

Abstract
The application of desymmetrization strategies in chemical synthesis has allowed fundamentally new synthetic sequences that efficiently create dense and polyfunctional stereochemical arrays. Enantiotopic group discrimination has become a well-established method of global desymmetrization, while the conceptually unique strategy of local desymmetrization by diastereotopic group discrimination has its own advantages. This microreview focuses on the application of local desymmetrization in natural product synthesis and places a particular emphasis on the efficiency engendered by diastereotopic group discrimination. Local desymmetrization is subdivided into three distinct manifolds; examples under each paradigm are presented and compared.
Keywords: desymmetrization, diastereotopic, enantiotopic, natural products, total synthesis
Graphical abstract
Three distinct modes of diastereotopic group selection in natural product synthesis are reviewed. Case studies that enable the rapid buildup of stereochemical complexity using local desymmetrization are emphasized.
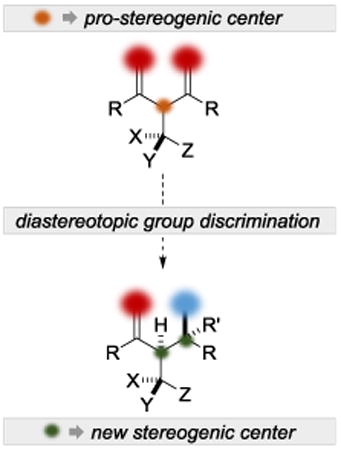
1. Introduction
The abundance of stereochemically rich molecules in natural products and drug molecules presents a plethora of synthetic challenges that can be attacked from many angles. The toolbox that synthetic organic chemists have at their disposal has evolved to provide reactions that efficiently construct stereochemical arrays. Of the myriad strategies available for stereoselective natural product synthesis, desymmetrization methods possess key advantages and simultaneously face unique challenges[1] Stereoselective desymmetrization as a synthetic tactic can be dissected into enantiotopic group discrimination (differentiation of enantiotopic groups in achiral molecules), and diastereotopic group discrimination. While global desymmetrization strategies that make use of enantiotopic group discrimination have been well studied and reviewed,[1] local desymmetrization strategies that apply diastereotopic group discrimination have received substantially less attention, despite their ability to input multiple stereocenters with high fidelity.[2]
The application of local desymmetrization as a strategy for the rapid construction of stereochemical complexity has been most evident in the field of natural product synthesis. The value afforded by this strategy is derived from its efficiency, which is a consequence of the multipurpose nature of diastereotopic group discrimination. For example, using this strategic manifold, it is possible to use achiral reagents/catalysts to carry out functional group manipulations while simultaneously controlling the outcome at a nascent stereocenter (Figure 1, i). Expanding on that theme, it is possible to introduce additional stereocenters if the diastereotopic groups are themselves prochiral (Figure 1, ii). If the diastereotopic group selection is occurring concomitantly with conversion of a chirotopic non-stereocenter into a stereocenter, then 1-2 stereocenters can be defined (Figure 1, iii). Cases (ii) and (iii) in particular offer possibilities for especially dramatic increases in stereochemical complexity. Desymmetrization tactics can become truly enabling in instances where it is markedly simpler to synthesize a symmetrical substrate (or fragment of a substrate) and apply desymmetrization methods than it is to construct the same motifs using pre-installed chirality. This microreview will focus on these themes, and specifically on cases where diastereotopic group discrimination (local desymmetrization) has successfully been applied to total synthesis. The microreview covers cases after 2010, with the inclusion of one earlier foundational example. Our aim is not to be comprehensive, but instead to provide the reader with the range of the current art.
Figure 1.
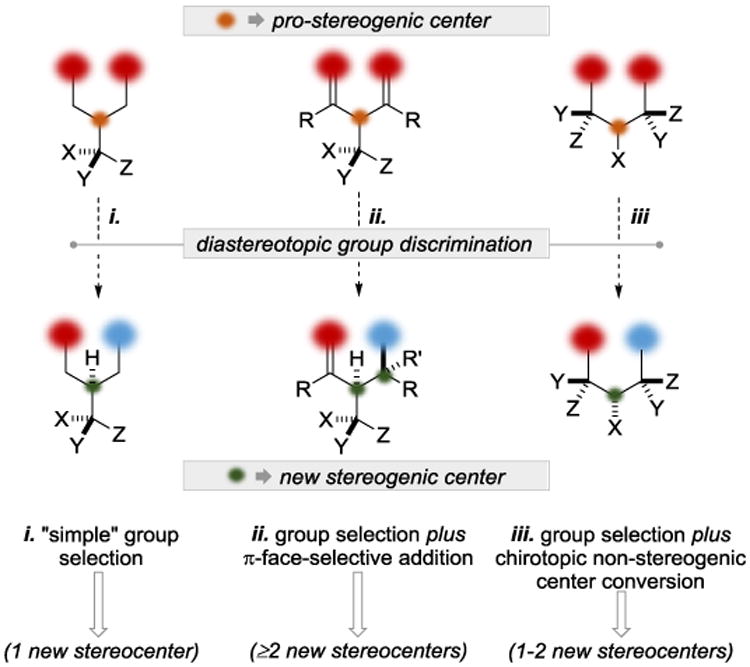
Complexity-building local desymmetrization modes enabled by diastereotopic group discrimination.
2. Simple Diastereotopic Group Selection
A. Koert's Synthesis of Awajanomycin (2011)
In 2011, the Koert group completed a total synthesis of (+)-awajanomycin (4) (Scheme 1).[3] The authors were able to synthesize enantioenriched alkene 1 rapidly from diethyl ketomalonate using an asymmetric allylboration. The diester 1 was treated with potassium osmate dihydrate to accomplish a substrate-controlled dihydroxylation; the authors attributed the stereochemical outcome of the dihydroxylation to the conformational bias enforced by the TMS group. The resulting diol (2) underwent a diastereotopic group discrimination via lactonization to form 3. By distinguishing the two ethyl esters, the authors were able to control the outcome at the new stereocenter defined during the lactonization. In addition to the requisite diasterotopic group selection, this case successfully navigates a potentially complicating constitutional isomer issue: the formation of a single γ -lactone occurs to the complete exclusion of alternative δ-lactones. By differentiating the diols during the diastereotopic group selection, the remaining alcohol could be functionalized in a manner that permitted the lactone intermediate 3 to be advanced through five additional synthetic steps to complete the total synthesis of (+)-awajanomycin (4).
Scheme 1.
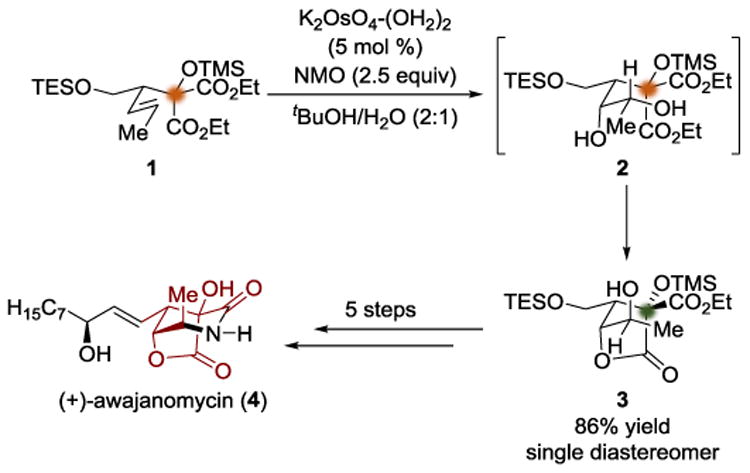
Total synthesis of awajanomycin by Koert.
