

Abstract
This work investigates the surface chemistry of H2O2 generation on a boron-doped ultrananocrystalline diamond (BD-UNCD) electrode. It is motivated by the need to efficiently disinfect liquid waste in resource constrained environments with limited electrical power. X-ray photoelectron spectroscopy was used to identify functional groups on the BD-UNCD electrode surfaces while the electrochemical potentials of generation for these functional groups were determined via cyclic voltammetry, chronocoulometry, and chronoamperometry. A colorimetric technique was employed to determine the concentration and current efficiency of H2O2 produced at different potentials. Results showed that preanodization of an as-grown BD-UNCD electrode can enhance the production of H2O2 in a strong acidic environment (pH 0.5) at reductive potentials. It is proposed that the electrogeneration of functional groups at oxidative potentials during preanodization allows for an increased current density during the successive electrolysis at reductive potentials that correlates to an enhanced production of H2O2. Through potential cycling methods, and by optimizing the applied potentials and duty cycle, the functional groups can be stabilized allowing continuous production of H2O2 more efficiently compared to static potential methods.
Keywords: boron-doped diamond, hydrogen peroxide, liquid disinfection, surface modification, catalysis, reactive oxygen species, reactive oxidative species, oxidized boron-doped diamond
1. Introduction
The primary routes for human exposure to microbial pathogens from wastewater arise in agricultural settings.1 Good results for pathogen removal or inactivation have been obtained by membrane filtration, UV irradiation, and pasteurization.2−6 However, such treatments have high investment costs and require maintenance that may not be feasible for developing countries. Other established disinfection techniques like chlorination and ozonation require storage of chemicals and generate harmful byproducts, which present a major drawback to small-scale treatment plant safety.2,3
Electrochemical disinfection presents an alternative technology that is effective, simple to operate, low cost, and energy efficient. Some key benefits provided by this method include in situ generation of disinfectants, no storage of chemicals, and a straightforward low maintenance process. This is especially the case for blackwater treatment, as the presence of urine provides sufficient electrical conductivity for electrochemical processes to be conducted without additional chemicals.7 Recent research in our group demonstrated that synthetic urine spiked with E. coli could be disinfected by means of electrochemically generated chlorine at a boron-doped diamond (BDD) anode.8
Although many electrochemical disinfection studies utilize dimensionally stable anodes or mixed metal oxides,9,10 BDD has gained considerable attention because of its high oxygen evolution overpotential in aqueous environments.11,12 This property enables the formation of reactive oxygen species (ROS) such as hydroxyl radicals, ozone, and hydrogen peroxide.11,13−15 Jeong et al.9 compared para-chlorobenzoic degradation at metal oxides (RuO2 and IrO2) and BDD electrodes. BDD was found to be more efficient because of its low electroactivity,16 allowing generated hydroxyl radicals to react in the bulk solution. In contrast, on metal oxide electrodes, hydroxyl radicals generally react at the electrode surface to form a higher oxide surface with a weak oxidation capacity.13 Schmalz et al.17 showed that the bacterial inactivation of wastewater could be increased by a faster generation of electrochemical oxidants using BDD. Their study illustrated the energy efficiency of BDD electrodes over other conventional electrode materials. Despite the promise of BDD electrodes for application in sanitation technologies, there is little information in literature that details the optimal conditions for ROS generation, which would allow production of these species with higher efficiency and at lower cost.
Thin-film BDD has been explored as an electrode material in electrochemical research and for water treatment applications.12,18−22 Though microcrystalline diamond (MCD) (>100 nm grain sizes), and nanocrystalline diamond (NCD) (<100 nm grain sizes) have been studied for water sanitation applications, there has been little published work on ultrananocrystalline diamond (UNCD) electrodes, a subcategory of NCD electrodes, despite their numerous electrochemical benefits.23−25 UNCD differs from MCD and NCD because its grain size is exceptionally small (3–5 nm) allowing for a large relative surface area per grain volume.22 The relative percentage of carbon atoms in a diamond grain that occur at a crystallite surface can be as high as 10%.26,27 This induces a high degree of disordered sp2 carbon bonding present at grain boundaries relative to MCD and NCD. As a result, roughly 5% of the total carbon present in UNCD films is disordered, sp2 bonded and at grain boundaries.26,27 Nondiamond carbon, such as sp2 carbon, forms at the grain boundaries of BDD crystallites during chemical vapor deposition (CVD). Subsequent removal of nondiamond carbon increases the inertness and operational voltage window of the BDD electrode.12 The high degree of amorphous sp2 increases the electrochemical activity of the boron-doped UNCD (BD-UNCD) electrode.12,28 Surface treatments, such as annealing at high temperature or in situ direct current (DC) polarization, can lead to functionalization of the electrode surface at these sp2 carbon sites.29−31 Depending on the surface treatment and subsequent functionalization, the electrochemical characteristics of the BD-UNCD electrode can change drastically from being active to inert, hydrophobic to hydrophilic, and even optically opaque to clear.12,21,28,32,33 These surface treatments are typically carried out with the intent of removing nondiamond carbon. Interestingly, it was reported that functionalization of the nondiamond carbon, rather than removal, can enhance the production of ROS for carbon based electrodes.34−36 Yano et al. showed that carbon–oxygen functional groups created on sp2 carbon can greatly increase the reduction of dissolved oxygen gas in both acidic18 and basic37 aqueous electrolytes. Carbon–oxygen functionalities are present in quinone species, which are widely used in industrial processes for the production of hydrogen peroxide because of their catalytic behavior and have been found on BDD electrodes.38−41
The present work investigates the generation of hydrogen peroxide (H2O2) using as-grown BD-UNCD electrodes, and in situ surface modifications. Given the relatively short recombination time of ROS,42,43 electrogeneration of H2O2 was monitored indirectly using a colorimetric technique.44−47 Combining electrochemical and physical characterization data, the evolving surface chemistry of BD-UNCD resultant from potentiometric testing was monitored. Static and potential cycling methods of generating H2O2 are compared to determine the nature of surface chemistry of BD-UNCD and its effect on the generation of H2O2. These results build on an evolving understanding of the electrochemical generation of ROS, such as H2O2, from BDD electrodes, and include the effects of potential cycling methods. Furthermore, these results provide an enhanced understanding of the kinetic properties of BD-UNCD electrodes and form the basis of the phenomenological model presented herein.
