be described by metabolic structure: a general framework and application to a seasonally variable, depth-stratified microbial community from the coastal West Antarctic Peninsula. PLoS One 10: e0135868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman JS, Vick-Majors TJ, Morgan-Kiss RM, Takacs-Vesbach C, Ducklow HW, Priscu JC. (2016). Microbial community dynamics in two polar extremes: the lakes of the McMurdo dry valleys and the West Antarctic Peninsula marine ecosystem. Bioscience 66: 830–847. [Google Scholar]
- Brum JR, Hurwitz BL, Schofield O, Ducklow HW, Sullivan MB. (2015). Seasonal time bombs: dominant temperate viruses affect Southern Ocean microbial dynamics. ISME J 10: 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church MJ, DeLong EF, Ducklow HW, Karner MB, Preston CM, Karl DM. (2003). Abundance and distribution of planktonic Archaea and Bacteria in the waters west of the Antarctic Peninsula. Limnol Oceanogr 48: 1893–1902. [Google Scholar]
- Cottrell MT, Kirchman DL. (2003). Contribution of major bacterial groups to bacterial biomass production (thymidine and leucine incorporation) in the Delaware estuary. Limnol Oceanogr 48: 168–178. [Google Scholar]
- Delmont TO, Hammar KM, Ducklow HW, Yager PL, Post AF. (2014). Phaeocystis antarctica blooms strongly influence bacterial community structures in the Amundsen Sea polynya. Front Microbiol 5: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducklow H, Schofield O, Vernet M, Stammerjohn S, Erickson M. (2012). Multiscale control of bacterial production by phytoplankton dynamics and sea ice along the western Antarctic Peninsula: a regional and decadal investigation. J Mar Syst 98–99: 26–39. [Google Scholar]
- Ducklow HW, Myers KMS, Erickson M, Ghiglione J, Murray AE. (2011). Response of a summertime Antarctic marine bacterial community to glucose and ammonium enrichment. Aquat Microb Ecol 64: 205–220. [Google Scholar]
- Fox J, Weisberg S. (2011) An R Companion to Applied Regression, 2nd edn. Sage: Thousand Oaks. [Google Scholar]
- Fuhrman JA, Suttle CA. (1993). Viruses in marine planktonic systems. Oceanography 6: 51–63. [Google Scholar]
- Garzio LM, Steinberg DK, Erickson M, Ducklow HW. (2013). Microzooplankton grazing along the Western Antarctic Peninsula. Aquat Microb Ecol 70: 215–232. [Google Scholar]
- Ghiglione JF, Murray AE. (2012). Pronounced summer to winter differences and higher wintertime richness in coastal Antarctic marine bacterioplankton. Environ Microbiol 14: 617–629. [DOI] [PubMed] [Google Scholar]
- del Giorgio P, Gasol JM, Vaqué D, Mura P, Agustí S, Duarte CM. (1996). Bacterioplankton community structure: Protists control net production and the proportion of active bacteria in a coastal marine community. Limnol Oceanogr 41: 1169–1179. [Google Scholar]
- Gonzalez JM, Sherr EB, Sherr BF. (1990). Size-selective grazing on bacteria by natural assemblages of estuarine flagellates and ciliates. Appl Environ Microb 56: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen T, Becks L. (2014). Consumer co-evolution as an important component of the eco-evolutionary feedback. Nat Commun 5: 5226. [DOI] [PubMed] [Google Scholar]
- Howard EC, Henriksen JR, Buchan A, Reisch CR, Bürgmann H, Welsh R et al. (2006). Bacterial taxa that limit sulfur flux from the ocean. Science 314: 649–652. [DOI] [PubMed] [Google Scholar]
- Jürgens K, Matz C. (2002). Predation as a shaping force for the phenotypic and genotypic composition of planktonic bacteria. Antonie van Leeuwenhoek 81: 413–434. [DOI] [PubMed] [Google Scholar]
- Kim H, Ducklow HW. (2016). A decadal (2002-2014) analysis of dynamics of heterotrophic bacteria in an Antarctic coastal ecosystem: variability and physical and biogeochemical forcing. Front Mar Sci 3: 214. [Google Scholar]
- Kohonen T. (2001) Self-Organzing Maps, 3rd edn. Springer: Berlin. [Google Scholar]
- Luria C, Amaral-Zettler L, Ducklow H, Rich J. (2016). Seasonal succession of free-living bacterial communities in coastal waters of the western Antarctic Peninsula. Front Microbiol 7: 1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria C, Ducklow H, Amaral-Zettler L. (2014). Marine Bacterial, Archaeal and Eukaryotic diversity and community structure on the continental shelf of the western Antarctic Peninsula. Aquat Microb Ecol 73: 107–121. [Google Scholar]
