

Abstract
Reliable transmission of symbionts between host generations facilitates the evolution of beneficial and pathogenic associations. Although transmission mode is typically characterized as either vertical or horizontal, the prevalence of intermediate transmission modes, and their impact on symbiont genome evolution, are understudied. Here, we use population genomics to explore mixed transmission modes of chemosynthetic bacterial symbionts in the bivalve Solemya velum. Despite strong evidence for symbiont inheritance through host oocytes, whole-genome analyses revealed signatures of frequent horizontal transmission, including discordant mitochondrial-symbiont genealogies, widespread recombination and a dynamic symbiont genome structure consistent with evolutionary patterns of horizontally transmitted associations. Population-level analyses thus provide a tractable means of ascertaining the fidelity of vertical versus horizontal transmission. Our data support the strong influence horizontal transmission can have on symbiont genome evolution, and shed light on the dynamic evolutionary pressures shaping symbiotic bacterial genomes.
Introduction
Bacterial symbioses have revolutionized eukaryotic lifestyles repeatedly throughout the history of life and these symbiotic associations have allowed a wide diversity of taxa to colonize formerly inaccessible niches (Moya et al., 2008). One striking example is chemosynthetic symbiosis, which enables invertebrate taxa to thrive in reducing environments from hydrothermal vents to coastal sediments. These bacterial symbionts produce nutrients for themselves and host species by oxidizing reduced chemicals, for example, sulfide, to fix carbon dioxide and generate ATP (Cavanaugh et al., 2013). These intimate relationships necessitate reliable mechanisms for host colonization each generation in order for the association to be maintained over evolutionary time.
Traditionally, two primary modes of transmission are recognized: vertical, in which symbionts are inherited directly from parents, and horizontal, in which symbionts are acquired from contemporary hosts or from free-living populations. Despite such rigid categorization, some associations exhibit evidence consistent with both modes (Bright and Bulgheresi, 2010; Ebert, 2013). However, this evidence is based on limited population and genetic sampling (for example, gene markers opposed to genomes (Itoh et al., 2014; Sipkema et al., 2015), which limits the detection of horizontal events, and it is unknown how prevalent and influential mixed modes of transmission are among symbiotic taxa. Hence, characterizing the transmission mode and dynamics of symbiotic associations is essential to understanding how these intimate relationships are maintained over evolutionary time.
Transmission mode plays a central role in symbiont genome evolution through influencing symbiont population sizes and gene flow. Horizontally transmitted symbionts experience evolutionary pressures similar to those of free-living bacteria, that is, the need to persist and/or reproduce out in the environment. Thus these bacteria maintain diverse functional elements in their genomes (Gomes et al., 2014; Salem et al., 2015). In contrast, stringent vertical transmission prevents symbiont exchange between hosts and the environment, reduces opportunities for horizontal gene transfer (HGT) to events that occur between microbes occupying the same host, and often imposes a genetic bottleneck at each host generation if only a small fraction of symbionts are transmitted (Moran and Bennett, 2014). Based on trends in vertically transmitted bacterial symbionts of insects, long-term inheritance leaves its mark through accumulation of deleterious mutations and gradual gene loss, resulting in extreme genome size reduction (<1 Mb) and genomic stasis (McCutcheon and von Dohlen, 2011). Even infrequent horizontal transmission events may be evident in the genomes of otherwise inherited associations (Brandvain et al., 2011). Mixed modes therefore may generate intermediate patterns of genome evolution, which would mitigate the consequences of vertical transmission while retaining the benefits of ensured symbiont acquisition and subsequent co-evolution of host–symbiont genotypes. However, additional data are needed from diverse symbiotic groups to assess the prevalence of mixed transmission modes and their impacts on symbiont genome evolution.
The bivalve Solemya velum and its intracellular chemosynthetic gammaproteobacterial gill symbionts exhibit evidence for and against vertical transmission, and is therefore an ideal system to test for a mixed transmission mode and characterize its impact on genome evolution. Until recently, symbionts of the genus Solemya were thought to be vertically transmitted based on detection in adult ovaries and in developing embryos/juveniles (Cary, 1994; Krueger et al., 1996). However, the S. velum symbiont genome is relatively large (2.7 Mb) and replete with genes typically lost from vertically transmitted bacteria (Dmytrenko et al., 2014). In addition, although Solemyidae is an ancient clade whose ancestors are inferred to be symbiotic (Stewart and Cavanaugh, 2006; Oliver et al., 2011; Sharma et al., 2013), Solemya symbionts are polyphyletic and closely related to free-living bacteria and other chemosynthetic symbionts (Supplementary Figure S1 and Cavanaugh et al., 2013). Thus, it is possible that the Solemya symbionts have been replaced repeatedly over evolutionary time and are distantly related to the original partners when the symbiosis evolved. While the symbiont genome is similar in size to many free-living sulfur oxidizers (for example, Thiomicrospira crunogena: 2.4 Mb, Thiovulum sp.: 2.1 Mb, Thiobacillus denitrificans: 2.9 Mb), the genome may be in the earliest stages of reduction, as some closely related taxa have much larger genome sizes (for example, Allochromatium vinosum: 3.6 Mb; Beller et al., 2006; Marshall et al., 2012; Dmytrenko et al., 2014). Collectively, these lines of evidence suggest dynamic transmission strategies in solemyid bivalves, and justify a detailed analysis of the effects of transmission mode on symbiont genome evolution.
We tested for evidence of mixed transmission modes in the S. velum symbiosis by looking for patterns of host–symbiont codiversification and characterizing symbiont genome evolution via population genomics. Multiple localities were sampled to account for the role geography can play in shaping the demographic processes influencing gene flow among symbiont and host populations. Specimens were collected from five geographic localities along the east coast of North America (Figure 1a). DNA from the symbiont-containing gills of 61 adult S. velum and three outgroup solemyid species was extracted and sequenced on the Illumina HiSeq platform to assemble consensus mitochondrial and symbiont genomes for each host specimen. Through population genetic and genealogical analyses (see Supplementary Figure S2), we present evidence that the S. velum symbiosis has experienced horizontal transmission events in its recent past and these events have enabled recombination and HGT, which has shaped the symbiont genome.
Figure 1.

Solemya velum mitochondria and symbiont genealogies are geographically subdivided, but discordant within localities. (a) Collection localities for S. velum subpopulations. (b) Whole-genome cladograms of S. velum mitochondria and symbionts inferred by maximum likelihood and Bayesian methodologies with support values (bootstrap/posterior probability). Nodes with less than 98% posterior probability are collapsed into polytomies and identical sequences are represented as triangles. Branches and clades are colored by locality. Lines connect symbiont and mitochondrial genomes from the same host individual, indicating concordant (dashed lines) or discordant (solid lines) relationships supported by resolved (black) or unresolved (gray) topologies.