B. Wu's Synthesis of 2-epidevinylantirhine, geissoschizol, isogeissoschizol, and dihydrocorynantheol (2013)
In 2013, the Wu group realized a catalytic asymmetric transfer hydrogenation/lactamization cascade that delivers the core of several indoloquinolizidine natural products (Scheme 2).[4] The initial asymmetric reduction of the imine moiety of 5 to 6 rendered the ethyl esters diastereotopic. Diastereoselection is realized in stage two of the cascade by virtue of the fact that the favored and disfavored six-membered transition states place the C2 substituent in an equatorial or axial position, respectively. The locally desymmetrizing lactamization is spontaneous after the enantioselective reduction, delivering two new stereocenters in one step. The approach is conceptually similar to the example from Koert above, which also utilized local desymmetrization of a diester. Reduction of the resulting amide 7 with LiAlH4 delivered 2-epidevinylantirhine (8) in 84% yield. Using this reaction manifold to efficiently access 7, the authors were also able to complete the total syntheses of geissoschizol (9), isogeissoschizol (10), and dihydrocorynantheol (11).
C. Sorensen's Synthesis of Jiadifenolide (2014)
The relative inertness of simple methyl groups renders them a priori as unlikely candidates for diastereotopic group selection; however, the advent of modern C–H activation methodologies has revealed opportunities for the generation of quaternary stereocenters by differentiation of gem-dimethyl groups. During their synthesis of jiadifenolide (14), the Sorensen group arrived at intermediate 12.[5] Their synthetic approach to fashion the all-carbon quaternary center at C5 was to use diastereotopic group discrimination (Scheme 3). By utilizing a C–H activation protocol developed by Sanford and coworkers,[6] they were able to achieve directed oxidation of the unactivated diastereotopic methyl groups at C5. Sanford's Pd(II)/PhI(OAc)2 conditions led to a 1:1 mixture of diastereomeric acetates (13a and 13b), presumably due to the equilibrating chair conformations (12a and 12b) that are similar in their stability and reactivity (Scheme 3), a circumstance that would lead to poor differentiation. While the observed selectivity was not optimal, the reaction fortunately did furnish separable diastereomers. The authors were then able to carry acetate 13b through eight synthetic steps to arrive at the target molecule (14).
D. Johnson's Synthesis of Paspaline (2015)
In 2015, the Johnson group described a total synthesis of the indole diterpenoid paspaline (17) which merged local and global desymmetrization strategies (Scheme 4).[7] In the course of the synthesis, intermediate 15 was synthesized by a route that took advantage of enantiotopic group discrimination via a biocatalytic reduction. Moving forward from 15, application of the Sanford C–H activation[6] allowed the completely diastereselective acetoxylation of the equatorial methyl group to give 16. In contrast to the jiadifenolide case, the tricycle 15 is a rigid system that places the diastereotopic methyl groups in very different environments in relation to the oxime directing group. The equatorial methyl group is approximately coplanar with the benzyl oxime, a circumstance that led to perfect diastereocontrol for the C–H acetoxylation. This local desymmetrization enabled the stereochemical outcome at an all-carbon quaternary center to be governed. This route was not “plan A” and the application of the local symmetry-breaking C–H functionalization in this synthesis averted a crisis by allowing a rather non-intuitive disconnection. Acetate 16 was then carried on to complete the total synthesis of paspaline (17).
E. Porco's Synthesis of (-)-6-epi- and (+)-30-epi-13,14-Didehydroxyisogarcinol (2016)
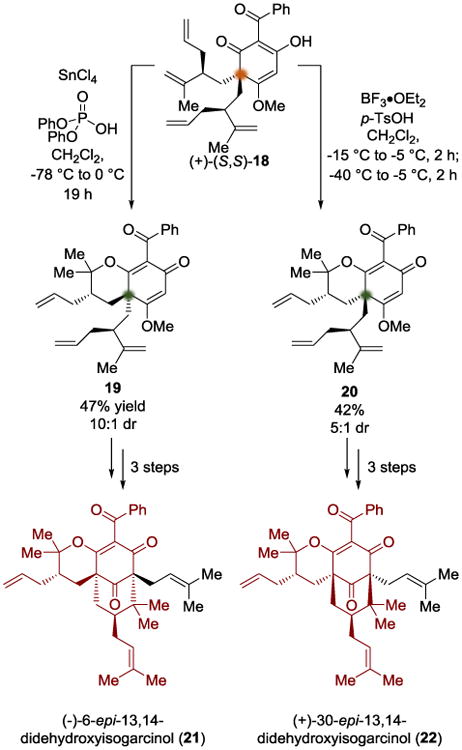
The Porco group recently reported a synthesis of four polycyclic polyprenylated acylphloroglucinol (PPAP) targets, two of which required the use of diastereotopic group differentiation (Scheme 5).[8a] Through a regioselective bis-alkylation of a phenol, the authors were able to synthesize polyene (+)-(S,S)-18. When exposed to SnCl4 and diphenyl phosphoric acid, the bicyclic pyran 19 was obtained in 47% yield and 10:1 dr. Interestingly, when they treated the same acylphloroglucinol starting material with BF3 and p-TsOH, the diastereomeric product 20 was obtained in 42% yield and 5:1 dr. It was proposed that the Lewis acid binds in a bidentate manner to the β-keto enol, resulting in a hexacoordinate Sn- or tetrahedral BF2 complex, depending on the reagent used.[8b-d] They argued, using B3LYP calculations comparing relative transition state energies, that the diastereoselectivity in these reactions arises from an intramolecular protonation event after complex formation. The authors went on to use 19 to complete the synthesis of (-)-6-epi-13,14-didehydroxyisogarcinol (21) and 20 to complete the synthesis of (+)-30-epi-13,14-didehydroxyisogarcinol (22) using the same three-step sequence.
