

Abstract
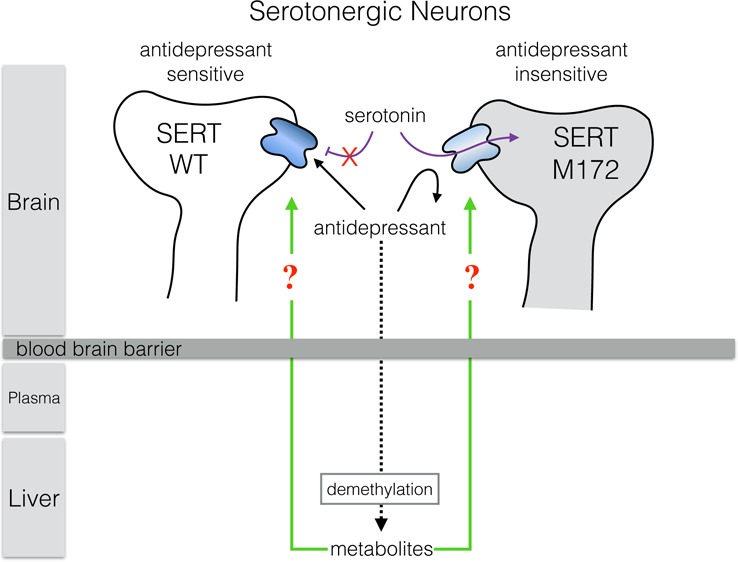
Previous studies of transgenic mice carrying a single isoleucine to methionine substitution (I172M) in the serotonin transporter (SERT) demonstrated a loss of sensitivity to multiple antidepressants (ADs) at SERT. However, the ability of AD metabolites to antagonize SERT was not assessed. Here, we evaluated the selectivity and potency of these metabolites for inhibition of SERT in mouse brain-derived synaptosomes and blood platelets from wild-type (I172 mSERT) and the antidepressant-insensitive mouse M172 mSERT. The metabolites norfluoxetine and desmethylsertraline lost the selectivity demonstrated by the parent compounds for inhibition of wild-type mSERT over M172 mSERT, whereas desvenlafaxine and desmethylcitalopram retained selectivity. Furthermore, we show that the metabolite desmethylcitalopram accumulates in the brain and that the metabolites desmethylcitalopram, norfluoxetine, and desvenlafaxine inhibit serotonin uptake in wild-type mSERT at potencies similar to those of their parent compounds, suggesting that metabolites may play a role in effects observed following AD administration in wild-type and M172 mice.
Keywords: Serotonin transporter, SSRI, antidepressants, metabolites, M172 mouse, mass spectrometry
The serotonin transporter (SERT) is a member of the solute carrier 6 (SLC6) family of neurotransmitter transporters. SERT is expressed in the brain and periphery,1,2 where it is the primary mechanism for termination of serotonergic signaling following vesicular release of serotonin (5-hydroxytryptamine, 5-HT). Altered activity of SERT has been implicated in multiple neurobehavioral disorders, including major depression, autism spectrum disorder, anxiety, post-traumatic stress disorder, and obsessive-compulsive disorder.3,4 Antidepressants (ADs) such as tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors (SNRIs) effectively block 5-HT and/or norepinephrine (NE) reuptake, thereby increasing synaptic levels of those neurotransmitters. Of these drugs, SSRIs and SNRIs are prescribed most often due to their superior safety profile in comparison to TCAs, which have multiple secondary targets.5,6 Although treatment with SSRIs and SNRIs effectively block 5-HT and/or NE reuptake almost immediately upon administration, chronic blockade is required for clinical efficacy.7,8 The mechanisms underlying the delay in symptom amelioration with AD treatment and the distinct roles of both the norepinephrine transporter (NET) and SERT as primary targets in depression are areas that remain poorly understood.9,10
Structurally, SERT and the related NET and dopamine transporters contain 12 transmembrane domains (TMs), of which TMs 1, 3, 6, and 8 form the majority of the substrate permeation pathway as well as key interactions with ions, substrates, and antagonists.11−14 Species scanning mutagenesis studies of SERT TMs have identified the well-conserved Ile172 (I172) residue as an important molecular determinant responsible for high-affinity AD binding in human and mouse SERT (hSERT and mSERT, respectively) as substitution to methionine (M172) resulted in a dramatic loss in inhibitory potencies for many SERT antagonists, such as citalopram, fluoxetine, sertraline, and cocaine, in both hSERT and mSERT.15−20 Importantly, the M172 substitution does not alter normal 5-HT transport or SERT surface expression. The unique properties of this mutation allow for the generation of an M172 SERT mouse that is virtually pharmacologically invisible to many ADs supported by studies showing M172 transgenic mice lose sensitivity to multiple SSRIs without altering 5-HT transport or extracellular levels of 5-HT.15,20 This is notable considering that the actions of SSRIs are often evaluated and characterized in studies by comparison to the SERT null mouse,21 which is generally inappropriate due to the effect of SERT loss on 5-HT levels and how this disruption of 5-HT homeostasis can impact development of both serotonergic and nonserotonergic systems.22 Chronic loss of appropriate serotonergic tone caused by the SERT null genotype results in alteration of multiple physiological and behavioral phenotypes that make any comparisons to the wild-type mouse during SSRI treatment confounding and problematic at best.23 The SERT M172 AD-insensitive mouse represents a significant step forward and a powerful new tool to define the role of SERT and 5-HT signaling following AD administration as this model, unlike the SERT null mouse, has normal 5-HT transport and no known phenotypic changes other than loss of SSRI sensitivity. Therefore, the utility of the SERT M172 mouse to serve as a better control in AD studies will likely result in its increased use making more detailed characterizations of this mouse model of interest to the field.