Materials and methods
Collections
Adult S. velum were collected from intertidal-subtidal sediments along the east coast of North America (see Figure 1a and Supplementary Table S1) in the spring and summer of 2012, using a shovel and sieve. Immediately after collection, specimens were rinsed with 0.2 mm filtered seawater, sterilely dissected, placed in 100% ethanol, flash frozen and stored at −80 °C. Gill and testis (for two RI specimens to test for the presence of double-uniparental mitochondrial inheritance) DNA was extracted with the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany).
Outgroup species specimens were obtained from various sources as follows: Solemya pervernicosa from the Santa Monica sewage outfall in 1992, and the Museum of Comparative Zoology at Harvard University provided Solemya velesiana (Malacology 379149; collected in 2008 by the BivAToL Team) and Solemya elarraichensis (Malacology 379147; collected in 2004 by Marina R Cunha) specimens (Supplementary Table S1). Specimens were stored at −80 °C until extraction.
Sequencing
Genomic DNA was sheared to 350 bp (Covaris S220; Covaris, Woburn, MA, USA) and Illumina paired-end libraries were either made on the Apollo 324 System (Wafergen, Fremont, CA, USA) using the PrepX ILM kit (IntergenX, Pleasanton, CA, USA), or manually with the Hyperprep kit (Kapa Biosystems, Wilmington, MA, USA) or a custom protocol (see Supplementary Methods). Library prep method caused no detectable bias in the data, based on PCA analysis with SNPRelate (Zheng et al., 2012; Supplementary Figure S3A). NEXTflex adapters (Bioo Scientific, Austin, TX, USA) were used in all protocols. Libraries were quantified for size by a bioanalyzer (Agilent Bioanalyzer 2100; Agilent Technologies, Santa Clara, CA, USA) and qPCR with the PerfeCta library quantification kit (Quanta Biosciences, Beverly, MA, USA). PCR was not performed on libraries unless they were low concentration (8/64), and in those cases, underwent no more than six cycles. Libraries with unique adapter barcodes were pooled and paired-end sequenced on the Illumina HiSeq2000 or HiSeq2500 platform (Bauer Core Facility, Harvard University), using heat denaturation. Sequence data from demultiplexed libraries were evaluated with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and trimmed/filtered with Trimmomatic v0.32 (Bolger et al., 2014; Supplementary Table S1).
Genome assembly
Reference-based genome assembly of S. velum symbionts and mitochondria
The mitochondrial and symbiont genomes were saved to a single file and the reads from each specimen were mapped to the two genomes (Plazzi et al., 2013 and Dmytrenko et al., 2014) with Stampy 1.0.18 (Lunter and Goodson, 2011) to prevent suboptimal mapping to paralogues in one or the other genome. Only the four largest symbiont scaffolds were used in these analyses, as they contain 98.9% of the sequence and 99.4% of the genes. Alignments were processed from Sam to Bam format and indexed with Samtools 1.0 (Li et al., 2009), duplicates were removed with Picardtools (http://broadinstitute.github.io/picard/) and coverage was calculated with Bedtools 2.18.1 (Quinlan and Hall, 2010). Indel realignment was performed with the Realigner module and variant calling was performed with the UnifiedGenotyper module of the Genome Analysis Toolkit (GATK, version 3.2-2; see Supplementary Methods for parameters; DePristo et al., 2011). Variant calling was done on haploid mode to call the consensus symbiont sequence for each host individual, as the more deeply sequenced specimens exhibited no evidence of containing more than one type of symbiont genome (Supplementary Figure S3B). Variant calls were hard-filtered to remove low-confidence variants according to the GATK Best Practices protocol (Van der Auwera et al., 2002). To avoid erroneous calls, single-nucleotide polymorphisms within five base pairs of an indel were removed. Mitochondrial and symbiont genome sequences were generated for each specimen/tissue by applying the filtered variants calls to the reference sequences with a custom script. Gene sequences were extracted for each specimen/tissue using the reference genome annotation coordinates.
De novo assembly of outgroup solemyid symbiont species and S. velum symbionts
Read alignments to the reference symbiont genome were used as a starting point for de novo genome assembly in order to estimate the copy number, and thus coverage, of the symbiont genome in each sample (see Supplementary Figures S4A–D). This allowed reads originating from the nuclear genome, sequencing error, or potential environmental contamination to be removed from the data set assembled. Utilizing relative coverage differences to identify symbiont reads is a reasonable strategy for these specimens because (1) symbiont genome coverage exceeds nuclear genome coverage in S. velum 50:1 and microscopy data suggest that symbionts display similar intracellular densities and distributions among solemyid species (Stewart and Cavanaugh, 2006; Taylor et al., 2008; Fujiwara et al., 2009), and (2) the S. velum symbiosis consists of a single 16S rRNA type across the host's geographic range, making each host more of a symbiont population pool than a metagenome.
Estimates for symbiont genome coverage were obtained by mapping reads from each species to the S. velum symbiont genome with the permissive polymorphism aligner, LAST (Kielbasa et al., 2011), calculating the kmer coverage distribution for symbiont and mitochondrial-mapped reads and unmapped pairs of mapped reads with Jellyfish 1.1.11 (Marcais and Kingsford, 2011; Supplementary Figures S4A–D). Higher kmer values produced sharper distributions (odd kmer values from 11 to 25 were tested), but increased RAM requirements exponentially, limiting the usable kmer size to 19. The lower bounds of the 19-mer distributions were used as cutoffs to filter the unmapped solemyid reads with Quake (Kelley et al., 2010), removing low coverage reads attributable to the host nuclear genome or sequencing errors.