2. Experimental Section
2.1. Electrolyte
To enable colorimetric detection of H2O2, we used 0.12 M Ti(SO4)2 as an electrolyte. The 0.12 M Ti(SO4)2 electrolyte was made following the procedure described by Eisenberg.44 1 g Ti(IV)O2 anatase powder (Sigma-Aldrich) was dissolved in 100 mL H2SO4 (J.T. Baker, UN 1830) by heating at 150 °C for 16 h and stirring at 350 rpm. The resulting solution was cooled to room temperature and left covered and unstirred for 8−12 hours until undissolved TiO2 settled out of solution. The supernatant was then filtered and diluted 1:39 with deionized water, resulting in an optically clear electrolyte with pH 0.5, and [Ti(SO4)2] of 0.12 M. Unless otherwise noted, the electrolyte was bubbled with ultrahigh purity O2 gas (Airgas Inc., PN# OX UHP 300) for 20 min prior to each use to ensure saturation of oxygen content.
2.2. H2O2 Measurement
Known concentrations of H2O2 (VWR, MK524002) were reacted with 0.12 M Ti(SO4)2 (Section 2.1) to form H2TiO4, which strongly absorbs visible light.42,44 The absorbance of H2TiO4 at each concentration of H2O2 was measured using a monochromated light source at 420 nm (Newport Oriel 300W 87005 lamp housing, Newport Oriel 6258 ozone free xenon lamp, Newport Oriel 74004 1/8 m Cornerstone Monochromator) and a photodetector (Newport Oriel, 479). The absorbance was then plotted with respect to the H2O2 concentration to create a linear absorbance–concentration standard curve in accordance with Beer’s Law (Figure S1). Electrochemically generated H2O2 concentrations reacted in 0.12 M Ti(SO4)2 were then determined by comparing the experimentally obtained absorbance value to the standard curve, similar to the technique of Eisenberg44 and Peralta et al.42
2.3. Electrode Fabrication
Boron-doped ultrananocrystalline diamond (BD-UNCD) films (thickness ∼2 μm) on SiOx/Si (500 μm) with geometric areas between 0.5–1.5 cm2 were cleaved from a 4 in. UL25 wafer purchased from Advanced Diamond Technologies (Romeoville, IL). Cleaved pieces from this wafer were then electrically connected to a thin copper wire using silver paste (Ted Pella, PN#16031). The contact was left to dry at room temperature for several hours. The copper wire and paste were then isolated from the electrolyte using a glass tube and nonconductive epoxy (Loctite EA 9462 Hysol).
2.4. Electrochemical Conditions
Unless otherwise mentioned, electrodes were tested as the working electrode in a 3-electrode 40 mL cell of 0.12 M Ti(SO4)2 where Ag/AgCl sat. KCl (Biologic RE-1CP) and Pt wire were used as the reference and counter electrodes, respectively. All measurements were made using a SP-200 BioLogic potentiostat. Prior to testing, samples were thoroughly rinsed with deionized water and blown dry with ultrahigh purity N2 gas (Airgas Inc., PN# NI UHP 300). The electrolysis for measurement of H2O2 (Section 3.4) was performed in a 2-electrode cell at varied voltages for 30 min in a 10 mL-solution of 0.12 M Ti(SO4)2. A 2-electrode cell was used due to space constraints. The Pt counter electrode was also used as a pseudoreference electrode. In our conditions, its potential remains constant, and equal to ∼0.5 V vs Ag/AgCl. Electrodes were anodized in acidic solutions prior to electrolysis, a method we refer to as “pre-anodization.”
2.5. X-ray Photoelectron Spectroscopy
Collection
X-ray photoelectron spectroscopy (XPS) was conducted using a Kratos Analytical Axis Ultra instrument with a monochromated Al Kα X-ray source (1486.69 eV) operated at 15 kV and 10 mA (150 W) in a 5 × 10–8 Torr chamber. Survey spectra were collected using an analyzer pass energy of 160 eV, and binding energies were collected from −5 to 1200 eV scanned at 1 eV increments. Each step was integrated for 500 ms and the entire spectrum was averaged across 5 sweeps. Regional C 1s spectra (276.9–300.0 eV) were collected using the same conditions as the survey spectra, except a pass energy of 20 eV (resolution of 0.4 eV) was used.
Analysis
Collected spectra were calibrated, analyzed, and deconvoluted using Casa XPS software following best practices as outlined in Briggs and Grant.48 All data reported were the average result of 3 measurements per electrode sample. The XPS O 1s to C 1s ratios seen in Figure 1 were determined by normalizing the integrated O 1s peak intensity centered at a binding energy of 532 eV to the integrated C 1s peak intensity centered at 285 eV. A Shirley fitted background subtraction was used for both peaks. The XPS C 1s regional spectra seen in Figure 2 were normalized against the bulk peak maximum located at 285 eV, with background subtraction carried out using a Shirley background fit. Following a similar protocol as Ayers et al.,49 peak assignments were made in reference to the characteristic sp3 diamond-like-carbon C–C bonding peak located at 285 eV. For nondiamond carbon bonds, the following assignments were made: sp2 C=C (graphitic carbon) was assigned to −1.1 eV, C–H (hydrocarbon) was assigned to +0.5 eV, C–OH (hydroxyls bonded to carbon) was assigned to +1 eV, C–O–C (ether groups) was assigned to +1.7 eV, C=O (carbonyl groups) was assigned to +3.4 eV, and COOH (carboxyl groups) was assigned to +4.1 eV. Deconvolution, and subsequent quantification of C 1s regional spectra, was carried out using Gaussian–Lorentzian peak shapes and ±0.2 eV constraints for fwhm and peak position, respectively, where the FWHW was set to 1 eV. The resulting deconvoluted peak area quantities (can be seen in Figure S4) were then summed to equal the relative amount of functional groups corresponding to the C–H, C–OH, C–O–C, C=O, and COOH content for each electrode and plotted as seen in Figure 3A. For details not mentioned herein, XPS spectra were analyzed following best practices as outlined in Briggs and Grant.48
Figure 1.
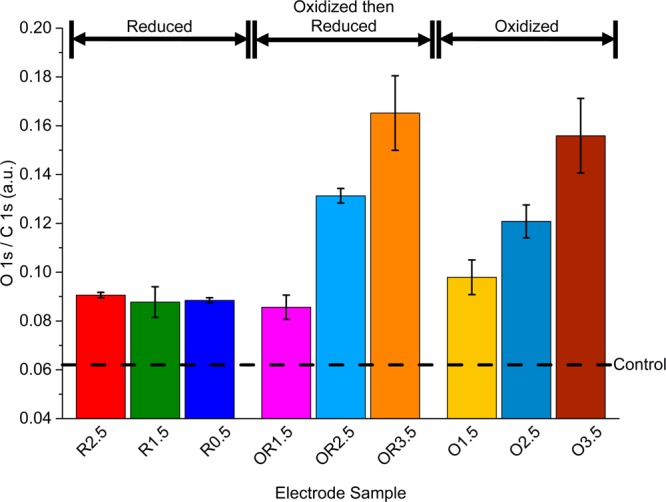
Oxygen content of each electrode given by the O 1s integrated peak area normalized to the C 1s integrated peak area. O 1s/C 1s values displayed were taken as the average of three measurements. The dashed line corresponds to the unpolarized control electrode oxygen content. The error bars correspond to the standard deviation of the measurements for each electrode.