Whereas many SSRIs have been evaluated for inhibition of the mSERT M172 mutant,15,16 there exists no analysis of SSRI metabolites in regards to potency and selectivity between the native and M172 SERT proteins. In humans and rodents, most SSRIs and SNRIs are metabolized to demethylated forms by cytochrome P450 enzymes, found primarily in the liver.24 Many of these demethylated metabolites are active antagonists of SERT mediated 5-HT transport.25 However, the selectivity and potency of these metabolites for wild-type mSERT (I172) versus M172 mSERT has not been evaluated. This is an important consideration for chronic drug studies using this model (or similar animal models),26−28 as some of these metabolites have very long half-lives leading to bioaccumulation of the compounds. If the M172 mSERT mouse were to retain sensitivity to these relatively long-lasting drug metabolites despite being insensitive to the parent compound, it could confound experimental outcomes. In this study, we investigate the sensitivity of M172 mSERT in brain-derived synaptosomes and platelets to antagonism by multiple SSRIs, SNRIs, and their metabolites. Furthermore, we measure the generation of the citalopram metabolite desmethylcitalopram in serum and brain tissue at 45 min and 2 h following intraperitoneal injections to determine if SSRI chronic treatment regimens could be impacted by bioactive metabolites that have equivalent or biologically relevant potencies at wild-type mSERT and M172 mSERT. This information is critical to ensuring that the physiological and behavioral effects observed with AD administration are due to the action of the parent compound and not to metabolites.
Results and Discussion
Our previous study revealed that in comparison to wild-type mice, the SSRIs citalopram and fluoxetine display significantly reduced potencies for inhibition of SERT mediated 5-HT uptake in brain-derived synaptosomes from M172 SERT mice.15 Similarly, in this study, the SSRIs citalopram, fluoxetine, and sertraline as well as the SNRI venlafaxine showed significant losses in potency for antagonism of M172 synaptosomes, reductions ranging from 16-fold (fluoxetine) to 633-fold (citalopram) (Figure 1, Table 1). As multiple SSRIs and SNRIs have active metabolites with half-lives long enough to make them biologically relevant,25 we also characterized the potency of the correlate metabolites desmethylcitalopram, norfluoxetine, desmethylsertraline, and desvenlafaxine. Comparison of potencies of norfluoxetine and desmethylsertraline to inhibit 5-HT uptake in wild-type mSERT and M172 mSERT derived synaptosomes revealed that the demethylated compounds no longer exhibited strong selectivity for inhibition of wild-type mSERT over M172 mSERT as the fold-change differentials between the IC50 values was reduced to 3.1- and 2.5-fold, respectively. However, desvenlafaxine and desmethylcitalopram retained selectivity for wild-type mSERT over M172 mSERT (49- and 80-fold, respectively) (Figure 1, Table 1). It is important to note that whereas the selectivity of desmethylsertraline between wild-type mSERT and M172 mSERT is 2.5-fold, the potency of the drug to inhibit wild-type mSERT is 39-fold less than the parent drug sertraline. Conversely, metabolites norfluoxetine, desvenlafaxine, and desmethylcitalopram inhibit 5-HT uptake in wild-type mSERT at potencies similar to their parent compounds (≤2.5-fold-change). For this reason, it is plausible that these metabolites in particular contribute to physiological blockade of transport and have a role in the effects observed following chronic AD administration.
Figure 1.

Competition 5-HT uptake analysis in brain-derived synaptosomes. Brain-derived synaptosomes were prepared from wild-type (filled circles, ●) and M172 (open boxes, □) mice and utilized to assess the ability of multiple SSRI class drugs and their metabolites to compete for [3H]5-HT uptake. All SSRIs showed decreased potency in the M172 synaptosomes, while some metabolites showed similar potency in both wild-type and M172 synaptosomes (n = 4–8 per drug; values represent the mean ± SE).