Mapped read pairs and unmapped kmer-filtered reads were assembled with IDBA 1.1.1 (Peng et al., 2012). Other assemblers were tested (MaSuRCA (Zimin et al., 2013) and Ray Meta (Boisvert et al., 2012). The three assemblies for each specimen were characterized with Quast (Gurevich et al., 2013) and compared by alignment with MUMmer (Kurtz et al., 2004). All assemblers produced highly contiguous and very similar assemblies (Supplementary Table S2). However, IDBA produced assemblies with fewer gaps and longer scaffolds in lower coverage specimens, so these assemblies were used for subsequent analyses. All reads were then mapped to the de novo assemblies with Stampy as described above for the S. velum reference-based assembly to calculate scaffold coverage. Mitochondrial chromosomes and candidate symbiont scaffolds were identified and isolated by coverage (average±1 s.e.), length (>1000 bp) and GC content (50 GC±10%). Mitochondrial chromosomes were annotated with MITOS (Bernt et al., 2013). Candidate symbiont scaffolds were annotated for coding sequences with Prodigal (Hyatt et al., 2012) and blastp (Camacho et al., 2009) against the NCBI non-redundant (nr), TrEMBL (Bairoch and Apweiler, 2000) and UniRef90 databases (Suzek et al., 2007; cutoff values: minimum coverage 50%, minimum 30% identity, e-value 1e-6) to annotate as many CDs as possible. Predicted CDSs with hits failing these criteria were annotated as hypothetical proteins. tRNAs were annotated with tRNAscan (Lowe and Eddy, 1997) and rRNAs with RNAmmer (Lagesen et al., 2007). Annotation, coverage and GC content were utilized to remove host nuclear scaffolds. We tested for evidence of chimeric sequence joining by comparing the coverage distribution of sequence segments aligned to the reference genome compared with novel segments, and found no differences (Supplementary Figure S4E). Draft assembly completeness was evaluated with Quast (Gurevich et al., 2013), by comparing genome size to expected values for other sulfur-oxidizing symbionts, checking for the presence of 31 core bacterial phylogenetic markers (Wu and Eisen, 2008), and evaluating the assemblies compared with the core Proteobacterial gene set with CheckM taxonomy_wf (Parks et al., 2014; Supplementary Table S3).
Genomic analyses
16S rRNA phylogenetic analysis
16S ribosomal RNA sequences from outgroup solemyid and S. velum reference assemblies were blasted against the NCBI non-redundant nucleotide database to obtain closely related 16S rRNA sequences from gammaproteobacterial sulfur-oxidizing symbionts, free-living bacteria and isolated environmental clones. Deltaproteobacterial and Epsilonproteobacterial 16S rRNA sequences served as outgroup taxa (Supplementary Table S4). Sequences were aligned with MUSCLE (Edgar, 2004) and manually inspected and trimmed in Geneious (Geneious 8.1 (http://www.geneious.com; Kearse et al., 2012). The 16S rRNA phylogeny was inferred by maximum likelihood with RAxML (version 8.1.5) using the GTRGAMMA model of nucleotide evolution and 1000 bootstrap replicates (Stamatakis, 2014).
Whole-genome genealogical inference
Mitochondrial and symbiont whole-genome alignments were performed with progressiveMauve, allowing for rearrangements (version 2015-2-13; default parameters; Darling et al., 2010). Regions of the outgroup symbiont sequences that failed to align to the S. velum symbiont genomes were removed, and the remaining alignment blocks were concatenated and converted to Phylip format. This approach is similar to a supermatrix method, which concatenates gene alignments, but has the added benefit of retaining non-protein-coding regions of the genome. Whole-genome genealogies were inferred by maximum likelihood with RAxML (version 8.1.5) using the GTRGAMMA model of nucleotide evolution and 1000 bootstrap replicates (Stamatakis, 2014) and by Bayesian inference with MrBayes (version 3.2.5; Ronquist et al., 2012), using three independent runs, each with one million Markov chain Monte Carlo iterations using the mixed GTR+gamma substitution model, sub-sampling every 1000 generations, and discarding the first 25% of samples as burn-in. Trees were viewed and converted to cladograms in FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Identical mitochondrial sequences were collapsed and nodes with <50% bootstrap support were represented as polytomies. Symbiont and mitochondrial genealogy topologies were compared visually and by topological distance using the R package APE (Paradis et al., 2004; R Core Development Team, 2012).
Symbiont gene and sliding window genealogies were inferred by maximum likelihood. Genes were selected by identifying orthologous open reading frames among the S. velum symbionts and the three outgroup symbionts by reciprocal best blast hits (see Supplementary Methods). Genes with less than 10% alignment coverage and identity to all other symbionts, e-values less than 1000 × better than the next best alignment and non-identical reciprocal best hits among genomes were excluded. S. velum symbiont gene sequences were aligned to outgroup sequences for each gene with MUSCLE (version 3.8.31; default parameters; Edgar, 2004). Sequences from 100 kb sliding windows of the S. velum symbiont genome were aligned to outgroup genomes with progressiveMauve and unaligned regions of the outgroup genomes were removed with a custom script. Gene and window genealogies were inferred with RAxML (Stamatakis, 2014) as described above. Genealogies were rooted and nodes with less than 50% bootstrap support were collapsed with the APE package in R (Paradis et al., 2004; R Core Development Team, 2012). Genealogies of sliding windows were plotted together and the summary tree calculated with DensiTree (Bouckaert, 2010).
Polymorphism analyses
Genetic diversity was measured for the entire population, each subpopulation, and pairwise at all sites, synonymous sites, and nonsynonymous sites by calculating pairwise nucleotide diversity (π) (Nei and Li, 1979) by site and averaging across all sites:
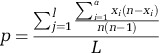 |
where n is the total number of individuals sampled, xi is the number of individuals with allele i, a is the total number of sampled alleles at site j of l total sites, and L is the total sequence length. Substitution effects were determined with snpEff (Cingolani et al., 2012). Calculations were performed on the snpEff annotated, GATK variant file for mitochondria and symbionts, removing sites not present in at least half of the specimens from the given population.
Linkage disequilibrium analyses
Linkage disequilibrium (LD) within the symbiont and mitochondrial genomes and interspecific disequilibrium between mitochondrial and symbiont genomes was measured by calculating the correlation coefficient (r2; Hill and Robertson, 1968; Awadalla et al., 1999) between all pairwise combinations of biallelic segregating sites with allele frequencies above 0.1 (to exclude recent mutations lacking the opportunity to recombine). Greater than half of all specimens or all specimens from a subpopulation were required to have a call at a given site for that site to be included in the analysis. For every pair of biallelic variant sites across the genome, linkage disequilibrium (D) and the correlation coefficient (r) between alleles was calculated as follows:
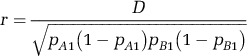 |
where pA1 and pB1 are the frequencies of the reference alleles of the A and B loci, respectively, and x11 is the frequency of the AB reference genotype. LD r2 values were binned by distance between sites and averaged, with the maximum distance between sites being half of the total genome size for circular genomes, and plotted in R (R Core Development Team, 2012). Spearman's rank correlation coefficient (ρ) was calculated between each (unbinned) r2 value and distance using the Statistics::RankCorrelation (http://search.cpan.org/~gene/Statistics-RankCorrelation-0.1204/lib/Statistics/RankCorrelation.pm, accessed 15 December 2014) perl module. Permutation tests were conducted by randomly shuffling r2 and distance values 5000 times with the Math::Random (http://search.cpan.org/~grommel/Math-Random-0.71/Random.pm, accessed 15 December 2014) perl module. Exact permutation P-values were calculated as P=(b+1)/(m+1), where b was the number of permutations in which r was less than or equal to the observed value and m was the number of permutations (Phipson and Smyth, 2010). Owing to low diversity within geographic localities, the relationship between LD and distance was unable to be ascertained within subpopulations with these data.