3. Diastereotopic Group Selection with π -Face Selective Addition
A. Leighton's Synthesis of Zincophorin Methyl Ester (2011)
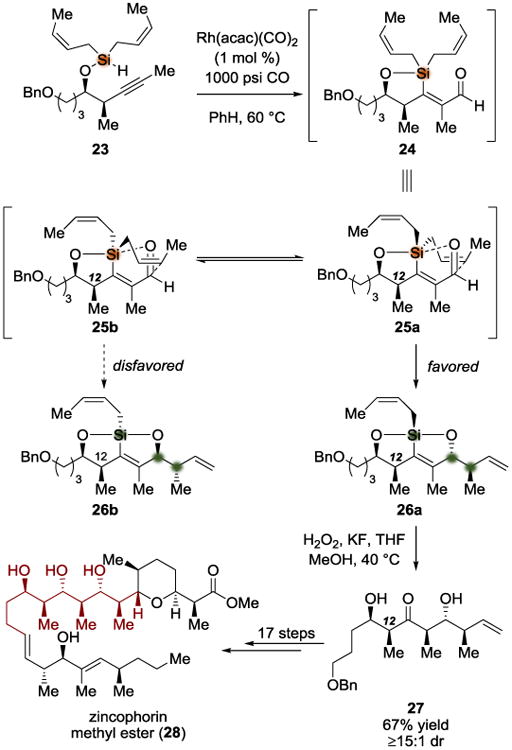
In 2011, Leighton and coworkers reported a convergent and stereoselective synthesis of zincophorin methyl ester (28) (Scheme 6).[9] Using Shi's asymmetric epoxidation,[10] Pagenkopf's epoxide-opening alkynylation,[11] and a NaH-promoted dehydrogenative silylation, the authors assembled the key substrate 23. The authors were then able to use Rh(acac)CO2 with 1000 psi CO to trigger a silylformylation-crotylsilylation cascade followed by a Tamao-Fleming oxidation and a diastereoselective tautomerization. The diastereotopic group discrimination in this case occurs during the crotylsilylation, where the crotyl groups were successfully distinguished. Based on an earlier model proposed by the same group,[9c] it feasible that attack of the crotyl group from the Si face of the aldehyde (as in 25b) would be disfavored compared to attack from the Re face (as in 25a) due to steric clashing with the C12 methyl group. In prior work,[9d] the authors described the enol intermediate formed by crotylsilylation and Tamao-Fleming oxidation to be conformationally controlled by A[1,3] strain and syn-pentane interactions; they argued that both secondary alcohols were able to participate in hydrogen bonding to allow delivery of a proton to the back face of the enol. The final product of this cascade, ketone 27, was obtained in ≥15:1 dr in a reaction sequence that rapidly provides five new contiguous stereocenters from a simple starting material. Ketone 27 was carried on and used to complete the total synthesis of zincophorin methyl ester (28). This sequence represents an unusual case where the three stereocenters that are established during the diastereotopic group selection do not all correspond to the three new stereocenters that appear in the final product. The silicon atom becomes stereogenic upon intramolecular crotylation but is lost during the Tamao-Fleming oxidation/diastereoselective tautomerization sequence that provides the third stereocenter (ketone α-center) in the final product. Thus, diastereotopic group selection in this case is leveraged exclusively to provide mutual π-facial selectivity in two distal prochiral functional groups.
B. Willis's Synthesis of Blepharocalyxin D (2013)
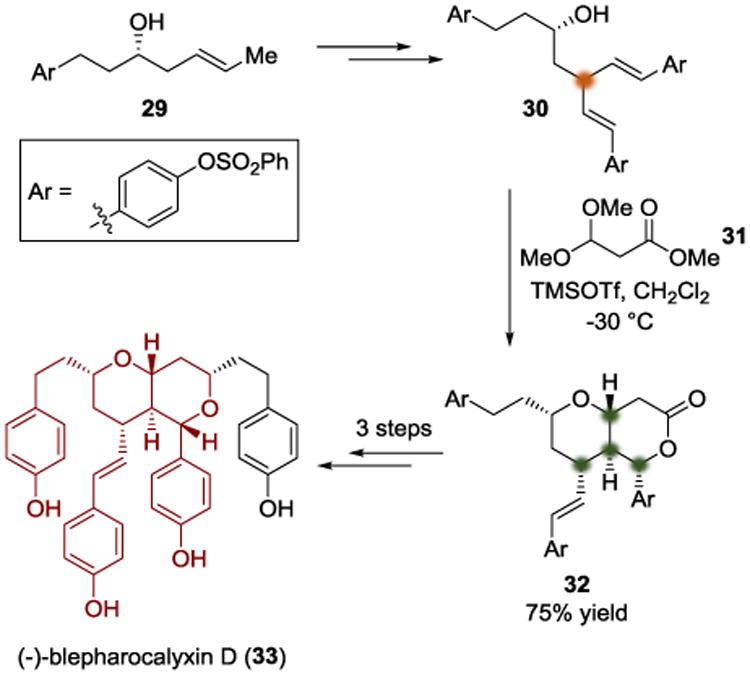
In 2013, the Willis group reported a total synthesis of (−)-blepharocalyxin D (33) (Scheme 7).[12] Using (S)-homoallylic alcohol 29, the authors were able to synthesize the desymmetrization substrate 30, which possesses diastereotopic styrenyl groups. The authors found that treating diene 30 with acetal 31 and TMSOTf led to the bicyclic lactone 32 completely diastereoselectively. Three additional steps led to the completion of the synthesis.
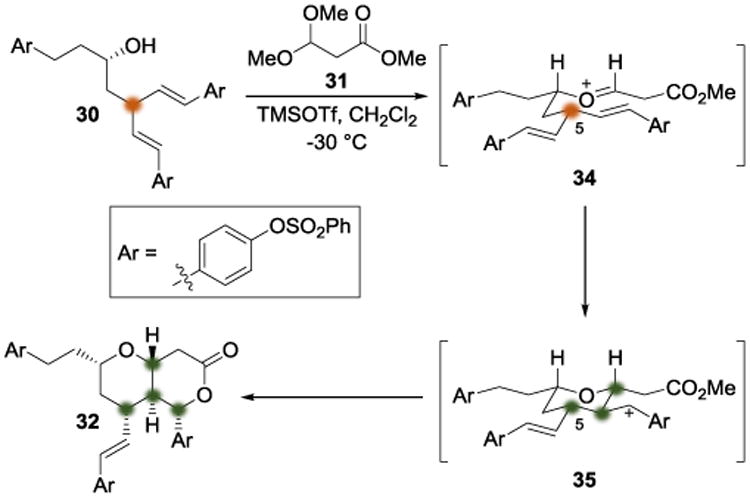
The authors explained their high diastereoselectivity in the transformation from 30 to 32 using a chair model for the transition state geometry (Scheme 8). They argued that, following oxocarbenium ion formation from the two reaction components to give 34, the lowest-energy reaction path would require the substituent at C5 to be equatorial. Subsequent trapping of the nascent benzylic carbenium ion (in 35) by the ester would then furnish lactone 32.
C. Zakarian's Synthesis of Tatanans B and C (2013)
Zakarian's approach to the synthesis of tatanans B (39) and C (40) takes advantage of diastereotopic group differentiation during a cyclodearomatization (Scheme 9).[13] α, β-Unsaturated ester 36 was used as a starting point to synthesize the desymmetrization substrate 37. Then, the allylic carbonate was converted into a palladium π-allyl species, which was found to undergo an intramolecular cyclodearomatization to give spirocyclohexadienone 38 in 84% yield as a mixture of diastereomers (it should be noted that one of these diastereomers arises from atropoisomerism). This reaction quickly allowed tatanans B (39) and C (40) to be completed after two additional steps.
The authors rationalized the stereochemical outcome of the reaction using four transition state-based arguments (Scheme 10). First, they argued that TS-41a is more favorable than TS-41b due to the steric clashing that would occur in the latter, between the β-methoxy substituent of the cyclohexadienone and the nearby aryl ring. By comparing TS-42a and TS-42b, they were able to explain which diastereotopic aryl group undergoes the cyclodearomatization; they pointed out that the axial positioning of the aryl group in TS-42b is likely to be disfavored compared to the equatorial position in TS-42a. The next pair of transition states, TS-43a and TS-43b, compare an attack on an equatorial π-allyl electrophile with an axial one; the former was predicted to be favored. Finally, with respect to the atropodiastereomeric structures TS-44a and TS-44b, the authors proposed that there would be a noticeable steric interaction occurring if the ortho-OMe group forced into the proximity of the fledgling cyclohexane ring. In this example, both the reactive functional group that is breaking symmetry and the π-faces of the diastereotopic groups are prochiral; therefore, three new stereogenic centers arise in the symmetry breaking event.