Table 1. IC50 Values for Inhibition of [3H]5-HT Uptakea.
| platelets |
synaptosomes |
|||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
IC50 (μM) |
|||||
| WT | M172 | fold shift | WT | M172 | fold shift | |
| fluoxetine | 0.040 [0.02, 0.12] | 0.63 [0.18, 2.75] | 16 | 0.10 [0.03, 0.24] | 1.6 [1.19, 3.12] | 16 |
| norfluoxetine | 0.10 [0.07, 0.13] | 0.20 [0.10, 0.49] | 2 | 0.16 [0.10, 0.21] | 0.50 [0.43, 0.58] | 3.1 |
| sertraline | 0.010 [0.01, 0.02] | 0.79 [0.28, 3.01] | 79 | 0.016 [0.003, 0.07] | 1.3 [1.04, 1.76] | 81 |
| desmethylsertraline | 0.050 [0.04, 0.07] | 0.40 [0.26, 0.95] | 8 | 0.63 [0.25, 1.91] | 1.6 [1.12, 3.52] | 2.5 |
| venlafaxine | 0.40 [0.23, 1.07] | 126 [49.43, 381.07] | 315 | 0.16 [0.09, 0.29] | 7.9 [2.44, 13.74] | 49 |
| desvenlafaxine | 0.50 [0.17, 2.55] | 50 [24.10, 72.78] | 100 | 0.16 [0.09, 0.25] | 7.9 [3.91, 13.30] | 49 |
| citalopram | 0.016 [0.01, 0.03] | 10 [8.34, 16.18] | 625 | 0.0079 [0.004, 0.02] | 5.0 [3.42, 6.65] | 633 |
| desmethylcitalopram | 0.025 [0.01, 0.15] | 10 [6.87, 13.09] | 400 | 0.020 [0.02, 0.03] | 1.6 [0.92, 3.06] | 80 |
Competition uptake analysis of platelet rich plasma (PRP) and brain synaptosomes derived from wild-type and M172 mice was performed to calculate the IC50 for multiple SSRIs and their active metabolites (mean with range given in brackets). Fold shift compares the rightward shift in potency for inhibiting SERT activity in M172 compared to wild-type.
NET and SERT are both significant targets of SNRIs. Although our 5-HT uptake assays should not be affected by the presence of NET, we also tested the potency of these drugs in platelets, which eliminates any contribution of NET. Platelets isolated from whole blood are often used as peripheral markers29 in human studies of SERT as they are easily obtained from subjects.30 In addition, platelets cannot synthesize 5-HT and are therefore entirely dependent on SERT-mediated uptake to accumulate 5-HT.2,31 Our inhibitory data from platelets are consistent with the synaptosomal results in that all drugs lose potency for the M172 mutant (Table 1). However, there were some differences in the magnitude of the fold-change of drug potencies between platelets and synaptosomes (Table 1, Figures 1 and 2). In addition, some compounds showed higher potency in platelets versus synaptosomes while other compounds were more potent in synaptosomes. This is not entirely unexpected as platelets likely lack some of the SERT interacting proteins present in neurons whose interactions can affect conformational states of the transporter and thereby selectively impact binding of each antagonist. Similarly, post-translational modifications of SERT such as phosphorylation status could be altered in synaptosomes versus platelets and these changes are known to impact potency of some inhibitors.32 These factors could account for changes in magnitude and cell-type specific potency differences for each drug. Nevertheless, the identical trends in potency loss from WT to M172 in each condition support our conclusions. Notably, we are primarily focused on metabolite action in the brain; the following analyses are from synaptosome assays unless specifically noted.
Figure 2.

Competition 5-HT uptake analysis in isolated platelets. Isolated platelet rich plasma (PRP), from wild-type (filled circles, ●) and M172 (open boxes, □) mice, was utilized to assess the ability of multiple SSRI class drugs and their metabolites to compete for [3H]5-HT uptake. All SSRIs showed decreased potency in the M172 platelets, while some metabolites showed similar potency in both wild-type and M172 platelets, comparable to what was seen in synaptosomes (n = 5–7 per drug; values represent the mean ± SE).
Analysis of SERT activity in brain synaptosomes derived from the M172 mouse reveals that the fluoxetine metabolite norfluoxetine shows a slight decrease in antagonist potency for wild-type mSERT, but gains potency for inhibiting M172 mSERT resulting in a significant reduction in selective inhibition of wild-type mSERT versus M172 mSERT from 16-fold to 3.1-fold (Figure 1, Table 1). In addition, norfluoxetine has been reported to return to the mouse brain where significant levels can accumulate due to its longer half-life (12.3 h) compared to fluoxetine (6.8 h).22,33,34 As a result, norfluoxetine is likely the major antagonist of 5-HT uptake by SERT in both wild-type mSERT and M172 mSERT following chronic administration of fluoxetine. Desmethylsertraline, the breakdown product of sertraline, also displays reduced selectivity for wild-type mSERT over M172 mSERT (81-fold to 2.5-fold) in synaptosomes but importantly is 39-fold less potent than sertraline for inhibiting wild-type mSERT and is therefore unlikely to significantly contribute to inhibition of 5-HT uptake. Venlafaxine and its metabolite desvenlafaxine maintain their selective potencies for inhibition of wild-type mSERT versus M172 mSERT in synaptosomes, as both exhibit a 49-fold selectivity for wild-type mSERT over M172 mSERT (Figure 1, Table 1). Therefore, if desvenlafaxine is capable of entering the brain, it would have the same antagonistic effect as venlafaxine in both wild-type mSERT and M172 mSERT. Desmethylcitalopram, the primary metabolite of citalopram, exhibits reduced potency compared to citalopram but retains robust selectivity for inhibition of wild-type mSERT over M172 mSERT (633-fold selectivity for citalopram versus 80-fold for desmethylcitalopram).