Figure 2.

Change in carbon bonding at surface of (A) reduced (R), (B) oxidized (O), and (C) oxidized then reduced (OR) BD-UNCD electrodes compared to the control electrode indicated by change in C 1s regional spectra taken from XPS.
Figure 3.

(A) Comparison of relative amount of functional groups between O and OR electrodes. The O electrodes were oxidized at the given oxidation voltage for 20 min. O electrodes that were subsequently reduced at −1.5 V vs Ag/AgCl for 20 min are renamed OR electrodes. The relative amount of functional groups before, and after reduction of the oxidized electrodes indicates presence, and stability dependent on the oxidation voltage. Oxidation voltage of 0.5 V corresponds to the relative amount of functional groups on the control electrode. The green arrows indicate instability of the oxidized surface in reducing environment at the given voltages. (B) Charge transfer for the R1.5 and OR electrodes at discrete times during reduction at −1.5 V vs Ag/AgCl sat. KCl for 20 min. The charge transferred for the OR1.5 and OR2.5 electrodes compared to R1.5 indicates there is greater charge transferred during reduction if BD-UNCD is “pre-anodized” at either 1.5 or 2.5 V. However, OR1.5 and OR2.5 compared to OR3.5 shows that too high of an oxidation voltage during “pre-anodization” removes this effect. Charge transfer is proportional to oxygen reduction, and therefore hydrogen peroxide production in acidic aqueous environments.
3. Results and Discussion
3.1. Electrochemical Surface Pretreatment
To better understand the role of surface chemistry and biasing history on H2O2 generation, we grouped electrodes and studied them in three categories. Table 1 summarizes these treatments, and provides a guide for the reader in electrode sample naming assignment. The treatments were chosen to elucidate the effects that oxidation, or “pre-anodization” of BD-UNCD has when it is then followed by cathodic potentials with respect to the open-circuit potential (0.5 V vs Ag/AgCl sat. KCl). The first group consisted of electrodes that were reduced (at −0.5, −1.5, and −2.5 V vs Ag/AgCl sat. KCl) for 20 min and then characterized. These are the “R” electrodes. The second group electrodes were oxidized (at 1.5, 2.5, and 3.5 V vs Ag/AgCl sat. KCl) for 20 min and then characterized. These are the “O” electrodes. The third group, an extension of the second group, was first oxidized (at 1.5, 2.5, and 3.5 V vs Ag/AgCl sat. KCl) before being reduced at −1.5 V vs Ag/AgCl sat. KCl. These are the “OR” (oxidized-reduced) electrodes. As a matter of comparison, an unpolarized control electrode was characterized, and results plotted along with the polarized electrodes. The effects of surface chemistry, and biasing history were studied by analysis of XPS and Raman spectra.
Table 1. Surface Pretreatments of 3 Groups of Electrodes Where R, O, and OR Prefixes Designate Electrodes That Were Tested at Reductive, Oxidative, or Oxidative Then Reductive Potentials, Respectively.
| name | oxidation voltage (V vs Ag/AgCl sat. KCl) | reduction voltage (V vs Ag/AgCl sat. KCl) |
|---|---|---|
| R2.5 | − | –2.5 |
| R1.5 | − | –1.5 |
| R0.5 | − | –0.5 |
| O1.5 | 1.5 | − |
| O2.5 | 2.5 | − |
| O3.5 | 3.5 | − |
| OR1.5 | 1.5 | –1.5 |
| OR2.5 | 2.5 | –1.5 |
| OR3.5 | 3.5 | –1.5 |
| controla | − | − |
The Control electrode was left at open-circuit voltage in 0.12 M Ti(SO4)2, which was measured to be 0.5 V vs. Ag/AgCl sat. KCl.
3.2. Surface Chemistry
Comparison of surface chemistry across BD-UNCD electrodes as a result of an applied voltage can be found in Figures 1–3. Figure 1 shows the relative O 1s to C 1s peak area intensity collected from XPS survey spectra across electrode samples polarized at different potentials. From Figure 1, there appears to be an increase in oxygen content occurring on the BD-UNCD surface across all pretreatment potentials with respect to the control electrode. This implies that even slight polarization in strong acidic environments is enough to oxidize BD-UNCD. Oxygen has previously been shown to terminate BDD lattices when oxidized by electrochemical treatments of greater than 1.5 V vs Ag/AgCl in aqueous-based electrolytes by bonding to amorphous sp2 content present at grain boundaries.29,30 At potentials <1.5 V vs Ag/AgCl, corresponding to the R2.5, R1.5 and R0.5 electrodes, there also appears to be an overall (but constant) increase in oxygen content which is independent of the applied voltage. Martin et al.29 ascribed this increase to 2 possible mechanisms: (i) adsorption of H2O to oxygen terminated sites on BD-UNCD crystallites after testing due to atmospheric effects, or (ii) carbon bound oxygen functionalities created during reduction. However, the particular species leading to the increase in oxygen content was not investigated. Since the control electrode does not exhibit this increase in oxygen content, even though it was left in 0.12 M Ti(SO4)2 for the same amount of time as the other electrodes, it seems likely that the second mechanism (ii) proposed by Martin et al. is the cause of increased oxygen content.
The C 1s regional spectra in Figure 2 show an apparent difference in carbon bonding resulting from the (A) reducing, (B) oxidizing, or (C) oxidizing then reducing test conditions. All the reduced (R) electrodes in Figure 2A show a substantial increase in carbon functionalization given by the widened shoulder of the C 1s peak shape at binding energies greater than 285 eV. This widened shoulder in the peak shapes is indicative of an increased presence of carbon–hydrogen (C–H), and carbon–oxygen (C–OH, C–O–C) bonding.40 The R1.5 electrode has a more prominent shoulder at higher binding energies (>285.5 eV) than the R2.5, R0.5 and control electrodes. This is likely the result of a higher proportion of C–OH, C–O–C, C=O, and COOH bonding occurring on the BD-UNCD surface. This change in carbon-bonding for BD-UNCD electrodes can be correlated to the creation of carbon functional groups occurring on the surface.28,30,40 The relative proportion of these functional groups across all the R electrode surfaces compared to the control electrode can be seen in Figures S3D–F and S4A, D. The maximum number of functional groups is seen in the R1.5 electrode. Because the electrodes were not exposed to oxidative potentials, it is unlikely that these functional groups are due to electrochemical oxidation of sp2 carbon bonds (C=C). Oxidation of nondiamond content on BDD is widely reported for BDD electrodes tested in aqueous electrolytes at oxidative potentials. However, it is less commonly reported for reductive potentials.12,28,31,49−52 Although the exact reductive reaction pathway is unknown, there is an increased amount of functionalization beyond the control electrode seen across the R electrodes in Figure 2A, C and Figures S3A–C and S4A that suggests an electrochemical mechanism.