Chi-square P-values for interspecific disequilibrium values were calculated by multiplying the correlation coefficient, r2, by the number of samples (Hill and Robertson, 1968), using the Statistics::Distributions (http://search.cpan.org/~mikek/Statistics-Distributions-1.02/Distributions.pm, accessed 5 May 2015) perl module, with one degree of freedom and a 5% significance cutoff.
Transposable element analyses
Mobile elements were detected and annotated in the symbiont reference genome by type. Insertion sequences were detected with ISsaga (www-is.biotoul.fr; Siguier et al., 2006). Integrated prophages were detected with PHAST (phast.wishartlab.com; Zhou et al., 2011). Plasmids, prophages, and viruses as well as integrative conjugative elements were detected by blastn searches against the ACLAME (Leplae et al., 2009) and ICEberg (Bi et al., 2011) databases, respectively (cutoff values: minimum alignment length of 250 nucleotides, 90% identity, e-value 0.0001).
Symbiont genomes were scanned for large-scale deletions by analyzing coverage files produced by Bedtools (see Reference-based genome assembly) for contiguous regions with zero coverage greater than 150 bp long. Deletions were annotated for functional genes (Dmytrenko et al., 2014) and mobile elements. For visualization, read coverage was averaged over 1000 bp windows and plotted with Circos (Krzywinski et al., 2009).
The S. velum symbiont transcriptome (Stewart et al., 2011) was analyzed for active, and thus likely functional, mobile elements. Transcripts were blasted against the annotated mobile elements with blastn (cutoff values: minimum coverage 50%, minimum 90% identity, e-value 1e–6).
Synteny analysis
Gene order in the de novo assembled symbiont genomes was compared with the symbiont reference by alignment of whole genomes and individual coding sequences. For whole-genome alignment, scaffolds in the draft assemblies were reordered to the S. velum symbiont reference sequence in Mauve (Rissman et al., 2009). The de novo assemblies were then aligned to the reference and outgroup genomes in progressiveMauve, allowing for rearrangements (Darling et al., 2010). Synteny along the genome was examined by mapping local collinear blocks with Circos (Krzywinski et al., 2009). The relative order of homologous genes was also examined. Homologs were identified by reciprocal best blast hits (see Whole-genome genealogical inference) and their relative positions calculated with a custom script. Relative gene order was visualized with Circos (Krzywinski et al., 2009) by plotting links between homologous genes on the reference and de novo scaffolds.
HGT analysis
All genes in the S. velum symbiont reference genome and de novo assembled genes that lacked homologs in the reference genome were blasted against the NCBI whole-genome shotgun database (best_hit_overhang 0.1, best_hit_score_edge 0.1, e-value 1e–6). Blast results were filtered to remove hits with less than 60% coverage and less than 60% identity. Genes on scaffolds containing fewer than five ORFs with homology to the S. velum symbiont reference were removed to eliminate the possibility that the scaffold was assembled from contaminating sequence at the same coverage and not filtered out during assembly. Sequences were identified as potential HGT events if they had greater than 98% identity to the bacterial species hit at the locus, but less than 90% identity at the 16S rRNA gene. As a more stringent filter against contamination, de novo assembled symbiont scaffolds that contained five or more of the core genes (present in all of the symbiont genomes mapped to the reference) were identified.
A subset of the putatively horizontally transferred genes was selected for phylogenic analysis. Sets of homologous genes were identified by blast hit identity and manual inspection of alignments generated with MUSCLE (Edgar, 2004). Sets with sequences in multiple samples at least several hundred bp in length with functional annotations (opposed to ‘hypothetical') were selected for phylogenetic inference. Sequences from other bacteria with high identity to the symbiont sequences were downloaded from NCBI and aligned to the symbiont sequences. Phylogenies were inferred from alignments with RAxML (Stamatakis, 2014), run as detailed above.
All perl scripts used in these analyses are available at https://github.com/shelbirussell/Russell-et-al-2016.
Results
Mitochondrial and symbiont populations were highly subdivided between geographic localities and exhibited consistent patterns of subdivision (Figure 1). All sequences in both of these populations shared high identity with S. velum reference sequences (99–99.9%), precluding the possibility of cryptic species. Mitochondria and symbiont populations exhibited low pairwise nucleotide diversity across the sampled localities and across both mitochondrial (average π=2.3 × 10−3) and symbiont (average π=1.7 × 10−3) genomes, suggesting that these subpopulations diverged relatively recently. Southern localities generally exhibited higher diversity than northern localities, and New Jersey (NJ) was the least diverse (Table 1). Monophyly within each geographic locality indicates that each of these locations harbors its own subpopulation of S. velum and symbionts, and suggests restricted gene flow with other sampled subpopulations.
Table 1. Nucleotide diversity statistics for mitochondria and symbionts sequenced from subpopulations collected along the east coast of North America.
| All (n=61) |
Subpopulation |
|||||
|---|---|---|---|---|---|---|
| MA (n=9) | RI (n=20) | NJ (n=7) | MD (n=10) | NC (n=15) | ||
| Mitochondrial genome | ||||||
| Segregating sites (S) | 262 | 32 | 76 | 2 | 25 | 131 |
| S synonymous | 164 | 14 | 41 | 0 | 15 | 90 |
| S nonsynonymous | 41 | 5 | 15 | 0 | 3 | 18 |
| S intergenic | 57 | 13 | 20 | 2 | 7 | 23 |
| Indels | 12 | 4 | 5 | 0 | 2 | 6 |
| Biallelic sites | 249 | 27 | 70 | 1 | 22 | 124 |
| Triallelic sites | 1 | 1 | 1 | 1 | 1 | 1 |
| Pairwise diversity (π) | 2.27E-03 | 5.49E-04 | 9.21E-04 | 1.82E-05 | 3.74E-04 | 3.40E-03 |
| π nonsynonymous | 2.82E-04 | 9.21E-05 | 1.38E-04 | 0.00E+00 | 3.83E-05 | 4.85E-04 |
| π synonymous | 1.50E-03 | 2.80E-04 | 5.54E-04 | 0.00E+00 | 2.65E-04 | 2.45E-03 |
| π intergenic | 5.68E-04 | 2.69E-04 | 3.00E-04 | 1.82E-05 | 9.64E-05 | 5.98E-04 |
| Deletions (>500 bp) | 0 | 0 | 0 | 0 | 0 | 0 |
| Symbiont genome | ||||||
| Segregating sites (S) | 28 836 | 2845 | 2667 | 1786 | 4760 | 9058 |
| S synonymous | 11 523 | 925 | 705 | 384 | 1217 | 3835 |
| S nonsynonymous | 9665 | 761 | 803 | 377 | 1855 | 2671 |
| S intergenic | 4493 | 681 | 672 | 574 | 969 | 1525 |
| indels | 4319 | 604 | 600 | 589 | 817 | 1375 |
| biallelic sites | 24 094 | 1869 | 1697 | 853 | 3567 | 7317 |
| triallelic sites | 423 | 372 | 370 | 344 | 376 | 366 |
| pairwise diversity (π) | 1.70E-03 | 2.81E-04 | 2.14E-04 | 1.54E-04 | 4.84E-04 | 5.78E-04 |
| π nonsynonymous | 7.55E-04 | 1.24E-04 | 7.63E-05 | 5.54E-05 | 1.51E-04 | 2.60E-04 |
| π synonymous | 6.80E-04 | 9.59E-05 | 8.50E-05 | 4.95E-05 | 2.33E-04 | 2.10E-04 |
| π intergenic | 3.84E-04 | 8.40E-05 | 6.72E-05 | 7.74E-05 | 1.22E-04 | 1.45E-04 |
| Deletions (>500 bp) | 50 | 27 | 23 | 34 | 33 | 38 |
Genome-wide statistics for all 61 specimens are given (All), as well as statistics by the subpopulation locality (state abbreviations as in Figure 1). 15 660 and 2 667 804 sites were confidently called in the mitochondrial and symbiont genomes, respectively. Many open reading frames in the symbiont genome overlap (1284 of 2699), making many positions synonymous in one gene and nonsynonymous in the other. These sites were excluded from calculations of S and π by type.