Having established that AD metabolites are capable of antagonizing SERT mediated 5-HT transport in ex vivo assays, we determined whether the metabolite desmethylcitalopram is capable of crossing the blood brain barrier, because re-entry into the brain following first-pass metabolism could contribute to the behavioral effects observed with chronic SSRI treatment. Desmethylcitalopram was selected due to its importance in our ongoing studies. Wild-type mice injected intraperitoneally with citalopram or its metabolite desmethylcitalopram were sacrificed after 45 min or 2 h then blood serum and brain tissue extracts were analyzed by liquid chromatography and mass spectrometry to quantify the levels of parent compounds and metabolites. Comparison of the 45 min and 2 h time points revealed that brain and serum citalopram levels decrease over time whereas desmethylcitalopram levels, metabolized from citalopram, increased in the brain (Figure 3, Table 2). In contrast, the concentration of desmethylcitalopram was not significantly altered in the serum of mice injected with citalopram consistent with the injected citalopram being metabolized over the 45 min to 2 h time period. Likewise, mice directly injected with desmethylcitalopram show an accumulation in the brain over time though there was a time dependent reduction in serum levels, which can be attributed to elimination. Importantly, desmethylcitalopram, unlike norfluoxetine and desmethylsertraline, maintains selectivity for wild-type SERT over M172 SERT, and therefore, its accumulation in the brain is not expected to interfere with chronic citalopram treatment studies in the M172 mouse.
Figure 3.

Time course analysis of brain and serum levels of citalopram and the metabolite desmethylcitalopram. Age-matched C57BL/6J mice were injected with either citalopram or desmethylcitalopram and sacrificed at 45 min or 2 h postinjection. The analytes were isolated from whole brain or serum and detected by LC-MS/MS. In the brain and serum, citalopram levels (filled circles, ●, Cit) decrease from 45 min to 2 h, whereas the metabolite desmethylcitalopram (open triangles, △, dCit from Cit) levels increase only in the brain over time. As a control, mice injected with the metabolite desmethylcitalopram (open boxes, □, dCit) show an accumulation of the metabolite in the brain, when comparing 45 min and 2 h, but a decreased concentration in the serum. (n ≥ 2 mice; values represent mean ± SE; one-way ANOVA with posthoc Holm–Šídák multiple comparison test was used to compare 45 min to 2 h for each compound, *, p < 0.05, **, p < 0.01).
Table 2. LC-MS/MS Quantification of Citalopram and the Metabolite Desmethylcitalopram in Brain and Seruma.
| brain (μM) |
serum (μM) |
|||||
|---|---|---|---|---|---|---|
| 45 min | 2 h | ANOVA | 45 min | 2 h | ANOVA | |
| citalopram | 5.52 ± 0.54 | 2.85 ± 0.38 | * | 0.76 ± 0.04 | 0.42 ± 0.05 | * |
| desmethylcitalopram | 0.38 ± 0.03 | 0.73 ± 0.12 | ** | 1.03 ± 0.04 | 0.51 ± 0.10 | * |
| desmethylcitalopram from citalopram | 0.14 ± 0.01 | 0.37 ± 0.05 | * | 0.33 ± 0.01 | 0.46 ± 0.09 | n.s. |
Age-matched C57BL/6J mice were injected with either citalopram or desmethylcitalopram and sacrificed at 45 min or 2 h post-injection. The analytes were isolated from whole brain or serum and detected by LC-MS/MS (n ≥ 2 mice; values represent mean ± S.E.; one-way ANOVA with post-hoc Holm-Šídák multiple comparison test was used to compare 45 min to 2 h, *p < 0.05, **p < 0.01; n.s. not significant).
In conclusion, synaptosomes and platelets derived from M172 SERT mice show a reduced sensitivity to multiple SERT antagonists, as was previously demonstrated.15 However, the ability of bioactive metabolites to antagonize 5-HT uptake by the M172 mSERT substitution had not previously been studied. Here, we explored the potency of these drugs for antagonism of wild-type mSERT versus M172 mSERT. Our results suggest that chronic drug administration studies, where it is important to maintain selective inhibition of wild-type mSERT over M172 mSERT, need to account for active metabolites and determine if there are changes in selectivity, inhibitory potency, and blood brain barrier passage and retention as these effects could lead to misinterpretation of the results. Over the course of days or weeks of treatment, metabolites could play a significant, if not, primary role in the blockade of SERT. Therefore, this study suggests that the levels of both parent drugs and their metabolites be assessed over the duration of the experimental paradigm to determine the potential contribution of each compound. Thus, compounds whose metabolites lose selectivity for wild-type mSERT versus M172 mSERT, but retain potencies comparable to the parent compound are problematic and should not be used in M172 mouse studies requiring differential inhibition of wild-type mSERT over M172 mSERT.