For the C 1s regional spectra seen in Figure 2B–C corresponding to the O and OR electrodes, respectively, there is an apparent creation of functional groups from oxidation that remain even after reduction. With increasing oxidation, formation of carbon–oxygen and carbon–hydrogen functional groups occurring at grain boundaries also increases, a trend that has been reported elsewhere.30,40,49 Comparison of the overall C 1s peak shape between the O2.5 and O1.5 electrodes (Figure 2B) shows only a slight increase of functional groups at higher binding energies (>286 eV). In contrast, the O3.5 electrode appears to show a notable increase in this same region. The difference is likely due to an increased overpotential that is able to sufficiently oxidize the nondiamond content present on BD-UNCD grain surfaces. Figure 1 further confirms this hypothesis. In Figure 2B, (also see Figures S4B and S4E) there is an apparent component of the C 1s peak shape due to C–H, C–OH, C–O–C, and C=O functional groups bonded to surface carbon of which a significant proportion is C=C (sp2).23,26,53Figure 2B also indicates an increasing C–OH and C–H content with increasing voltage, while the other components appear to be constant. Hydroxyl groups created on a BD-UNCD surface within a certain oxidation potential range (2–3 V vs Ag/AgCl sat. KCl) have been reported elsewhere.54−56 However, an increasing hydrocarbon C–H content is not as widely reported. Raman spectroscopy of the oxidized electrodes (seen in Figure S5B) did not give direct insight into a mechanism. This is likely because the functional groups are adsorbed on the surface and Raman spectroscopy is a bulk characterization technique. Chaplin et al.57 indicated that at such elevated potentials in aqueous electrolytes, hydroxyl radicals can radicalize carbon. This usually results in the removal of bonded hydrogen, but in some instances leaves additional C–H behind. It is also possible that at such elevated potentials, sufficient oxidation of the electrodes leads to etching of the surface atoms to expose the underlying unoxidized C–Hx bonds created by CVD growth conditions. Because it is widely reported that sufficient oxidation of the BDD surface is irreversible by electrochemical methods,30,49,58 reduction of these electrodes should give insight as to what species could cause the C–H signature and whether or not they are stable.
Figure 2C provides greater understanding of functional groups created on the BD-UNCD surface during oxidation, which we refer to as “pre-anodization,” and how they respond upon reduction at −1.5 V vs Ag/AgCl sat. KCl. Comparison of the C 1s regional spectra in Figure 2B with 2C show marked differences in functional groups present on the surface of the oxidized O1.5 and O2.5 electrodes compared to the oxidized then reduced OR1.5 and OR2.5 electrodes. Functional groups created on the O electrodes are largely reduced to C–O–C and C–H bonds on the OR electrodes. This trend is further supported in Figures S4B, C. Because of the complexity of XPS spectra deconvolution, it is hard to definitively state the particular functional groups on the O, and OR electrodes. Again, it can be stated that the functionalization of the BD-UNCD electrodes occurs only at the surface because Raman spectroscopy (Figure S4) showed little change across electrodes compared to the XPS data. Moreover, it appears that functionalization of the surface is proportional to the magnitude of the oxidation voltage. Upon reduction, these functional groups are either reduced, leading to reconstruction of the carbon bonds at the grain boundaries, or are removed entirely. Figure S4D–F also show a strong correlation between the relative amount of sp2 (C=C) bonding, and the relative amount of functional group bonding. The correlation further suggesting that the functional groups are linked to the presence of nondiamond content.
Figure 3 shows the preanodization effect on the following: (A) the stability of functional groups on BD-UNCD surfaces and (B) the charge transferred upon subsequent reduction at −1.5 V vs Ag/AgCl sat. KCl. The relative concentration of carbon bonds that are not C–C (sp3) or C=C (sp2), (i.e., functional groups on the surface), appear to remain constant or increase on the OR1.5 and OR2.5 electrodes following reduction. In contrast to this trend, the OR3.5 electrode appears to lose functional groups compared to its O counterpart. Figure 3B indicates the charge transferred at discrete times for the OR and R1.5 electrodes during reduction at −1.5 V vs Ag/AgCl sat. KCl. Clearly, the OR1.5 and OR2.5 have a greater amount of charge transfer compared to the R1.5 and OR3.5 electrodes. Comparison of all four electrodes in Figure 3B indicates that preanodization has a proportional impact on the reducing current, which is optimal between pre-anodized voltages of 1.5 and 3.5 V
Because the R1.5 electrode was never oxidized, yet shows low charge transfer, it can be stated that oxidized functional groups on the OR1.5 and OR2.5 lead to the observed increase. Correlation of the C 1s peak shape of R1.5 in Figure 2A with Figure 3B indicates that the functional groups must be oxidized to create the difference in charge transfer observed among the OR1.5, OR2.5, and R1.5 electrodes. This relationship does not hold for the OR3.5 electrode (Figure 3B). In contrast, Figure 3A shows a decrease in the relative concentration of functional groups between the O3.5 and OR3.5 electrodes. This decrease can be ascribed to sufficient overpotentials that destabilize the observed catalytic functional groups leaving them more prone to reductive etching at negative potentials. Reductive etching of BD-UNCD functional groups is commonly cited in literature, and brought upon by wide voltage window cycling.12,28,29,31,49−51,57,59
As will be shown in the electrochemical section of this paper, the increased charge transfer shown in Figure 3B is quasi-reversible. Hence, surface bonded or adsorbed species on the electrodes oxidized at 1.5 and 2.5 V appear to possess catalytic properties. The surface sensitivity of XPS gives us detail as to how these functional groups are altering surface chemistry.