D. Johnson's Synthesis of Pactamycin (2013)
In 2013, Johnson and coworkers disclosed a successful total synthesis of pactamycin (Scheme 11).[14b, 14c] This approach used 2,4-pentanedione-derived urea 45 in an enantioselective Mannich reaction with imine diene 46, which provided 47 in 70% yield and 98:2 er. A diastereoselective reduction using Li(OtBu)3AlH (LTBA) afforded the secondary alcohol 49 with three contiguous stereocenters in 72% yield. The stereochemical model (48) that we proposed for the hydride addition invoked chelation[15] of the lithium to the carbamate nitrogen as well as the carbonyl of the ketone that is undergoing the reduction. In this way, the stereochemical information from the Mannich addition was parlayed in a diastereotopic group differentiation, and 47 was locally desymmetrized. This is a good example of how prochiral diastereotopic groups can be differentiated in order to generate two new adjacent stereocenters. Keto alcohol 49 was then elaborated into pactamycin (50) in eleven steps. The brevity of this approach facilitated creation of a library of pactamycin analogs and assessment of their biological activity in cancer cell lines.[14d]
E. Shair's Synthesis of Hyperforin, Nemorosone, and Secohyperforin (2013, 2015)
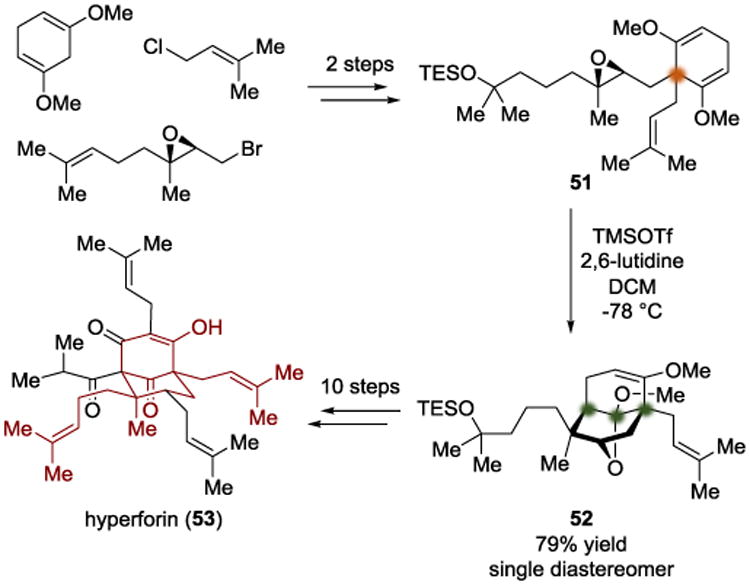
In 2013, the Shair group disclosed an asymmetric total synthesis of the PPAP natural product hyperforin (53) (Scheme 12).[16] The crux of their synthetic strategy was to differentiate diastereotopic enol ethers to define an all-carbon quaternary center. The group developed a two-step synthesis of the local desymmetrization substrate 51 from simple starting materials. Their intention was to use a Lewis acid to activate the epoxide in 51 and allow a nucleophilic attack by an enol ether to form an oxonium intermediate (55). Subsequent intramolecular attack by the nascent secondary alkoxide would then form a cyclic ketal (Scheme 13). By screening Lewis acids that were capable of engaging the epoxide in 51, TMSOTf was identified as the optimal Lewis acid, providing the desired tricyclic product in 79% yield. The authors were then able to use desymmetrized intermediate 52 to finish the total synthesis of hyperforin (53) after ten additional steps.
Scheme 12.
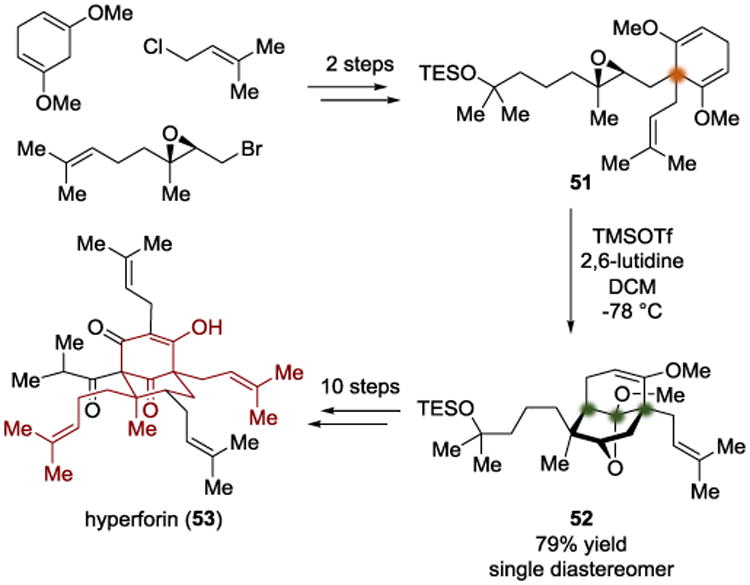
Total synthesis of hyperforin by Shair.
Their stereochemical outcome of the Lewis acid-mediated epoxide opening cascade was consistent with the chairlike transition state (54b). Epoxide-opening by the alternative enol ether would require a boatlike transition state (54a), which was suggested to be less favorable. Their approach also imposed sufficient geometric constraints to favor 6-endo-tet cyclization (at the more substituted epoxide carbon) over the potentially competitive 5-exo-tet mode.
After completing their synthesis of hyperforin (53), the Shair group went on to apply their local desymmetrization approach to two other PPAP natural products: nemorosone (58) and secohyperforin (59) (Scheme 14).[17] In a departure from their approach to hyperforin (53), the authors sought to eliminate the need for a C-O bond cleavage step (primarily due to the use of Me2BBr in that step). With that goal in mind, the authors developed conditions to prevent the type of ring closure (55 → 52) that led to cyclic ketal 52 in the hyperforin synthesis. It was found that TIPSOTf and 2,6-lutidine in DCM, warming from -78 °C to room temperature, gave a 95% yield of the TIPS-protected secondary alcohol resulting from epoxide opening (57). This intermediate (57) was converted into a common intermediate which the authors used to access both target molecules. They were able to install a benzoyl (in the case of nemorosone (58)) or an isobutyryl (in the case of secohyperforin (59)) group at the bridgehead by deprotonating with LiTMP and trapping with the desired acyl chloride. This strategy allowed the authors to expand the desymmetrization motif used for hyperforin and enabled the total synthesis of both targets.
Scheme 14.