Methods
Animals
Serotonin transporter (SERT) M172 knock-in mice were previously generated on a 129S6 background.15 The M172 knock-in and I172 wild-type littermates (8–12 weeks old) from heterozygous breeding litters were used for the ex vivo serotonin transport inhibition assays. Age-matched I172 wild-type C57BL/6J (Jackson Laboratory) mice from homozygous breeding litters were used for mass spectrometry (MS) analysis. Food and water were provided ad libitum, and mice were maintained on a 12 h light/dark schedule. All animal procedures follow protocols approved by the UTHSCSA and/or UND IACUC animal use and care committees.
Synaptosome and Platelet-Rich Plasma (PRP) Preparations
Samples were prepared as previously described,15 but adapted to obtain both synaptosomes and PRP from the same animal to minimize the number of animals used. Briefly, mice were deeply anesthetized using Euthasol (500 μL/animal) (Virbac AH, TX) and effectiveness assessed using foot pinch. Following ophthalmectomy for PRP preparation, orbital blood was collected with heparinized capillary tubes and the brain (forebrain and midbrain) dissected on ice for synaptosome preparation.
Brain blocks were homogenized in a buffer solution (containing 0.32 M sucrose and 4.2 mM HEPES, pH 7.4). Synaptosomal pellets were obtained through two-stage centrifugation: the homogenate was spun at 1500 rpm for 15 min at 4 °C and then the supernatant at 1.5 × 10 000g for 15 min at 4 °C. Synaptosomal pellets were resuspended with Krebs-Ringer HEPES buffer (KRH; 130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.8 g/L d-glucose, 10 mM HEPES, pH 7.4, 100 mM pargyline, and 100 mM ascorbic acid). Synaptosomal suspensions were assessed for protein content using a spectrophotometer (Thermo Scientific) and then diluted to 667 μg/mL with KRH buffer. The prepared synaptosomes were maintained on ice until dispensed for serotonin transport assays.
Blood was collected as described above and anticoagulated with Acid Citrate Dextrose Solution A (BD Franklin Lakes, NJ) at a ratio of 100 μL per 500 μL and then centrifuged at 150g for 20 min at 22 °C to obtain PRP (the upper, clear supernatant). The PRP was collected and platelet counts/μL were determined using a hemocytometer. Platelets were diluted to achieve a final concentration of 3 × 106 platelets/assay. To prevent platelet activation, PRP was maintained at room temperature until use in competition 5-HT uptake assays as described below.
5-HT Competition Uptake Assays
Transport of 5-HT was assayed as described previously.35 Briefly, [3H]5-HT (100 nM) and serial dilutions of SERT inhibitors were prepared separately in KRH for synaptosomes or a platelet buffer solution (150 mM NaCl, 50 mM Tris HCl, 25 mM EDTA, pH 7.4, 100 mM pargyline, and 100 mM ascorbic acid) for platelets. The assays were set up on ice in a final volume of 250 μL (150 μL synaptosome or PRP, 50 μL [3H]5-HT, and 50 μL antagonist). The assay was initiated by incubation in a water bath for 10 min at 37 °C followed by immediate filtration over 0.3% polyethylenimine-coated glass fiber filters (Brandel) using a cell harvester (Brandel). Filter-bound radioactivity was quantified by a Beckman LS 6000 liquid scintillation counter. All transport assays were performed in duplicate. Radioactivity was normalized as a percentage of inhibition and plotted to determine the IC50 of the tested antagonist.
Drugs
Antidepressants and metabolites used for 5-HT competition uptake assays were purchased as follows: citalopram (cat # C7861), fluoxetine (cat #F132), and norfluoxetine (cat #F133) from Sigma-Aldrich; venlafaxine (cat #sc-201102), desvenlafaxine (cat #sc-255071), sertraline (cat #sc-201104), S-desmethylcitalopram (cat #sc-220017), and rac-trans-N-desmethylsertraline (cat #sc-219920) from Santa Cruz Biotechnology.
Animal Injections and Measurement of Brain and Serum Drug Concentrations
VK05-19.Oxalate S-Citalopram, a gift from Amy Newman (NIH-NIDA), and the metabolite (S)-N-desmethylcitalopram (Toronto Research Chemicals Inc., cat #D291571) were administered via intraperitoneal injection in 0.9% NaCl at 10 mg/kg, 5 mL/kg, respectively. For mass spectrometry analysis, imipramine (Sigma-Aldrich, cat #I7379) was used as an internal standard while citalopram and desmethylcitalopram were used as positive controls and were reconstituted in 100% methanol with a final concentration of 5 ng.
Brain Tissue and Serum Preparations
At 45 min and 2 h postinjection, mice were decapitated. Whole brains were frozen and stored in liquid nitrogen (N2). Trunk blood was collected and centrifuged for 10 min at 4 °C, 10 000 rpm, and then the supernatant transferred to cryovials for storage in N2. Brain tissue was pulverized to a fine powder under N2 temperatures to allow for a homogeneous mixing of different brain compartments and extracted using a one-step protocol.36 For extraction, 10 mg of brain tissue or 10 μL of plasma was spiked with the internal standard imipramine and then mixed with 90 μL of 100% methanol. Samples were briefly sonicated on ice, vortexed, and centrifuged for 10 min at 4 °C, 10 000 rpm. Collected supernatant was diluted 100-fold with 90% methanol in water.