3.3. Electrochemical Characterization
Voltammograms performed at 500 mV/s in oxygen-saturated H2SO4 (0.5 M) (Figure 4A) exhibit two anodic peaks, Pa1 and Pa2, at 1.85 and 2.15 V, respectively. The capacitive current at this scan-rate was measured to be 0.11 μA by taking the average current ±0.1 V from the open-circuit potential (0.5 V) in Figure 4A. SEM images (Figure S2) showed that the surface is well polished and therefore the capacitive current was found to be negligible throughout the electrochemical experiments. Pa1 and Pa2 are attributed to sp2 surface-bonded carbon oxidation and the formation of oxygenated functional groups based on previous studies.12,49,60,61 A cathodic plateau, Pc , is observed during the reverse bias in the cathodic potential region between −0.35 and −0.6 V following a sufficient anodic overpotential. As a result, Pc is directly related to the anodic vertex potential (i.e., the potential at which the CV scan is reversed), as highlighted in Figure 4B. In fact, no cathodic plateau is observed when the anodic vertex potential is lower than 1.0 V. Yano et al. made a similar observation, both assigning Pc to oxygen reduction and ruling out the current contribution of functional groups during reduction.61 In a control experiment, with no oxygen in solution, we did not see any cathodic peak between 0 and −1 V on voltammograms, which partially confirms the statement made by Yano et al.61 Additionally, as shown in Figure 4B, Pc intensity increases with the anodic vertex potential for three possible reasons: (i) more functional groups are generated anodically, and therefore can be reduced, (ii) an increased number of functional groups allow more oxygen to be electrocatalytically reduced, and (iii) the anodic sweep generates oxygen in the near electrode vicinity (i.e., in the electrode porosity), which leads to a locally increased O2 concentration. However, when this anodic vertex potential is higher than 2.5 V, the subsequent reverse bias shows no cathodic peak between 0.0 V and −0.5 V (Green curve, Figure 4B). It is believed that the oxygenated functional groups formed between 1.85 V, and 2.15 V can be irreversibly overoxidized.12,29,30,57,61 Therefore, it can be concluded that both oxygen and oxygenated functional groups contribute to the reduction peak (Pc), consistent with the discussion in Section 3.2.
Figure 4.

CV scans at 500 mV/s in oxygen-saturated H2SO4 (0.5 M): (A) between −0.7 and 2.5 V, (B) between −0.5 V and an increasing anodic vertex potential.
Hereafter, attention is drawn to the evolution of the aforementioned peaks over successive scans as a function of the cathodic vertex potential (Figure 5). When the cathodic vertex potential is more anodic than −0.7 V, successive cathodic peaks retain the same current intensity (Figure 5A), whereas the successive anodic peaks exhibit an intensity decrease (Figure 5B). When the cathodic vertex is more cathodic than −0.7 V, Pc peak intensity increases over successive scans along with a similar increase of Pa2 intensity (Figure 5C–D). Pa1 appears to be less sensitive to the cathodic vertex potential than Pa2. Potentials beyond −0.7 V correspond to the onset of hydrogen evolution (Figure 5C). It is hypothesized that hydrogen adsorption favors the reduction of additional oxygenated functional groups, which leads to an increase of Pc and Pa2 intensity over successive CV scans. In order to successfully functionalize BD-UNCD electrodes in situ and also repeatedly regenerate the catalytic oxygenated functional groups, the anodic potential has to be chosen between 1 and 3 V vs Ag/AgCl, whereas the cathodic potential has to be more cathodic than −0.7 V vs Ag/AgCl.
Figure 5.

CV scans at 500 mV/s in oxygen-saturated H2SO4 (0.5 M). The anodic vertex potential is 2.5 V and the cathodic vertex potential is (A, B) higher than −0.7 V and (C, D) lower than −0.7 V.
3.4. H2O2 Generation
As described in Sections 3.2 and 3.3, preanodization allows oxygenated functional groups to be present at the BD-UNCD surface. There are two primary electrochemical pathways to create H2O2 in oxygen-saturated water. Equations 1 and 2 below correspond to the two-step anodic generation process of H2O2, whereas eq 3 corresponds to the direct cathodic generation of H2O2 from dissolved oxygen. Figure 6A shows an increase in H2O2 generation when the applied potential increases (for anodic generation) or decreases (for cathodic generation). Anodic Coulombic efficiency (CE) decreases for higher voltages (Figure 6A, blue curves), likely because of the competition with oxygen evolution. Cathodic CE has a maximum at −2 V. The decrease is due to competition with hydrogen evolution. However, the H2O2 concentration keeps increasing with more negative cathodic voltages as a result of the regeneration of oxygenated functional groups, highlighted in the electrochemical characterization section (Figure 5C, D).
Figure 6.

(A) H2O2 concentration produced and corresponding Coulombic efficiency after 30 min of electrolysis at different static voltages in a two-electrode cell. (B) H2O2 concentration produced using a potential cycling method for 30 min where the anodic cycle was constant at 100 s and the cathodic cycle, given by the horizontal-axis, was varied. When the cathodic cycle was equal to 1800 s, there was no anodic cycle. (C) H2O2 concentration produced using a potential cycling method for 30 min where the cathodic cycle was constant at 100 s and the anodic cycle, given by the horizontal-axis, was varied. When the anodic cycle was equal to 1800 s, there was no cathodic cycle.
Anodic H2O2 Generation
| 1 |
| 2 |
Cathodic H2O2 Generation
| 3 |
To study the influence of a potential cycling, experiments in the same electrolyte as before were performed as before by cycling the voltage between 2 V and −2 V vs Pt. In the first series of experiments, the anodic cycle time was kept constant at 100 s and the cathodic time was increased from 100 to 800 s. Additionally, one electrode was not cycled and was kept at −2 V for 1800 s without cycling. From Figure 6B, one can see that decreasing the cathodic cycle time increases the amount of H2O2 produced. With electrolysis time held constant, a longer cathodic polarization time implies a shorter anodic polarization time. As a result, less O2 is generated in the electrode vicinity and the chemical oxidative regeneration of oxygenated functional groups is shortened. Figure 6A highlights the higher CE at −2 V compared to 2 V. It is expected that the longer the cathodic polarization time (−2 V) and the correspondingly shorter the anodic polarization time (2 V), the higher the CE (Figure 6B, blue curve)
In a similar manner, the influence of the anodic cycle time on the overall H2O2 generation was studied. From Figure 6C, one can see that cycling the potential between 2 and −2 V increases both the generation of H2O2 and the corresponding CE, compared to anodizing at 2 V for 1800 s without cycling. These results suggest that reverse biasing allows for the reduction of surface accumulated oxygen nanobubbles formed in the electrode pores from water oxidation. Creation of nanobubbles on nanotextured electrodes from electrolysis has been previously reported.62,63 A long anodization time allows a substantial formation of oxygenated functional groups and oxygen nanobubbles, which increase the current and H2O2 generation during the cathodic polarization. Figure 7 confirms that the longer anodic cycle time correlates to a more gradual saturation of the total current during the cathodic step (i.e., due to greater availability of functional groups at the surface and oxygen in the electrode proximity) and leads to a higher overall current (i.e., greater reduction kinetics). As a result, more H2O2 can be generated from the reduction of dissolved oxygen.
Figure 7.