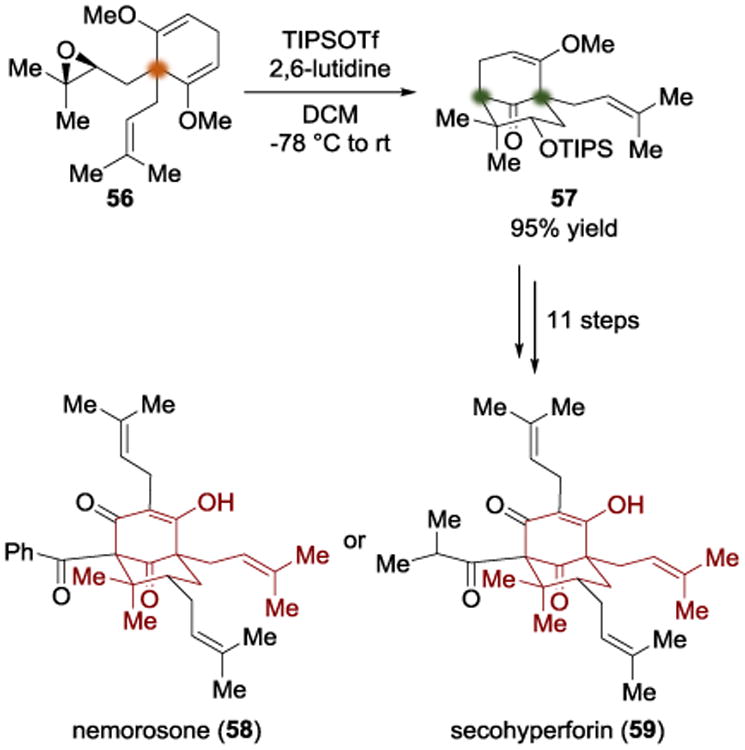
Total synthesis of nemorosone and secohyperforin by Shair.
4. Diastereotopic Group Selection and Configurational Definition of a Chirotopic Non-stereogenic Group
A. Schreiber's Synthesis of FK506 (1990)
An early example of local desymmetrization comes from Schreiber and coworkers, who applied a diastereoselective discrimination of two terminal methyl esters in the synthesis of the FKBP-binding agent FK506 (62) (Scheme 15).[18] The authors synthesized the pseudo-C2-symmetric substrate 60 from arabitol through a nine-step sequence. Treatment of 60 with Pd(OH)2 unveiled the chirotopic non-stereogenic free alcohol, which the authors used in a diastereoselective intramolecular lactonization (dr not reported) with pyridinium p-toluenesulfonate (PPTS) to arrive at 61.
Using ground-state conformational analysis similar to that of Hoye,[19] the stereochemical outcome of this desymmetrization can be analyzed by considering the relative populations of 63a and 63b at equilibrium (Scheme 16). Considering the quasi 1,3-diaxial interactions present in 63b, the equilibrium likely lies to the right, providing a plausible explanation for the preferential lactonization that gives 61. The benefits that the local desymmetrization strategy offered in this context were control of the incipient fifth stereocenter in 61, as well as differentiation of the terminal methyl esters. The authors were then able to use 61 to construct the C10-C19 domain of FK506 (62).
B. Sammakia's Synthesis of Dermostatin A (2011)
The oxopolyene macrolide dermostatin A (66) was synthesized by the Sammakia group using an early local desymmetrization of 64 (Scheme 17).[20] The authors found that subjecting 64 to PPTS in toluene at 110 °C allowed a trans-ketalization to give 65 with perfect diastereoselectivity. Although they observed a 9:1 mixture of internal acetal to terminal acetal, they were able to isolate 65a in 82% yield.
The authors rationalized their impressive stereoselectivity using the ground-state conformations of 65a and 65b (Scheme 18). They argued that the axial substituent present in 65b makes it less thermodynamically stable than 65a, which results in a population imbalance favoring the formation of 65a at thermodynamic equilibrium. Additionally, they emphasized the elevated impact of the A-value in 1,3-dioxanes compared to cyclohexanes.[21] By taking advantage of the relative stability of 65a, the authors were able to set a stereocenter and differentiate the termini. Using 65a to form the polyol core of the final product, the authors completed the total synthesis of dermostatin A (66).
C. Krische's Synthesis of Zincophorin Methyl Ester (2015)
The Krische group employed a distinct diastereotopic group selection in their synthesis of zincophorin methyl ester (28). The key building block was prepared through hydrogen borrowing catalysis (Scheme 19).[22] Using the simple, cheap, achiral starting material 67, the authors were able to use an (S)-Ir-SEGPHOS complex to catalyze a double anti-crotylation with 3-butenyl-2-acetate to give 68. From that point, an iodoetherification allowed a diastereotopic discrimination between the two terminal alkenes to provide 69a in high yield and stereoselectivity, forming a “propionate based triketide motif”.[22] Using an analysis of the conformer equilibrium between 70a and 70b, we can understand that prohibitively strong 1,3-diaxial interactions arise when the 1-buten-3-yl group is placed in the axial position, which results in the observed diastereomer (Scheme 20). This reaction sequence installed six contiguous stereocenters in only two synthetic operations. The iodopyran 69a served to temporarily protect one alkene so that the other olefinic terminus could be homologated via a cross metathesis reaction; reductive fragmentation immediately after the metathesis restored the alkene and alcohol functionality for ultimate elaboration to zincophorin methyl ester (28).
5. Outlook and Conclusions
The application of desymmetrization strategies to natural product synthesis continues to have a positive impact on the field in terms the efficiency with which difficult stereochemical patterns can be constructed. While enantiotopic group discrimination (global desymmetrization) remains a cornerstone for synthetic chemists seeking to control absolute stereochemistry, diastereotopic group discrimination (local desymmetrization) continues to evolve as a tactic that has its own benefits: 1) achiral reagents can be used to control the stereochemical outcome at prostereogenic carbons, 2) there is the realistic possibility of separating the products of the desymmetrization (which typically more challenging if the products are enantiomers, as in enantiotopic differentiation), and 3) if diastereotopic groups can be considered differentiable, it can greatly simplify retrosynthetic analysis of complex targets. For these reasons, we envisage that diastereotopic group differentiation will continue to have a burgeoning role in asymmetric synthesis and will find new applications in the field going forward.
Scheme 2.
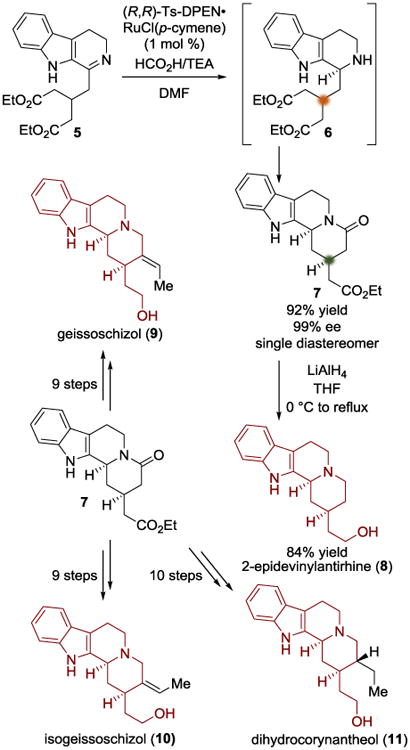
Total synthesis of 2-epivinylantirhine, geissoschizol, isogeissoschizol, and dihydrocorynantheol.
Scheme 3.