UPLC-MS/MS Analysis for Citalopram and Desmethylcitalopram
Compounds were resolved on a Waters ACUITY UPLC HSS T3 column (1.8 μM, 100 Å pore diameter, 2.1 × 150 mm, Waters, Milford, MA) with an ACUITY UPLC HSS T3 precolumn (1.8 μM, 100 Å pore diameter, 2.1 × 5 mm, Waters) at a temperature of 55 °C. Analytes were eluted using a Waters ACUITY UPLC pump with a well plate autosampler at 8 °C. A volume of 10 μL of sample was injected on the column.
The separation was based on the previously described method.37,38 Solvent A was water containing 0.1% formic acid and solvent B was acetonitrile with 0.1% formic acid. The flow rate was maintained at 0.45 mL/min. The initial conditions of 39% B were held for 0.5 min. Solvent B was increased to 40.5% over 6.88 min, followed by an increase to 70% over 1.62 min, then increased to 75% over 3 min, and further increased to 98 over 1.5 min where it was held for 5.3 min to elute hydrophobic molecules from the column. At 20 min, solvent B was returned to initial conditions over 0.2 min to re-equilibrate the column for 2 min.
Compounds were analyzed using a triple quadrupole mass spectrometer (Xevo TQ-S, Waters) with electrospray ionization optimized in a positive mode. The capillary voltage was 3.29 kV and cone voltage was 14 V. The desolvation temperature was 500 °C and the source temperature 150 °C. The desolvation gas flow was 1000 L/h, the cone gas flow was 150 L/h, and the nebulizer gas was at 5.0 bar. The collision gas was set at 0.18 mL/min. MassLynx V4.1 software (Waters) was used for instrument control, acquisition, and sample analysis.
Citalopram and desmethylcitalopram were quantified using imipramine as an internal standard. Each compound’s MS/MS parameters were optimized for multiple reactions monitoring (MRM) mode by direct infusion of a standard solution of each analyte. Citalopram was quantified using 325.18/109.04 mass transition (collision energy (CE) 14 V) with 325.18/234.14 conformational ion (CE 24 V); desmethylcitalopram was quantified using 311.16/109.04 mass transition (CE 14 V) with 311.16/262.15 conformational ion (CE 16 V); and imipramine was quantified using 281.20/86.09 mass transition (CE 14 V) with 281.20/208.11 conformational ion (CE 36 V). The mass transitions for analytes and the selection of internal standard were consistent with previous reports.39 The data are represented as the log of the concentration. This log scaling is used to reduce the impact of scaling for metabolite intensity differences during MS/MS analysis.40
Acknowledgments
We thank Svetlana A. Golovko, Department of Biomedical Sciences, University of North Dakota, for her excellent assistance with the mass spectrometry analysis and Dr. Vivek Kumar, Medicinal Chemistry Section, NIDA-Intramural Research Program, for the synthesis of VK05-19.Oxalate S-Citalopram.
Glossary
Abbreviations
- SERT
serotonin transporter
- ADs
antidepressants
- SSRIs
selective serotonin reuptake inhibitors
- SNRIs
serotonin-norepinephrine reuptake inhibitors
- mSERT
mouse SERT
- hSERT
human SERT
- serotonin
5-hydroxytryptamine (5-HT)
- TCAs
tricyclic antidepressants
- NE
norepinephrine
- NET
norepinephrine transporter
- TMs
transmembrane domains
Author Contributions
D.K., M.R., S.A.B., M.Y.G., L.K.H., and B.J.T. designed research; D.K., M.R., S.A.B., and B.J.T. performed research; D.K., M.Y.G., L.K.H., and B.J.T. analyzed data; and D.K., M.R., L.K.H., and B.J.T. wrote the paper.
This work was supported by NIH funded COBRE Grant P20 GM104360 to L.K.H. and an NIH funded COBRE Mass Spec Core Facility Grant 5P30GM103329-04 to M.Y.G.
The authors declare no competing financial interest.