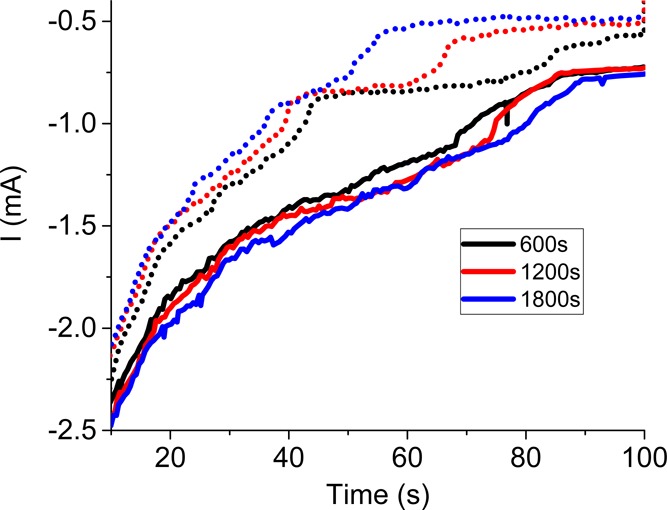
Chronoamperometric curves recorded at different time of the electrolysis during the cathodic step. The cathodic step lasted 100 s, whereas the anodic step lasted either 100 s (dash line) or 500 s (solid line).
From Figure 7, it can be seen that 100 s of anodic polarization is not long enough to regenerate all the catalytic oxygenated functional groups. Consequently, the cathodic current plateaus (between 30 and 85 s), and corresponding current intensity decreases with time (Figure 7, dashed line). For longer anodization, the cathodic plateaus either superimpose or are no longer present as a result of higher currents (Figure 7, solid line). This confirms that the oxygenated functional groups (OFG) (i) can be regenerated anodically and (ii) catalyze reduction processes60 including H2O2 generation as shown in Figure 6C.
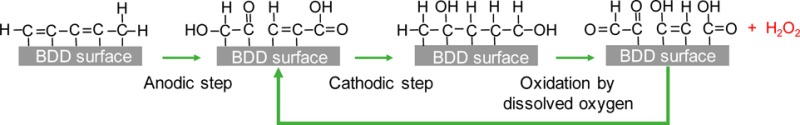
A description of the current versus time profile is proposed in Figure 8A. It is suggested that 4 successive steps occur during the reduction step: (i) The current decreases because of the reduction of oxygen concentration in close vicinity to the electrode surface. (ii) Although the OFG generated anodically are reduced, part of them are regenerated by dissolved oxygen. The regeneration process of these groups is schematized in Figure 8B. This leads to a steady-state evidenced by a quasi- plateau. (iii) Once oxygen is fully depleted in the electrode vicinity, OFG are then reduced, which leads to a current intensity decrease. (iv) Finally, a plateau is observed because of the constant electrolysis of the supporting electrolyte.
Figure 8.

(A) Proposed schematic of current vs time curve profiles during oxygen reduction. (B) Suggested mechanism for oxygenated functional group enhancement of H2O2 generation.
In controlled-potential electrolysis, the current decreases over time as a result of oxygen depletion in the electrode vicinity, which results in decreasing rates of H2O2 generation. Cycling to an anodic potential allows oxygen to be produced again, where long anodization coupled with short reduction times seems to be the optimal solution for increased H2O2 generation.
4. Summary and Conclusions
We have shown that anodic functionalization, or “pre-anodization” of an as-grown BD-UNCD electrode can enhance the cathodic production of H2O2 in a strong acidic environment (pH 0.5). It is proposed that the electrogeneration of functional groups at anodic potentials allows for an increase of current density during reductive potentials, resulting an increased production of H2O2. Through the proper choice of potential cycling conditions, it is shown that these functional groups can be stabilized, and used to continuously produce H2O2 more efficiently than static potential methods. Surface characterization measurements before and after testing of the BD-UNCD electrodes determined that sp2 carbon occurring at BD-UNCD grain boundaries can be oxidized into carbon–oxygen or carbon–hydrogen functionalities that influence the electron-transfer kinetics to catalyze the production of H2O2. Two important aspects of BD-UNCD surface chemistry and its response to an applied voltage in an acidic aqueous environment are proposed. First, sp2 and nondiamond content present at BD-UNCD grain boundaries serve as bonding sites for carbon–oxygen and carbon–hydrogen functional groups when an oxidative or reductive potential is applied. Second, there is a voltage window (−2.5 ≤ V vs Ag/AgCl sat. KCl ≤ 3.5) at which these functionalities can remain bonded to the surface before being destabilized by sufficient overpotential. Thus, it follows that there exists redox cycling conditions at which these functionalities are both optimized and stabilized in their redox processes. Moreover, it is likely that they can be oxidized and reduced repeatedly so long as they are not exposed to excessive overpotentials (lesser than −1.5 V; greater than 2.5 V). Future work will look at how these functional groups can enhance the production of other ROS species to more efficiently sanitize wastewater.
Acknowledgments
Partial funding for this work was provided by the Bill & Melinda Gates Foundation through the Reinvent the Toilet Program and NSF Award ECCS-1344745. The authors also thank the Shared Materials Instrumentation Facility (SMiF) at Duke University for their assistance and instrumentation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.7b01614.