- Matsen FA, Kodner RB, Armbrust EV. (2010). pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moline MA, Claustre H, Frazer TK, Schofield O, Vernet M. (2004). Alteration of the food web along the Antarctic Peninsula in response to a regional warming trend. Glob Chang Biol 10: 1973–1980. [Google Scholar]
- Montes-Hugo M, Doney SC, Ducklow HW, Fraser W, Martinson D, Stammerjohn SE et al. (2009). Recent changes in phytoplankton communities associated with rapid regional climate change along the Western Antarctic Peninsula. Science 323: 1470–1473. [DOI] [PubMed] [Google Scholar]
- Moorthi S, Caron DA, Gast RJ, Sanders RW. (2009). Mixotrophy: A widespread and important ecological strategy for planktonic and sea-ice nanoflagellates in the Ross Sea, Antarctica. Aquat Microb Ecol 54: 269–277. [Google Scholar]
- Morán AG, Alonso-Saez L, Nogueira E, Ducklow HW, Gonzalez N, Lopez-Urruntia A et al. (2015). More, smaller bacteria in response to ocean's warming? Proc Biol Sci 282: pii: 20150371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morán XAG, Bode A, Suárez LÁ, Nogueira E. (2007). Assessing the relevance of nucleic acid content as an indicator of marine bacterial activity. Aquat Microb Ecol 46: 141–152. [Google Scholar]
- Morán XAG, Ducklow HW, Erickson M. (2011). Single-cell physiological structure and growth rates of heterotrophic bacteria in a temperate estuary (Waquoit Bay, Massachusetts). Limnol Oceanogr 56: 37–48. [Google Scholar]
- Palmer LTER Science Team. Palmer LTER Datazoo http://pal.lternet.edu/data.
- R Core Team.. (2014). R: A language and environment for statistical computing http://www.r-project.org/ (Accessed 1 December 2012).
- Sherr EB, Sherr BF, Longnecker K. (2006). Distribution of bacterial abundance and cell-specific nucleic acid content in the Northeast Pacific Ocean. Deep Res Part I Oceanogr Res Pap 53: 713–725. [Google Scholar]
- Sommer U. (1989). The role of competition for resources in phytoplankton succession. Plankton Ecol pp 57–106.
- Stammerjohn SE, Martinson DG, Smith RC, Yuan X, Rind D. (2008). Trends in Antarctic annual sea ice retreat and advance and their relation to El Niño–Southern Oscillation and Southern Annular Mode variability. J Geophys Res 113: 1–20. [Google Scholar]
- Steig EJ, Schneider DP, Rutherford SD, Mann ME, Comiso JC, Shindell DT. (2009). Warming of the Antarctic ice-sheet surface since the 1957 International Geophysical Year. Nature 457: 459–462. [DOI] [PubMed] [Google Scholar]
- Sun J, Steindler L, Thrash JC, Halsey KH, Smith DP, Carter AE et al. (2011). One carbon metabolism in SAR11 pelagic marine bacteria. PLoS One 6: e23973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA. (2007). Marine viruses—major players in the global ecosystem. Nat Rev Microbiol 5: 801–812. [DOI] [PubMed] [Google Scholar]
- Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336: 608–611. [DOI] [PubMed] [Google Scholar]
- Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ. (2008). SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452: 741–744. [DOI] [PubMed] [Google Scholar]
- Vila-Costa M, Gasol JM, Sharma S, Moran MA. (2012). Community analysis of high- and low-nucleic acid-containing bacteria in NW Mediterranean coastal waters using 16S rDNA pyrosequencing. Environ Microbiol 14: 1390–1402. [DOI] [PubMed] [Google Scholar]
- Wehrens R, Buydens LMC. (2007). Self- and super-organizing maps in R: the kohonen package. J Stat Softw 21. [Google Scholar]
- Whitaker D, Christman M. (2014). clustsig: Significant Cluster Analysis. http://cran.r-project.org/package=clustsig (accessed 1 March 2016).
- Yoshida T, Ellner SP, Jones LE, Bohannan BJM, Lenski RE, Hairston NG. (2007). Cryptic population dynamics: rapid evolution masks trophic interactions. PLoS Biol 5: 1868–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeileis A, Grothendieck G. (2005). zoo: S3 infrastructure for regular and irregular time series. J Stat Softw 14: 27. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.