References
- Baganz N. L.; Blakely R. D. (2013) A Dialogue between the Immune System and Brain, Spoken in the Language of Serotonin. ACS Chem. Neurosci. 4, 48–63. 10.1021/cn300186b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M.; Gray J. A.; Roth B. L. (2009) The Expanded Biology of Serotonin. Annu. Rev. Med. 60, 355–366. 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws L. C.; Gould G. G. (2011) Ontogeny and regulation of the serotonin transporter: Providing insights into human disorders. Pharmacol. Ther. 131, 61–79. 10.1016/j.pharmthera.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki I. (1998) The spectrum of behaviors influenced by serotonin. Biol. Psychiatry 44, 151–162. 10.1016/S0006-3223(98)00139-5. [DOI] [PubMed] [Google Scholar]
- Gillman P. K. (2007) Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 151, 737–748. 10.1038/sj.bjp.0707253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens M. J.; Morgan W. N.; Plott S. J.; Nemeroff C. B. (1997) Neurotransmitter Receptor and Transporter Binding Profile of Antidepressants and Their Metabolites. J. Pharmacol. Exp. Ther. 283, 1305–1322. [PubMed] [Google Scholar]
- Schloss P.; Henn F. A. (2004) New insights into the mechanisms of antidepressant therapy. Pharmacol. Ther. 102, 47–60. 10.1016/j.pharmthera.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Andersen J.; Kristensen A. S.; Bang-Andersen B.; Strømgaard K. (2009) Recent advances in the understanding of the interaction of antidepressant drugs with serotonin and norepinephrine transporters. Chem. Commun. 3677–3692. 10.1039/b903035m. [DOI] [PubMed] [Google Scholar]
- Wong M.-L.; Licinio J. (2001) Research and Treatment Approaches to Depression. Nat. Rev. Neurosci. 2, 343–351. 10.1038/35072566. [DOI] [PubMed] [Google Scholar]
- Nemeroff C. B.; Owens M. J. (2002) Treatment of mood disorders. Nat. Neurosci. 5 (Suppl), 1068–1070. 10.1038/nn943. [DOI] [PubMed] [Google Scholar]
- Yamashita A.; Singh S. K.; Kawate T.; Jin Y.; Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature 437, 215–223. 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90. 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J. R.; Henry L. K.; Blakely R. D. (2010) Transmembrane domain 6 of the human serotonin transporter contributes to an aqueously accessible binding pocket for serotonin and the psychostimulant 3,4-methylene dioxymethamphetamine. J. Biol. Chem. 285, 11270–11280. 10.1074/jbc.M109.093658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry L. K.; Adkins E. M.; Han Q.; Blakely R. D. (2003) Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I. J. Biol. Chem. 278, 37052–37063. 10.1074/jbc.M305514200. [DOI] [PubMed] [Google Scholar]
- Thompson B. J.; Jessen T.; Henry L. K.; Field J. R.; Gamble K. L.; Gresch P. J.; Carneiro A. M.; Horton R. E.; Chisnell P. J.; Belova Y.; McMahon D. G.; Daws L. C.; Blakely R. D. (2011) Transgenic elimination of high-affinity antidepressant and cocaine sensitivity in the presynaptic serotonin transporter. Proc. Natl. Acad. Sci. U. S. A. 108, 3785–3790. 10.1073/pnas.1011920108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry L. K.; Field J. R.; Adkins E. M.; Parnas M. L.; Vaughan R. A.; Zou M.-F.; Newman A. H.; Blakely R. D. (2006) Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. J. Biol. Chem. 281, 2012–2023. 10.1074/jbc.M505055200. [DOI] [PubMed] [Google Scholar]
- Barker E. L.; Perlman M. A.; Adkins E. M.; Houlihan W. J.; Pristupa Z. B.; Niznik H. B.; Blakely R. D. (1998) High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis. An aromatic residue in transmembrane domain I dictates species-selective recognition of citalopram and mazindol. J. Biol. Chem. 273, 19459–19468. 10.1074/jbc.273.31.19459. [DOI] [PubMed] [Google Scholar]
- Andersen J.; Taboureau O.; Hansen K. B.; Olsen L.; Egebjerg J.; Strømgaard K.; Kristensen A. S. (2009) Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J. Biol. Chem. 284, 10276–10284. 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Yarravarapu N.; Lapinsky D. J.; Perley D.; Felts B.; Tomlinson M. J.; Vaughan R. A.; Henry L. K.; Lever J. R.; Newman A. H. (2015) Novel Azido-Iodo Photoaffinity Ligands for the Human Serotonin Transporter Based on the Selective Serotonin Reuptake Inhibitor (S)-Citalopram. J. Med. Chem. 58, 5609–5619. 10.1021/acs.jmedchem.5b00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackenoff A. G.; Moussa-Tooks A. B.; McMeekin A. M.; Veenstra-VanderWeele J.; Blakely R. D. (2016) Essential Contributions of Serotonin Transporter Inhibition to the Acute and Chronic Actions of Fluoxetine and Citalopram in the SERT Met172 Mouse. Neuropsychopharmacology 41, 1733–1741. 10.1038/npp.2015.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengel D.; Murphy D. L.; Andrews A. M.; Wichems C. H.; Feltner D.; Heils A.; Mössner R.; Westphal H.; Lesch K.-P. (1998) Altered brain serotonin homeostasis and locomotor insensitivity to 3, 4-methylenedioxymethamphetamine (“Ecstasy”) in serotonin transporter-deficient mice. Mol. Pharmacol. 53, 649–655. [DOI] [PubMed] [Google Scholar]