H2O2 concentration versus absorbance calibration curve, SEM images of BD-UNCD, X-ray photoelectron spectroscopy, and Raman spectroscopy (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- WHO Guidlines for the Safe Use of Wastewater, Excreta and Greywater; World Health Organization: Geneva, 2006. [Google Scholar]
- Chittoor Jhansi S.; Kumar Mishra S. Wastewater Treatment and Reuse: Sustainability Options. Consilience: J. Sustain. Dev. 2013, 10, 1–15. 10.7916/D8JQ10Q1. [DOI] [Google Scholar]
- Lazarova V. Advanced Wastewater Disinfection Technologies: State of the Art and Perspectives. Water Sci. Technol. 1999, 40, 203–213. 10.1016/S0273-1223(99)00502-8. [DOI] [Google Scholar]
- Bourrouet A.; García J.; Mujeriego R.; Peñuelas G. Faecal Bacteria and Bacteriophage Inactivation in a Full-Scale UV Disinfection System Used for Wastewater Reclamation. Water Sci. Technol. 2001, 43, 187–194. [PubMed] [Google Scholar]
- Jefferson B.; Laine A. L.; Stephenson T.; Judd S. J. Advanced Biological Unit Processes for Domestic Water Recycling. Water Sci. Technol. 2001, 43, 211–218. [PubMed] [Google Scholar]
- Rajala R. L.; Pulkkanen M.; Pessi M.; Heinonen-Tanski H. Removal of Microbes from Municipal Wastewater Effluent by Rapid Sand Filtration and Subsequent UV Irradiation. Water Sci. Technol. 2003, 47, 157–162. [PubMed] [Google Scholar]
- Fazil Marickar Y. M. Electrical Conductivity and Total Dissolved Solids in Urine. Urol. Res. 2010, 38, 233–235. 10.1007/s00240-009-0228-y. [DOI] [PubMed] [Google Scholar]
- Raut A. S.; Cunningham G. B.; Parker C. B.; Klem E. J. D.; Stoner B. R.; Deshusses M. A.; Glass J. T. Disinfection of E. Coli Contaminated Urine Using Boron-Doped Diamond Electrodes. J. Electrochem. Soc. 2014, 161, G81–G85. 10.1149/2.1121410jes. [DOI] [Google Scholar]
- Jeong J.; Kim C.; Yoon J. The Effect of Electrode Material on the Generation of Oxidants and Microbial Inactivation in the Electrochemical Disinfection Processes. Water Res. 2009, 43, 895–901. 10.1016/j.watres.2008.11.033. [DOI] [PubMed] [Google Scholar]
- Chen G. Electrochemical Technologies in Wastewater Treatment. Sep. Purif. Technol. 2004, 38, 11–41. 10.1016/j.seppur.2003.10.006. [DOI] [Google Scholar]
- Fujishima A.; Einaga Y.; Rao T. N.; Tryk D. A.. Diamond Electrochemistry; Elsevier Science: Amsterdam, 2005. [Google Scholar]
- Macpherson J. V. A Practical Guide to Using Boron Doped Diamond in Electrochemical Research. Phys. Chem. Chem. Phys. 2015, 17, 2935–2949. 10.1039/C4CP04022H. [DOI] [PubMed] [Google Scholar]
- Jeong J.; Kim J. Y.; Yoon J. The Role of Reactive Oxygen Species in the Electrochemical Inactivation of Microorganisms. Environ. Sci. Technol. 2006, 40, 6117–6122. 10.1021/es0604313. [DOI] [PubMed] [Google Scholar]
- Michaud P. Preparation of Peroxodisulfuric Acid Using Boron-Doped Diamond Thin Film Electrodes. Electrochem. Solid-State Lett. 1999, 3, 77. 10.1149/1.1390963. [DOI] [Google Scholar]
- Palmas S.; Polcaro A. M.; Vacca A.; Mascia M.; Ferrara F. Influence of the Operating Conditions on the Electrochemical Disinfection Process of Natural Waters at BDD Electrodes. J. Appl. Electrochem. 2007, 37, 1357–1365. 10.1007/s10800-007-9368-3. [DOI] [Google Scholar]
- Cañizares P.; Martínez F.; Díaz M.; García-Gómez J.; Rodrigo M. a. Electrochemical Oxidation of Aqueous Phenol Wastes Using Active and Nonactive Electrodes. J. Electrochem. Soc. 2002, 149, D118. 10.1149/1.1490359. [DOI] [Google Scholar]
- Schmalz V.; Dittmar T.; Haaken D.; Worch E. Electrochemical Disinfection of Biologically Treated Wastewater from Small Treatment Systems by Using Boron-Doped Diamond (BDD) Electrodes - Contribution for Direct Reuse of Domestic Wastewater. Water Res. 2009, 43, 5260–5266. 10.1016/j.watres.2009.08.036. [DOI] [PubMed] [Google Scholar]
- Yano T. Electrochemical Behavior of Highly Conductive Boron-Doped Diamond Electrodes for Oxygen Reduction in Acid Solution. J. Electrochem. Soc. 1999, 146, 1081. 10.1149/1.1391724. [DOI] [Google Scholar]
- Granger M. C.; Witek M.; Xu J.; Wang J.; Hupert M.; Hanks A.; Koppang M. D.; Butler J. E.; Lucazeau G.; Mermoux M.; Strojek J. W.; Swain G. M. Standard Electrochemical Behavior of High-Quality, Boron-Doped Polycrystalline Diamond Thin-Film Electrodes. Anal. Chem. 2000, 72, 3793–3804. 10.1021/ac0000675. [DOI] [PubMed] [Google Scholar]
- Einaga Y. Diamond Electrodes for Electrochemical Analysis. J. Appl. Electrochem. 2010, 40, 1807–1816. 10.1007/s10800-010-0112-z. [DOI] [Google Scholar]
- Williams O. A.; Nesladek M.; Daenen M.; Michaelson S.; Hoffman A.; Osawa E.; Haenen K.; Jackman R. B. Growth, Electronic Properties and Applications of Nanodiamond. Diamond Relat. Mater. 2008, 17, 1080–1088. 10.1016/j.diamond.2008.01.103. [DOI] [Google Scholar]
- Williams O. A. Nanocrystalline Diamond. Diamond Relat. Mater. 2011, 20, 621–640. 10.1016/j.diamond.2011.02.015. [DOI] [Google Scholar]
- Gruen D. M. Ultrananocrystalline Diamond in the Laboratory and the Cosmos. MRS Bull. 2001, 26, 771–776. 10.1557/mrs2001.204. [DOI] [Google Scholar]
- Carlisle J. A.; Auciello O. Ultrananocrystalline Diamond. Properties and Applications in Biomedical Devices. Electrochem. Soc. Interface 2003, 12, 28–31. [Google Scholar]
- Wang J.; Firestone M. A.; Auciello O.; Carlisle J. A. Surface Functionalization of Ultrananocrystalline Diamond Films by Electrochemical Reduction of Aryldiazonium Salts. Langmuir 2004, 20, 11450–11456. 10.1021/la048740z. [DOI] [PubMed] [Google Scholar]
- Qin L. C.; Zhou D.; Krauss A. R.; Gruen D. M. Tem Characterization of Nanodiamond Thin Films. Nanostruct. Mater. 1998, 10, 649–660. 10.1016/S0965-9773(98)00092-0. [DOI] [Google Scholar]
- Michaelson S.; Ternyak O.; Hoffman A.; Williams O. A.; Gruen D. M. Hydrogen Bonding at Grain Surfaces and Boundaries of Nanodiamond Films Detected by High Resolution Electron Energy Loss Spectroscopy. Appl. Phys. Lett. 2007, 91, 103104. 10.1063/1.2779848. [DOI] [Google Scholar]