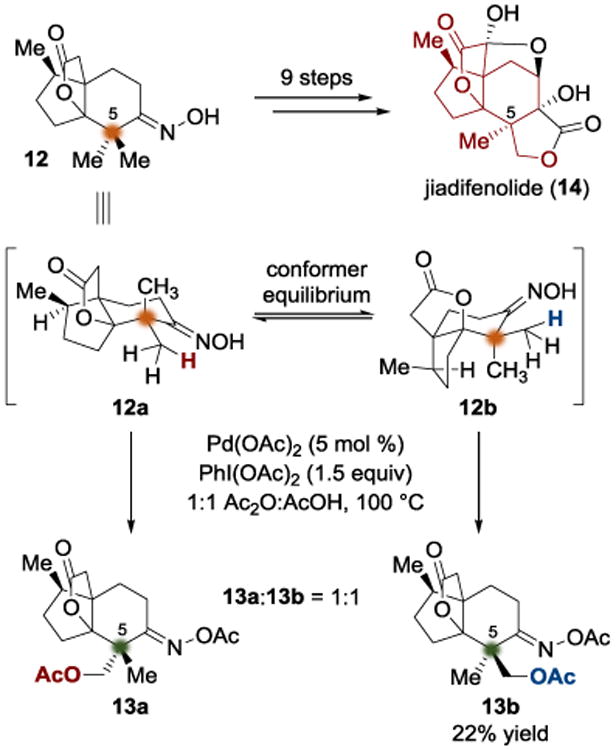
Total synthesis of jiadifenolide by Sorensen.
Scheme 4.
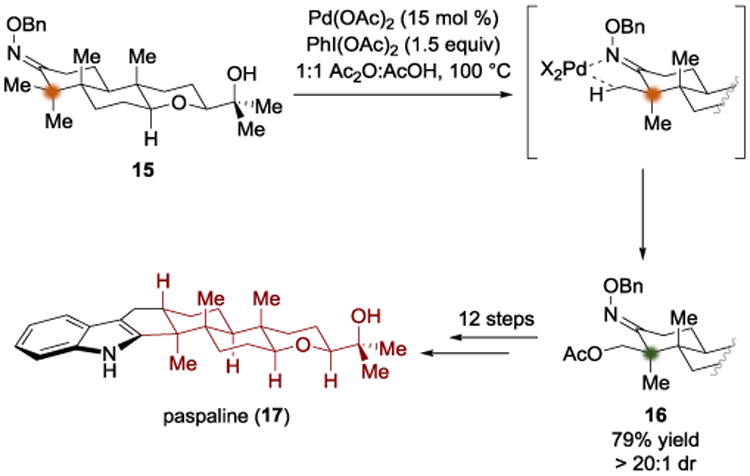
Total synthesis of paspaline by Johnson.
Scheme 5.
Porco's synthesis of two PPAP targets.
Scheme 6.
Total synthesis of zincophorin methyl ester by Leighton.
Scheme 7.
Total synthesis of (-)-blepharocalyxin D by Willis.
Scheme 8.
Rationale for observed diasteroetopic group differentiation.
Scheme 9.
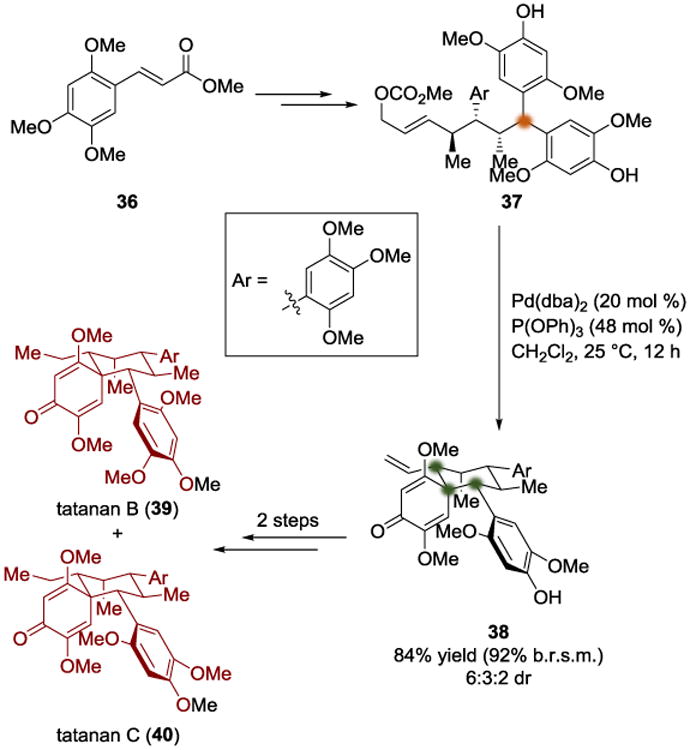
Total synthesis of tatanans B and C by Zakarian.
Scheme 10.

Transition state comparisions for cyclodearomatization.
Scheme 11.
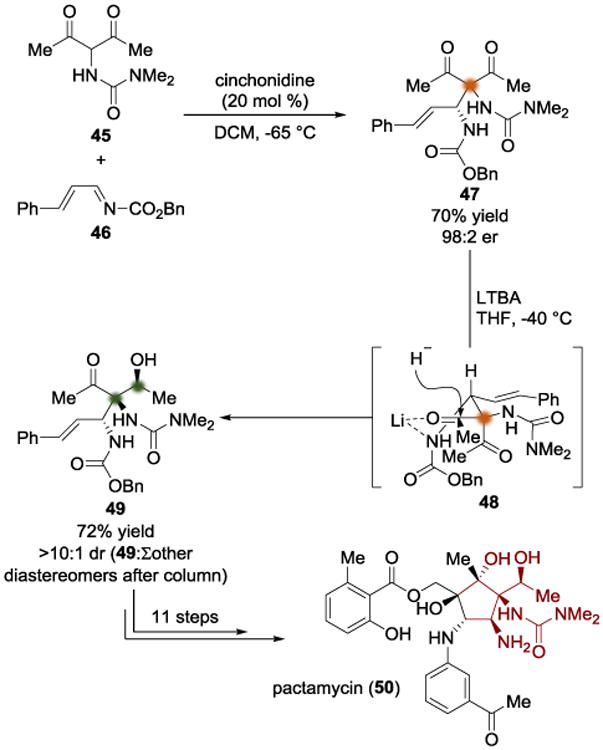
Total synthesis of pactamycin by Johnson.
Scheme 13.
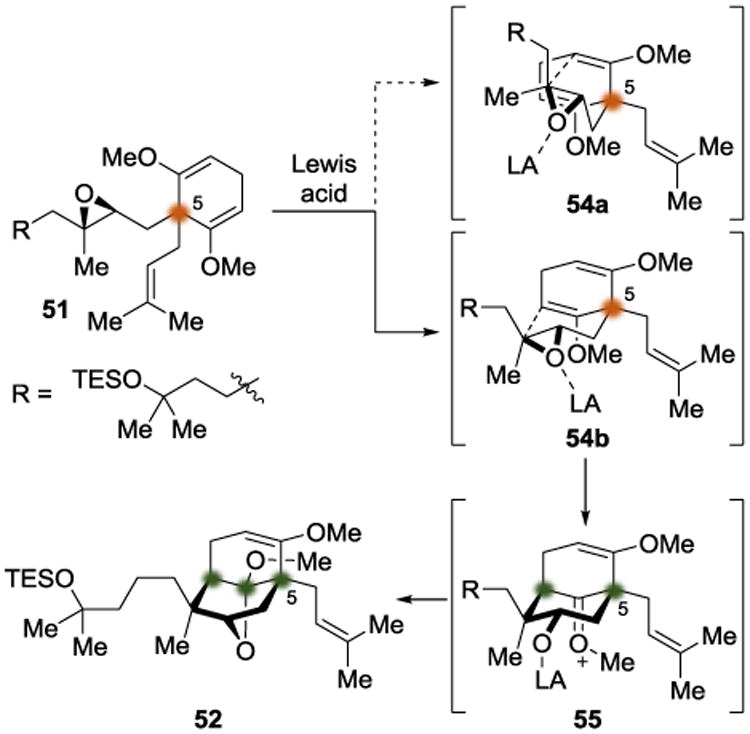
Stereochemical model for Lewis acid-mediated desymmetrization.