Despite strong and concordant geographic subdivision, mitochondrial and symbiont evolutionary histories were decoupled within localities (Figure 1b), supporting horizontal transmission. At 2.7 Mb, the symbiont genome is significantly larger than the mitochondrial genome (16 Kb), thus it contained more segregating sites, permitting higher resolution of the symbiont genealogy compared with the mitochondrial genealogy. Enough nodes could be resolved to reject the hypothesis of strict maternal transmission, although the exact rate of alternate routes of transmission could not be estimated. Comparing well-supported nodes, we found that of the 54 total internal nodes in the symbiont tree, a minimum of 35 nodes conflict with the mitochondrial tree. Furthermore, within a locality, no part of the symbiont genome is in strong LD with any part of the mitochondrial genome (Supplementary Table S5 and Supplementary Figures S5A and B). The only possible cases of concordance are among subsets of specimens containing identical mitochondrial haplotypes, and are likely artifacts of the unresolved mitochondrial topology (Supplementary Figure S5C). This extent of discordance is possible only if symbiont and/or mitochondrial genomes are sometimes acquired independently. Discordance due to paternal transmission of mitochondria (double-uniparental inheritance) is unlikely because the testis were found to contain the same mitochondrial genotype in the two individuals tested (Supplementary Figure S6A). Collectively, these data indicate that the S. velum symbionts are not strictly transmitted with the mitochondria through the eggs.
In further support of an alternative transmission route, we found extensive evidence for recombination within the symbiont genomes. Divergent genotypes must come into contact either as mixed infections within hosts or out in the environment for recombination to occur. In a recombining population, LD (the correlation between pairs of alleles) is inversely related to the distance between sites (Figure 2a). In contrast, there will be no association between LD and distance in clonal genomes (Halkett et al., 2005). In the symbiont genome, we found that LD is significantly negatively correlated with genomic distance (Figure 2b; Spearman's ρ=−0.185; P<0.0002), indicating that recombination events occur frequently in symbiont populations. No pattern of LD decay with distance was found in the mitochondrial genome (Supplementary Figure S6B; Spearman's ρ=−0.003; P=0.344), as expected for a clonal uniparentally inherited genome. Genealogies sampled from different regions of the symbiont genome have unique topologies, further reinforcing the idea that symbiont genomes are comprised of a mosaic of evolutionary histories (Supplementary Figure S7). Taken together, pervasive recombination combined with low pairwise nucleotide diversity suggest that recombination occurs relatively frequently in the symbiont genome compared with other intracellular bacteria (Vos and Didelot, 2008).
Figure 2.

Solemya velum symbiont genomes exhibit evidence of recombination. (a) Linkage disequilibrium (LD, measured by r2) measures the non-random association between alleles relative to what would be expected by chance. Recombination rate between sites in a genome is influenced by the number of base pairs between them. Thus, in a recombining genome, sites closer together co-occur more than those far apart. (b) Relationship between LD and distance between sites in the S. velum symbiont genome (averaged over windows of 100 bp and plotted by density). Symbiont LD decays with genomic distance. Inset plot magnifies the distance over which LD exponentially decays.
Mobile elements appear to drive abundant structural genomic variation within geographic subpopulations of S. velum symbionts, suggesting that symbionts have recently been in contact with the environment. Models of intracellular bacterial genome evolution indicate that mobile elements are strongly correlated with transmission mode and age of the symbiosis (McCutcheon and Moran, 2011; Newton and Bordenstein, 2011). Finding 2.6% of the symbiont genome to consist of mobile elements (Dmytrenko et al., 2014) suggests that either the symbionts encounter the environment or they did in the recent past, as this is a relatively large amount compared with other intracellular bacteria (see Newton and Bordenstein 2011). Consistent with ongoing TE movement, reads mapped to the symbiont reference genome (Figure 3a) and de novo genome assemblies (Figure 3b) revealed large insertions and deletions among specimens and conserved synteny within shared genomic intervals. Many indels corresponded to annotated transposable elements surrounding protein-coding genes (Supplementary Table S6). In data from a metatranscriptomic study of S. velum symbionts (Stewart et al., 2011), 25 of these elements were transcribed, indicating that they are functional (Supplementary Tables S6 and S7). Once inserted, mobile elements quickly become nonfunctional (Cerveau et al., 2011), suggesting many of these were recently acquired. However, on short evolutionary timescales, functionality is retained (for example, Kleiner et al., 2013). Furthermore, the lack of rearrangements among copies of these elements suggests that little time has passed since they entered the subpopulations. However, we cannot exclude the possibility of rearrangements between distant parts of the genome with these data, as the longest scaffold assembled was 450 kb long or ~17% of the genome. Either losses of elements in the genome upon host restriction or ongoing flux from environmental sources (for example, other bacteria, exogenous DNA, or viruses) could explain mobile element activity in the S. velum symbiont populations. In both cases, the abundance and functionality of the elements suggests that contact with the environment was relatively recent on an evolutionary scale.
Figure 3.