- Ansorge M. S.; Morelli E.; Gingrich J. A. (2008) Inhibition of serotonin but not norepinephrine transport during development produces delayed, persistent perturbations of emotional behaviors in mice. J. Neurosci. 28, 199–207. 10.1523/JNEUROSCI.3973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A.; Murphy D. L.; Crawley J. N. (2003) Abnormal behavioral phenotypes of serotonin transporter knockout mice: parallels with human anxiety and depression. Biol. Psychiatry 54, 953–959. 10.1016/j.biopsych.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Velasquez J. C.; Goeden N.; Herod S. M.; Bonnin A. (2016) Maternal Pharmacokinetics and Fetal Disposition of (±)-Citalopram during Mouse Pregnancy. ACS Chem. Neurosci. 7, 327–338. 10.1021/acschemneuro.5b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez C.; Hyttel J. (1999) Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell. Mol. Neurobiol. 19, 467–489. 10.1023/A:1006986824213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.; Han D. D.; Gu H. H. (2005) A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate. J. Neurochem. 94, 352–359. 10.1111/j.1471-4159.2005.03199.x. [DOI] [PubMed] [Google Scholar]
- Chen R.; Tilley M. R.; Wei H.; Zhou F.; Zhou F.-M.; Ching S.; Quan N.; Stephens R. L.; Hill E. R.; Nottoli T.; Han D. D.; Gu H. H. (2006) Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc. Natl. Acad. Sci. U. S. A. 103, 9333–9338. 10.1073/pnas.0600905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill B.; Tilley M. R.; Han D. D.; Thirtamara-Rajamani K.; Hill E. R.; Bishop G. A.; Zhou F.-M.; During M. J.; Gu H. H. (2014) Behavior of knock-in mice with a cocaine-insensitive dopamine transporter after virogenetic restoration of cocaine sensitivity in the striatum. Neuropharmacology 79, 626–633. 10.1016/j.neuropharm.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oreland L.; Hallman J. (1989) Blood platelets as a peripheral marker for the central serotonin system. Nord. Psykiatr. Tidsskr 43, 43–51. 10.3109/08039488909100833. [DOI] [Google Scholar]
- Yubero-Lahoz S.; Robledo P.; Farré M.; de laTorre R. (2013) Platelet SERT as a peripheral biomarker of serotonergic neurotransmission in the central nervous system. Curr. Med. Chem. 20, 1382–1396. 10.2174/0929867311320110003. [DOI] [PubMed] [Google Scholar]
- Ni W.; Watts S. W. (2006) 5-hydroxytryptamine in the cardiovascular system: focus on the serotonin transporter (SERT). Clin. Exp. Pharmacol. Physiol. 33, 575–583. 10.1111/j.1440-1681.2006.04410.x. [DOI] [PubMed] [Google Scholar]
- Moritz A. E.; Foster J. D.; Gorentla B. K.; Mazei-Robison M. S.; Yang J.-W.; Sitte H. H.; Blakely R. D.; Vaughan R. A. (2013) Phosphorylation of Dopamine Transporter Serine 7 Modulates Cocaine Analog Binding. J. Biol. Chem. 288, 20–32. 10.1074/jbc.M112.407874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw P. F.; Guimaraes A. R.; Fava M.; Rosenbaum J. F.; Pearlman J. D.; Flood J. G.; Puopolo P. R.; Clancy K.; Gonzalez R. G. (1992) Accumulation of fluoxetine and norfluoxetine in human brain during therapeutic administration. Am. J. Psychiatry 149, 1592–1594. 10.1176/ajp.149.11.1592. [DOI] [PubMed] [Google Scholar]
- Preskorn S. H., Stanga C. Y., Feighner J. P., and Ross R. (2012) Antidepressants: Past, Present and Future, Springer Science & Business Media, Berlin. [Google Scholar]
- Zhu C.-B.; Blakely R. D.; Hewlett W. A. (2006) The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 31, 2121–2131. 10.1038/sj.npp.1301029. [DOI] [PubMed] [Google Scholar]
- Brose S. A.; Baker A. G.; Golovko M. Y. (2013) A fast one-step extraction and UPLC-MS/MS analysis for E2/D 2 series prostaglandins and isoprostanes. Lipids 48, 411–419. 10.1007/s11745-013-3767-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose S. A.; Golovko M. Y. (2013) Eicosanoid post-mortem induction in kidney tissue is prevented by microwave irradiation. Prostaglandins, Leukotrienes Essent. Fatty Acids 89, 313–318. 10.1016/j.plefa.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose S. A.; Golovko S. A.; Golovko M. Y. (2016) Brain 2-Arachidonoylglycerol Levels Are Dramatically and Rapidly Increased Under Acute Ischemia-Injury Which Is Prevented by Microwave Irradiation. Lipids 51, 487–495. 10.1007/s11745-016-4144-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T.; Rong Z.; Peng L.; Chen B.; Xie Y.; Chen C.; Sun J.; Xu Y.; Lu Y.; Chen H. (2010) Simultaneous determination of citalopram and its metabolite in human plasma by LC–MS/MS applied to pharmacokinetic study. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 878, 615–619. 10.1016/j.jchromb.2010.01.004. [DOI] [PubMed] [Google Scholar]
- Xi B.; Gu H.; Baniasadi H.; Raftery D. (2014) Statistical analysis and modeling of mass spectrometry-based metabolomics data. Methods Mol. Biol. 1198, 333–353. 10.1007/978-1-4939-1258-2_22. [DOI] [PMC free article] [PubMed] [Google Scholar]