Scheme 15.
Total synthesis of FK506 by Schreiber.
Scheme 16.
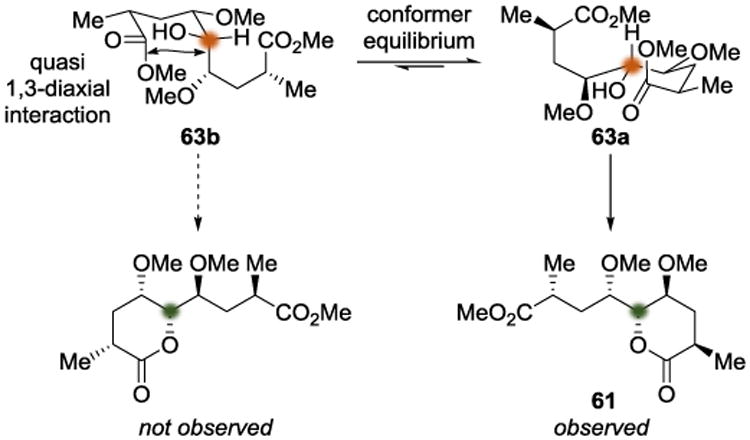
Conformational equilibrium leading to the observed lactonization.
Scheme 17.
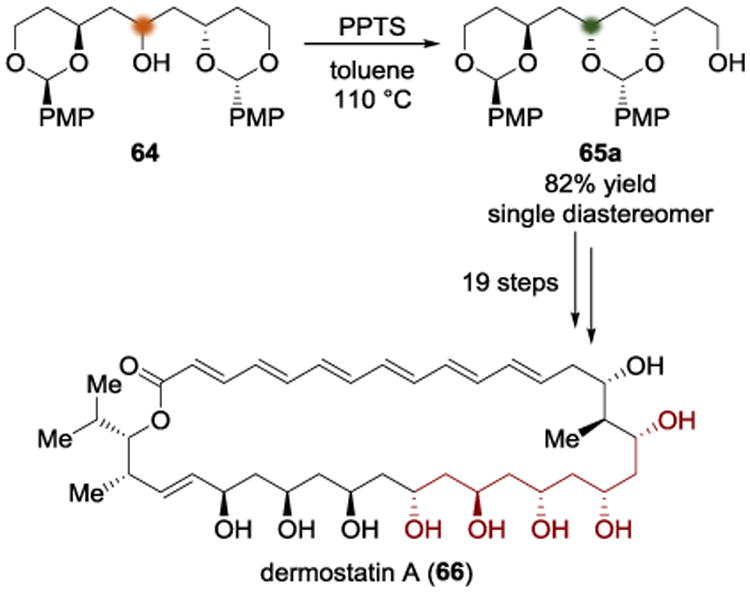
Total synthesis of dermostatin A by Sammakia.
Scheme 18.
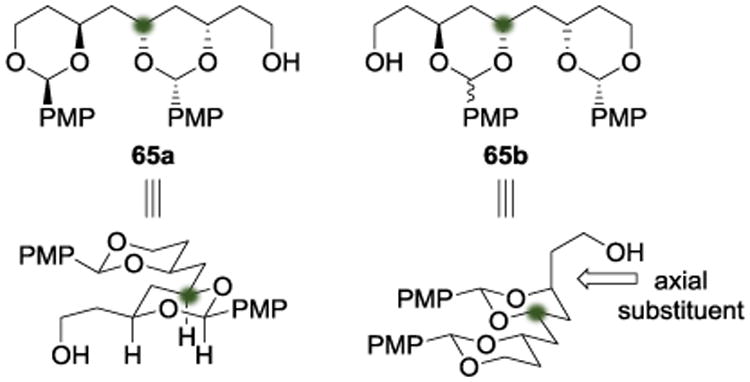
Total synthesis of dermostatin A by Sammakia.
Scheme 19.
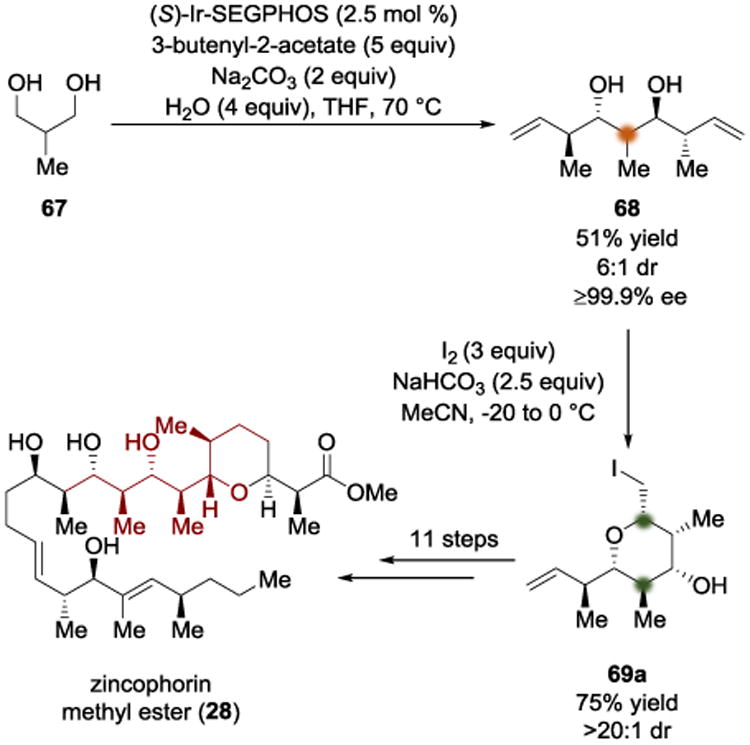
Total synthesis of zincophorin methyl ester by Krische.
Scheme 20.

Rationalization of stereochemical outcome in iodoetherification.
Acknowledgments
The authors acknowledge Award R35 GM118055 from the National Institute of General Medical Sciences for support of our work on desymmetrization in natural product synthesis.
Biographies
Jeffrey S. Johnson was born in Emporia, Kansas (USA) in 1971. He studied chemistry at the University of Kansas where he obtained his B.S. in Chemistry in 1994. Jeff earned his Ph.D. in 1999 from Harvard University under the direction of David A. Evans and conducted postdoctoral research from 1999-2001 in the laboratories of Robert G. Bergman at the University of California at Berkeley. He has been a faculty member at the University of North Carolina at Chapel Hill since 2001 and has held the A. Ronald Gallant Distinguished Professorship since 2014. His research interests are in chemical synthesis, catalysis, mechanism, new reaction development, and natural products.