Mobile elements drive structural genomic variation and HGT in S. velum symbiont genomes within and among localities. (a) Circos plot of resequenced symbiont read coverage by locality. Inner gray circle depicts scaffolds 1–4 of the S. velum symbiont reference genome with black lines marking annotated transposable elements. Plots outside of the reference show the depth of reads mapped to the symbiont reference genome for three representative specimens from each locality, colored as in Figure 1. Genomic positions are marked around the edge in kbp. (b) De novo assembled S. velum symbiont genomes from each geographic subpopulation are plotted in concentric circles around the reference (from center to outer edge: MA16, RI38, NJ6, MD7 and NC17) with Circos. Genome assembly alignments colored by homology and sequence order (color gradient). The S. velum symbiont reference sequence is colored in a white–black gradient from 5′-to-3′ in clockwise order: scaffold 2, 4, 1 and 3. Remaining novel regions unique to the de novo S. velum symbiont assemblies are colored in a rainbow gradient. (c, d) Maximum likelihood phylogenies for a putatively horizontally transferred DNA-binding protein (c) and zinc-binding primase/helicase (d). Bootstrap support values greater than 50% are displayed at the nodes. (e) Genomic region that genes in (c) and (d) occur. Blocks indicate coding sequences and are proportionally sized. Red=putative HGT; yellow=core symbiont gene; brown=annotated mobile element; blue=non-core symbiont gene; gray=other novel genes; white with dashed line=deletion relative to another sequence.
The de novo and reference symbiont genomes exhibit evidence of HGT, providing direct evidence of recent contact with the environment. Blasting large indel regions polymorphic among the genomes to NCBI's database of whole bacterial genome sequences revealed that many of these regions exhibit >99% identity to protein-coding genes from distantly related free-living gammaproteobacteria (78–88% identical at the 16S rRNA; Supplementary Table S8). These regions matched mobile element segments of genomes from 14 different bacterial taxa, with Vibrio cholerae being the most common (77 regions), followed by Idiomarina sp. (56 regions) and Aliiglaciecola lipolytica (39 regions). More than half of the regions matched uncharacterized or hypothetical proteins. The remaining regions exhibited high sequence identity to genes involved in functions ranging from DNA binding to signal transduction. The majority of the scaffolds bearing these regions contained five or more genes in the ‘core' symbiont genome (see Supplementary Table S8); however, the shorter scaffolds contained too few genes to meet this criterion, but were included to not discard these data. Within each locality, specimen sequences matched many of the same insertion products and taxa, and these regions exhibited conserved order and common ancestry, suggesting that they arose by a limited set of transfer events in the ancestors of the specimens (Figures 3c–e,Supplementary Figure S8 and Supplementary Table S8). Compared with RI, NJ and MD localities, specimens collected from NC and MA had far more variable sets of putatively transferred genes. Although the absence of elements from de novo sequenced genomes cannot be confirmed with these data because Illumina sequencing alone could not close the genomes, the pattern of detected indels demonstrates that there is an active flux of horizontally transmitted elements in the symbiont genome.
Discussion
Collectively, these population genomic data suggest that horizontal transmission occurs or has occurred recently in S. velum symbiont populations and has dramatically impacted genome evolution. Mitochondria and symbionts have not co-diversified and symbiont genomes exhibit evidence of frequent recombination, consistent with observations for horizontally transmitted bacterial symbionts (for example, Mouton et al., 2012; Ros et al., 2012). While these data could potentially be explained by paternal transmission or mitochondrial double-uniparental inheritance, finding structural genomic variation associated with evidence of HGT and active mobile elements suggests that symbionts experience some amount of horizontal transmission or have in the recent past. While it is not known precisely how common horizontal transmission is among S. velum individuals, these data indicate that effective rates are higher than symbiont mutation or host migration rates. Taken together, these results reveal the striking interplay between symbiont transmission mode and genome evolution.
We propose a mixed transmission mode for the S. velum symbionts, in which they undergo regular vertical transmission via spawned oocytes, but also experience horizontal transmission events. Mixed transmission modes would provide a functional compromise: Vertical transmission enables the evolution of coadapted host–symbiont genotypes, homogenizes intra-host–symbiont populations and ensures host offspring are colonized each generation. Horizontal transmission ameliorates deleterious mutations and genome stasis by facilitating recombination and HGT. Using both modes would allow a flexible transmission strategy, enabling populations to respond to long and short-term evolutionary pressures (Kaltz and Koella, 2003; Byler et al., 2013). Given that symbionts are highly divergent among Solemya species (Supplementary Figure S1) and evidence supporting vertical transmission has been reported for both S. velum and S. pervernicosa (Cary, 1994; Krueger et al., 1996), it is likely that transmission modes in these taxa are evolutionarily flexible on longer timescales as well. This finding is relevant to symbioses broadly, as rare horizontal events have been detected in a number of vertically transmitted associations (for example, Stewart et al., 2008; Sipkema et al., 2015), and may represent ongoing processes.
Our finding that symbionts and hosts exhibit concordant subdivision between geographic subpopulations is surprising in light of the genealogical discordance seen within subpopulations. Symbiont geographic subdivision mirrors that of mitochondria for three possible reasons, although other explanations may exist. First, symbionts and hosts may have identical dispersal patterns. However, this seems unlikely because S. velum has large dense eggs and no free-swimming stage (Gustafson and Lutz, 1992), while the bacteria should experience fewer dispersal barriers outside of their host. Second, symbiont and host populations may be genetically subdivided because they are locally adapted. Although local adaptation may play an important role, we would expect to observe the effects of local adaptation in a subset of genealogies, not genome-wide as seen in our data. Third, symbiont reproduction is tied to the host. We favor this explanation because in an environmental qPCR survey that included controls for symbiont detection threshold, symbionts were detected at only 3.65±9.38 × 103 copies per g sediment and 0.14±0.24 × 10−1 copies per ml seawater. These amounts are five orders of magnitude lower than total eubacterial 16S counts, which were 3.57±3.91 × 108 copies per g sediment and 8.54±0.12 × 104 copies per ml seawater. Furthermore, S. velum mitochondrial sequences were also detected in the majority of sediment samples, and at 4.17±7.80 × 103 copies per g of sediment, were present at a similar abundance, indicating that at least some of the symbionts arose from host tissues (Russell, 2016). Taken together, this suggests that symbiont reproduction occurs primarily within hosts and that horizontal transmission occurs between contemporary hosts within a geographic subpopulation.
The strong geographic subdivision among hosts and symbionts may be due to factors occurring on different timescales, and can obscure evidence of horizontal transmission. In geological time, southern populations (for example, NC) may have persisted as refugia during periods of glaciation and upon glacier retreat, reseeded the populations in the north (Hewitt, 1996). This works in conjunction with broadcast spawning, which can be described as ‘sweepstakes-style recruitment' (Eldon and Wakeley, 2009), to produce subpopulations composed of related individuals. These possible scenarios raise crucial considerations for existing practices for characterizing symbiont transmission modes. Specifically, tests of co-speciation may falsely support strict vertical transmission when only a few individuals are sampled from diverse geographic subpopulations. We therefore emphasize the need for ample sampling of hosts and symbionts when determining transmission modes using genetic data.
An important determinant of transmission mode may be the medium in which the symbiosis exists, for example, water versus air. For example, vertical transmission is seldom reported in the marine environment, whereas it is exceedingly common in terrestrial habitats (Normark and Ross, 2014). Consistent with our findings for the S. velum symbiosis, most of the marine vertically transmitted associations that have been reported bear evidence of horizontal transmission events at some point in the past (for example, Schmitt et al., 2008; Stewart et al., 2008; Byler et al., 2013; Decker et al., 2013; Sipkema et al., 2015). Taken together, these observations suggest that horizontal transmission may be an inherent aspect of symbiont transmission in the majority of marine associations. Elucidation of the factors that influence transmission modes is vital to understanding the evolutionary pressures that determine how symbiotic associations are maintained and perpetuated over millions of years. Whether the persistence of horizontal transmission is a constraint of the environmental medium or other parameters, such as the taxa involved, reproductive strategy, or symbiosis function, remains to be tested.
Now that population-level whole-genome sequencing is achievable, hypotheses of strict vertical or horizontal transmission and mixed transmission modes can be tested broadly across eukaryote–microbe associations to assess the prevalence of these strategies and their influence on genome evolution. Provided specimens are obtained from host populations with limited barriers to gene flow, these analyses can resolve co-evolutionary processes obscured by demographic factors affecting both host and symbiont populations. In addition, the relatively high frequency of recombination observed in the S. velum bacterial symbionts indicates that sexual population genetic theory, which assumes regularly occurring recombination, may be applicable in symbiont populations, for example, for detection of selective sweeps. Given the utility of population genomics to resolve fine-scale evolutionary events, investigations of the fidelity and dynamics of symbiont transmission in diverse symbiotic associations, ranging from deep-sea chemosynthetic symbioses to the human microbiome, will provide a better understanding of the forces driving symbiont–host co-evolution.
Acknowledgments
We thank Frank Stewart, Gonzalo Giribet, Peter Girguis, Cassandra Extavour, members of the Cavanaugh Lab, and three anonymous reviewers for valuable suggestions and comments. We gratefully acknowledge support from Harvard University's William F. Milton Fund, Department of Organismic and Evolutionary Biology, and Microbial Sciences Initiative. Jonathan Finlay provided valuable help with collection of the S. velum population samples. We thank Harvard's Museum of Comparative Zoology for the S. velesiana specimen (BivAToL project) and the S. elarraichensis specimen (collected by Marina R Cunha supported by project HERMES (contract GOCE-CT-2005-511234) and project HERMIONE (contract 226354) and donated to the BivAToL project). This work was supported from Harvard University's William F. Milton Fund, Department of Organismic and Evolutionary Biology, and Microbial Sciences Initiative.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
The authors declare no conflict of interest.
Supplementary Material
References
- Awadalla P, Eyre-Walker A, Smith JM. (1999). Linkage disequilibrium and recombination in hominid mitochondrial DNA. Science 286: 2524–2525. [DOI] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28: 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beller HR, Chain PSG, Letain TE, Chakicherla A, Larimer FW, Richardson PM et al. (2006). The genome sequence of the obligately chemolithoautotrophic facultatively anaerobic bacterium Thiobacillus denitrificans. J Bacteriol 188: 1473–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69: 313–319. [DOI] [PubMed] [Google Scholar]
- Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X et al. (2011). ICEberg: a web-based resource for integrative and conjugative elements found in bacteria. Nucleic Acids Res 40: D621–D626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert S, Raymond F, Godzaridis É, Laviolette F, Corbeil J. (2012). Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol 13: R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR. (2010). DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26: 1372–1373. [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Goodnight C, Wade MJ. (2011). Horizontal transmission rapidly erodes disequilibria between organelle and symbiont genomes. Genetics 189: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright M, Bulgheresi S. (2010). A complex journey: transmission of microbial symbionts. Nat Rev Microbiol 8: 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byler KA, Carmi-Veal M, Fine M, Goulet TL. (2013). Multiple symbiont acquisition strategies as an adaptive mechanism in the coral Stylophora pistillata. PLoS One 8: e59596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary S. (1994). Vertical transmission of a chemoautotrophic symbiont in the protobranch bivalve. Solemya reidi. Mol Mar Biol Biotech 3: 121. [PubMed] [Google Scholar]
- Cavanaugh CM, McKiness ZP, Newton I, Stewart FJ. (2013) Marine chemosynthetic symbioses. In: Rosenberg E (ed.). The Prokaryotes – Prokaryotic Biology and Symbiotic Associations. Springer-Verlag: Berlin, Heidelberg, pp 579–607. [Google Scholar]
- Cerveau N, Leclercq S, Leroy E, Bouchon D, Cordaux R. (2011). Short- and long-term evolutionary dynamics of bacterial insertion sequences: insights from Wolbachia endosymbionts. Genome Biol Evol 3: 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker C, Olu K, Arnaud-Haond S, Duperron S. (2013). Physical proximity may promote lateral acquisition of bacterial symbionts in vesicomyid clams. PLoS One 8: e64830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmytrenko O, Russell SL, Loo WT, Fontanez KM, Liao L, Roeselers G et al. (2014). The genome of the intracellular bacterium of the coastal bivalve, Solemya velum: a blueprint for thriving in and out of symbiosis. BMC Genomics 15: 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D. (2013). The epidemiology and evolution of symbionts with mixed-mode transmission. Annu Rev Ecol Evol Syst 44: 623–643. [Google Scholar]
- Edgar RC. (2004). Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1797–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldon B, Wakeley J. (2009). Coalescence times and FST under a skewed offspring distribution among individuals in a population. Genetics 181: 615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Okutani T, Yamanaka T, Kawato M. (2009). Solemya pervernicosa lives in sediment underneath submerged whale carcasses: its biological significance. Venus 68: 27–37. [Google Scholar]
- Gomes D, Stefânia da Silva Batista J, Rolla AAP, Paulina Da Silva L, Bloch C, Galli-Terasawa LV, Hungria M. (2014). Proteomic analysis of free-living Bradyrhizobium diazoefficiens: highlighting potential determinants of a successful symbiosis. BMC Genomics 15: 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson RG, Lutz RA. (1992). Larval and early post-larval development of the protobranch bivalve Solemya velum (Mollusca: Bivalvia). J Mar Biol Ass UK 72: 383–402. [Google Scholar]
- Halkett F, Simon J-C, Balloux F. (2005). Tackling the population genetics of clonal and partially clonal organisms. Trends Ecol Evol 20: 194–201. [DOI] [PubMed] [Google Scholar]
- Hewitt GM. (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linnean Soc 58: 247–276. [Google Scholar]
- Hill WG, Robertson A. (1968). Linkage disequilibrium in finite populations. Theor Appl Genet 38: 226–231. [DOI] [PubMed] [Google Scholar]