Matthew A. Horwitz was born in Hendersonville, North Carolina (USA), in 1992. He studied chemistry at Columbia University where he obtained his B.A. in Biochemistry in 2013. Matt entered the Ph.D. program at University of North Carolina at Chapel Hill in the same year and joined the research group of Jeffrey S. Johnson.

Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.For reviews on global desymmetrization via enantiotopic group selection, see: Ward RS. Chem Soc Rev. 1990;19:1–19.Magnuson SR. Tetrahedron. 1995;51:2167–2213.Willis MC. J Chem Soc, Perkin Trans. 1999;1:1765–1784.García-Urdiales E, Alfonso I, Gotor V. Chem Rev. 2005;105:313–354. doi: 10.1021/cr040640a.Wang M, Feng M, Tang B, Jiang X. Tetrahedron Lett. 2014;55:7147–7155.Borissov A, Davies TQ, Ellis SR, Fleming TA, Richardson MSW, Dixon DJ. Chem Soc Rev. 2016;45:5474–5540. doi: 10.1039/c5cs00015g.Zeng XP, Cao ZY, Wang YH, Zhou F, Zhou J. Chem Rev. 2016;116:7330–7396. doi: 10.1021/acs.chemrev.6b00094.
- 2.For reviews on local desymmetrization via diastereotopic group selection: Schreiber SL, Poss CS. Acc Chem Res. 1994;27:9–17.Hoffmann RW. Synthesis. 2004;13:2075–2090.Studer A, Schleth F. Synlett. 2005;20:3033–3041.Nakahara K, Fujioka H. Symmetry. 2010;2:437–454.
- 3.Wohlfahrt M, Harms K, Koert U. Angew Chem Int Ed. 2011;50:8404–8406. doi: 10.1002/anie.201103679. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Wang Q, Zhang Y, Huang J, Nie L, Chen J, Cao W, Wu X. J Org Chem. 2013;78:12009–12017. doi: 10.1021/jo4020547. [DOI] [PubMed] [Google Scholar]
- 5.Siler DA, Mighion JD, Sorensen EJ. Angew Chem Int Ed. 2014;53:5332–5335. doi: 10.1002/anie.201402335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; b) Neufeldt SR, Sanford MS. Org Lett. 2010;12:532–535. doi: 10.1021/ol902720d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936–946. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Sharpe RJ, Johnson JS. J Am Chem Soc. 2015;137:4968–4971. doi: 10.1021/jacs.5b02631. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sharpe RJ, Johnson JS. J Org Chem. 2015;80:9740–9766. doi: 10.1021/acs.joc.5b01844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Boyce JH, Eschenbrenner-Lux V, Porco JA., Jr J Am Chem Soc. 2016;138:14789–14797. doi: 10.1021/jacs.6b09727. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishihara K, Nakashima D, Hiraiwa Y, Yamamoto H. J Am Chem Soc. 2003;125:24–25. doi: 10.1021/ja021000x. [DOI] [PubMed] [Google Scholar]; c) Maeda H, Haketa Y. Org Biomol Chem. 2008;6:3091–3095. doi: 10.1039/b806161k. [DOI] [PubMed] [Google Scholar]; d) Štephane B. Org Lett. 2010;12:2900–2903. [Google Scholar]
- 9.Harrison TJ, Ho S, Leighton JL. J Am Chem Soc. 2011;133:7308–7311. doi: 10.1021/ja201467z. [DOI] [PMC free article] [PubMed] [Google Scholar]; for a related application, see: Bolshakov S, Leighton JL. Org Lett. 2005;7:3809–3812. doi: 10.1021/ol0515006.; for model of diastereoselective crotylsilylation, see: Zacuto MJ, O'Malley SJ, Leighton JL. J Am Chem Soc. 2002;124:7890–7891. doi: 10.1021/ja026511y.; for model of diastereoselective protonation, see Spletstoser JT, Zacuto MJ, Leighton JL. Org Lett. 2008;10:5593–5596. doi: 10.1021/ol802489w.
- 10.a) Wang ZX, Tu Y, Frohn M, Zhang JR, Shi Y. J Am Chem Soc. 1997;119:11224–11235. [Google Scholar]; b) Shi Y. Acc Chem. 2004;37:488–496. doi: 10.1021/ar030063x. [DOI] [PubMed] [Google Scholar]
- 11.Zhao H, Engers DW, Morales CL, Pagenkopf BL. Tetrahedron. 2007;63:8774–8780. [Google Scholar]
- 12.Cons BD, Bunt AJ, Bailey CD, Willis CL. Org Lett. 2013;15:2046–2049. doi: 10.1021/ol400736w. [DOI] [PubMed] [Google Scholar]
- 13.Xiao Q, Jackson JJ, Basak A, Bowler JM, Miller BG, Zakarian A. Nat Chem. 2013;5:410–416. doi: 10.1038/nchem.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For an initial study, see: Malinowski JT, McCarver SJ, Johnson JS. Org Lett. 2012;14:2878–2881. doi: 10.1021/ol301140c.; for the total synthesis of pactamycin, see: Malinowski JT, Sharpe RJ, Johnson JS. Science. 2013;340:180–182. doi: 10.1126/science.1234756.Sharpe RJ, Malinowski JT, Johnson JS. J Am Chem Soc. 2013;135:17990–17998. doi: 10.1021/ja409944u.; for the biological assays with nanoparticle encapsulation, see: Sharpe RJ, Malinowski JT, Sorana F, Luft JC, Bowerman CJ, DeSimone JM, Johnson JS. Bioorg Med Chem. 2015;23:1849–1857. doi: 10.1016/j.bmc.2015.02.022.
- 15.Davis FA, Gaspari PM, Nolt BM, Xu P. J Org Chem. 2008;73:9619–9626. doi: 10.1021/jo801653c. [DOI] [PubMed] [Google Scholar]
- 16.Sparling BA, Moebius DC, Shair MD. J Am Chem Soc. 2013;135:644–647. doi: 10.1021/ja312150d. [DOI] [PubMed] [Google Scholar]
- 17.Sparling BA, Tucker JK, Moebius DC, Shair MD. J Am Chem Soc. 2015;17:3398–3401. doi: 10.1021/acs.orglett.5b01121. [DOI] [PubMed] [Google Scholar]
- 18.Nakatsuka M, Ragan JA, Sammakia T, Smith DB, Uehling DE, Schreiber SL. J Am Chem Soc. 1990;112:5583–5601. [Google Scholar]
- 19.a) Hoye TR, Peck DR, Swanson TA. J Am Chem Soc. 1984;106:2738–2739. [Google Scholar]; b) Hoye TR, Ryba TD. J Am Chem Soc. 2005;127:8256–8257. doi: 10.1021/ja051604b. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Arpin CC, Cullen AJ, Mitton-Fry MJ, Sammakia T. J Org Chem. 2011;76:7641–7653. doi: 10.1021/jo2012658. [DOI] [PubMed] [Google Scholar]
- 21.Eliel EL, Sr, Knoeber MC. J Am Chem Soc. 1968;90:3444–3468. [Google Scholar]
- 22.Kasun ZA, Gao X, Lipinski RM, Krische MJ. J Am Chem Soc. 2015;137:8900–8903. doi: 10.1021/jacs.5b05296. [DOI] [PMC free article] [PubMed] [Google Scholar]