- Hyatt D, LoCascio PF, Hauser LJ, Uberbacher EC. (2012). Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28: 2223–2230. [DOI] [PubMed] [Google Scholar]
- Itoh H, Aita M, Nagayama A, Meng X-Y, Kamagata Y, Navarro R et al. (2014). Evidence of environmental and vertical transmission of Burkholderia symbionts in the oriental chinch bug, Cavelerius saccharivorus (Heteroptera: Blissidae). Appl Envir Microb 80: 5974–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltz O, Koella JC. (2003). Host growth conditions regulate the plasticity of horizontal and vertical transmission in Holospora undulata, a bacterial parasite of the protozoan Paramecium caudatum. Evolution 57: 1535–1542. [DOI] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL. (2010). Quake: quality-aware detection and correction of sequencing errors. Genome Biol 11: R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. (2011). Adaptive seeds tame genomic sequence comparison. Genome Res 21: 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner M, Young JC, Shah M, VerBerkmoes NC, Dubilier N. (2013). Metaproteomics reveals abundant transposase expression in mutualistic endosymbionts. mBio 4: e00223-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DM, Gustafson RG, Cavanaugh CM. (1996). Vertical transmission of chemoautotrophic symbionts in the bivalve Solemya velum (Bivalvia: Protobranchia). Biol Bull 190: 195–202. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C et al. (2004). Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35: 3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R, Lima-Mendez G, Toussaint A. (2009). ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res 38: D57–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Goodson M. (2011). Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21: 936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais G, Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27: 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall IPG, Blainey PC, Spormann AM, Quake SR. (2012). A single-cell genome for Thiovulum sp. Appl Envir Microb 78: 8555–8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, von Dohlen CD. (2011). An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr Biol 21: 1366–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. (2011). Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26. [DOI] [PubMed] [Google Scholar]
- Moran NA, Bennett GM. (2014). The tiniest tiny genomes. Annu Rev Microbiol 68: 195–215. [DOI] [PubMed] [Google Scholar]
- Mouton L, Thierry M, Henri H, Baudin R, Gnankine O, Reynaud B et al. (2012). Evidence of diversity and recombination in Arsenophonus symbionts of the Bemisia tabaci species complex. BMC Microbiol 12: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A, Peretó J, Gil R, Latorre A. (2008). Learning how to live together: genomic insights into prokaryote–animal symbioses. Nat Rev Genet 9: 218–229. [DOI] [PubMed] [Google Scholar]
- Nei M, Li WH. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci 76: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton ILG, Bordenstein SR. (2011). Correlations between bacterial ecology and mobile DNA. Curr Microbiol 62: 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normark BB, Ross L. (2014). Genetic conflict, kin and the origins of novel genetic systems. Philos Trans R Soc B 369: 20130364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver G, Rodrigues CF, Cunha MR. (2011). Chemosymbiotic bivalves from the mud volcanoes of the Gulf of Cadiz, NE Atlantic, with descriptions of new species of Solemyidae, Lucinidae and Vesicomyidae. ZooKeys 113: 1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. (2014). Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, Chin FYL. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Phipson B, Smyth GK. (2010). Permutation P-values should never be zero: calculating exact P-values when permutations are randomly drawn. Stat Appl Genet Mol Biol 9: 39. [DOI] [PubMed] [Google Scholar]
- Plazzi F, Ribani A, Passamonti M. (2013). The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics 14: 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Development Team. (2012) R: A Language and Environment for Statistical Computing. The R Foundation for Statistical Computing: Vienna, Austria. [Google Scholar]
- Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT. (2009). Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics 25: 2071–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros VID, Fleming VM, Feil EJ, Breeuwer JAJ. (2012). Diversity and recombination in Wolbachia and Cardinium from Bryobia spider mites. BMC Microbiol 12: S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SL Mode and fidelity of bacterial symbiont transmission and its impact on symbiont genome evolution. PhD thesis. Harvard University, Cambridge, MA, pp 123–139.
- Salem H, Florez L, Gerardo N, Kaltenpoth M. (2015). A out-of-body experience: the extracellular dimension for the transmission of mutualistic bacteria in insects. Proc R Soc B 282: 20142957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S, Angermeier H, Schiller R, Lindquist N, Hentschel U. (2008). Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl Envir Microbiol 74: 7694–7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma PP, Zardus JD, Boyle EE, González VL, Jennings RM, McIntyre E et al. (2013). Into the deep: a phylogenetic approach to the bivalve subclass Protobranchia. Mol Phylogenet Evol 69: 188–204. [DOI] [PubMed] [Google Scholar]
- Siguier P, Filée J, Chandler M. (2006). Insertion sequences in prokaryotic genomes. Curr Opin Microb 9: 526–531. [DOI] [PubMed] [Google Scholar]
- Sipkema D, de Caralt S, Morillo JA, Al-Soud WA, Sørensen SJ, Smidt H et al. (2015). Similar sponge-associated bacteria can be acquired via both vertical and horizontal transmission. Environ Microbiol 17: 3807–3821. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FJ, Cavanaugh CM. (2006). Bacterial endosymbioses in Solemya (Mollusca: Bivalvia)—model systems for studies of symbiont–host adaptation. Antonie van Leeuwenhoek 90: 343–360. [DOI] [PubMed] [Google Scholar]
- Stewart FJ, Dmytrenko O, Delong EF, Cavanaugh CM. (2011). Metatranscriptomic analysis of sulfur oxidation genes in the endosymbiont of Solemya velum. Front Microbiol 2: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FJ, Young CR, Cavanaugh CM. (2008). Lateral symbiont acquisition in a maternally transmitted chemosynthetic clam endosymbiosis. Mol Biol Evol 25: 673–687. [DOI] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. (2007). UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23: 1282–1288. [DOI] [PubMed] [Google Scholar]
- Taylor JD, Glover EA, Williams ST. (2008). Ancient chemosynthetic bivalves: systematics of Solemyidae from eastern and southern Australia (Mollusca: Bivalvia). Mem Queensl Mus 54: 75–104. [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A et al. (2002) From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. John Wiley and Sons, Inc.: Hoboken, NJ, USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Didelot X. (2008). A comparison of homologous recombination rates in bacteria and archaea. ISME J 3: 199–208. [DOI] [PubMed] [Google Scholar]
- Wu M, Eisen JA. (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol 9: R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. (2012). A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28: 3326–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. (2011). PHAST: a fast phage search tool. Nucleic Acids Res 39: W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. (2013). The MaSuRCA genome assembler. Bioinformatics 29: 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.