- Hutton L. A.; Iacobini J. G.; Bitziou E.; Channon R. B.; Newton M. E.; Macpherson J. V. Examination of the Factors Affecting the Electrochemical Performance of Oxygen-Terminated Polycrystalline Boron-Doped Diamond Electrodes. Anal. Chem. 2013, 85, 7230–7240. 10.1021/ac401042t. [DOI] [PubMed] [Google Scholar]
- Martin H. B. Voltammetry Studies of Single-Crystal and Polycrystalline Diamond Electrodes. J. Electrochem. Soc. 1999, 146, 2959. 10.1149/1.1392035. [DOI] [Google Scholar]
- Martin H. B. Hydrogen and Oxygen Evolution on Boron-Doped Diamond Electrodes. J. Electrochem. Soc. 1996, 143, L133. 10.1149/1.1836901. [DOI] [Google Scholar]
- Goeting C. H.; Marken F.; Gutiérrez-Sosa A.; Compton R. G.; Foord J. S. Electrochemically Induced Surface Modifications of Boron-Doped Diamond Electrodes: An X-Ray Photoelectron Spectroscopy Study. Diamond Relat. Mater. 2000, 9, 390–396. 10.1016/S0925-9635(99)00267-8. [DOI] [Google Scholar]
- Baldan M. R.; Azevedo A. F.; Couto A. B.; Ferreira N. G. Cathodic and Anodic Pre-Treated Boron Doped Diamond with Different Sp 2 Content: Morphological, Structural, and Impedance Spectroscopy Characterizations. J. Phys. Chem. Solids 2013, 74, 1830–1835. 10.1016/j.jpcs.2013.07.015. [DOI] [Google Scholar]
- Show Y.; Witek M. A.; Sonthalia P.; Swain G. M. Characterization and Electrochemical Responsiveness of Boron-Doped Nanocrystalline Diamond Thin-Film Electrodes. Chem. Mater. 2003, 15, 879–888. 10.1021/cm020927t. [DOI] [Google Scholar]
- Sarapuu A.; Vaik K.; Schiffrin D. J.; Tammeveski K. Electrochemical Reduction of Oxygen on Anthraquinone-Modified Glassy Carbon Electrodes in Alkaline Solution. J. Electroanal. Chem. 2003, 541, 23–29. 10.1016/S0022-0728(02)01311-6. [DOI] [Google Scholar]
- Vaik K.; Sarapuu A.; Tammeveski K.; Mirkhalaf F.; Schiffrin D. J. Oxygen Reduction on Phenanthrenequinone-Modified Glassy Carbon Electrodes in 0.1 M KOH. J. Electroanal. Chem. 2004, 564, 159–166. 10.1016/j.jelechem.2003.08.024. [DOI] [Google Scholar]
- Sarapuu A.; Helstein K.; Schiffrin D. J.; Tammeveski K. Kinetics of Oxygen Reduction on Quinone-Modified HOPG and BDD Electrodes in Alkaline Solution. Electrochem. Solid-State Lett. 2005, 8, E30. 10.1149/1.1848294. [DOI] [Google Scholar]
- Yano T.; Popa E.; Tryk D. A.; Hashimoto K.; Fujishima A. Electrochemical Behavior of Highly Conductive Boron-Doped Diamond Electrodes for Oxygen Reduction in Alkaline Solution. J. Electrochem. Soc. 1998, 145, 1870. 10.1149/1.1838569. [DOI] [Google Scholar]
- Goor G.; Glenneberg J.; Jacobi S.. Hydrogen Peroxide. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley–VCH: Weinheim, Germany, 2000; pp 394–427. [Google Scholar]
- Duo I.; Levy-Clement C.; Fujishima A.; Comninellis C. Electron Transfer Kinetics on Boron-Doped Diamond Part I: Influence of Anodic Treatment. J. Appl. Electrochem. 2004, 34, 935–943. 10.1023/B:JACH.0000040525.76264.16. [DOI] [Google Scholar]
- Goeting C. H.; Marken F.; Gutiérrez-Sosa A.; Compton R. G.; Foord J. S. Electrochemically Induced Surface Modifications of Boron-Doped Diamond Electrodes: An X-Ray Photoelectron Spectroscopy Study. Diamond Relat. Mater. 2000, 9, 390–396. 10.1016/S0925-9635(99)00267-8. [DOI] [Google Scholar]
- Kapałka A.; Fóti G.; Comninellis C. Investigations of Electrochemical Oxygen Transfer Reaction on Boron-Doped Diamond Electrodes. Electrochim. Acta 2007, 53, 1954–1961. 10.1016/j.electacta.2007.08.066. [DOI] [Google Scholar]
- Peralta E.; Vatividad R.; Roa G.; Marin R.; Romero R.; Pavon T. A Comparative Study on the Electrochemical Production of H 2 O 2 between BDD and Graphite Cathodes. Sustain. Environ. Res. 2013, 23, 259–266. 10.1016/j.watres.2015.05.019. [DOI] [Google Scholar]
- Komatsu M.; Rao T. N.; Fujishima A. Detection of Hydroxyl Radicals Formed on an Anodically Polarized Diamond Electrode Surface in Aqueous Media. Chem. Lett. 2003, 32, 396–397. 10.1246/cl.2003.396. [DOI] [Google Scholar]
- Eisenberg G. Colorimetric Determination of Hydrogen Peroxide. Ind. Eng. Chem., Anal. Ed. 1943, 15, 327–328. 10.1021/i560117a011. [DOI] [Google Scholar]
- Rynasiewicz J. Hydrogen Peroxide Determination in Presence of Chromate. Anal. Chem. 1954, 26, 355–358. 10.1021/ac60086a023. [DOI] [Google Scholar]
- Ohguri N.; Nosaka A. Y.; Nosaka Y. Detection of OH Radicals as the Effect of Pt Particles in the Membrane of Polymer Electrolyte Fuel Cells. J. Power Sources 2010, 195, 4647–4652. 10.1016/j.jpowsour.2010.02.010. [DOI] [Google Scholar]
- Salazar-Gastélum M. I.; Lin S. W.; Pina-Luis G. E.; Pérez-Sicairos S.; Félix-Navarro R. M. Electrochemical and Spectrometric Studies for the Determination of the Mechanism of Oxygen Evolution Reaction. J. Electrochem. Soc. 2016, 163, G37–G43. 10.1149/2.0251605jes. [DOI] [Google Scholar]
- Briggs D.; Grant J. T.. Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy; IM Publications: Chichester, U.K., 2003; Vol. pp 31–56. [Google Scholar]
- Ayres Z. J.; Borrill A. J.; Newland J. C.; Newton M. E.; Macpherson J. V. Controlled Sp 2 Functionalization of Boron Doped Diamond as a Route for the Fabrication of Robust and Nernstian pH Electrodes. Anal. Chem. 2016, 88, 974–980. 10.1021/acs.analchem.5b03732. [DOI] [PubMed] [Google Scholar]
- Foord J.; Hu J. P. Electrochemical Oxidation and Reduction Processes at Diamond Electrodes of Varying Phase Purity. Phys. Status Solidi A 2006, 203, 3121–3127. 10.1002/pssa.200671117. [DOI] [Google Scholar]
- Girard H.; Simon N.; Ballutaud D.; Herlem M.; Etcheberry A. Effect of Anodic and Cathodic Treatments on the Charge Transfer of Boron Doped Diamond Electrodes. Diamond Relat. Mater. 2007, 16, 316–325. 10.1016/j.diamond.2006.06.009. [DOI] [Google Scholar]
- Girard H. A.; Simon N.; Ballutaud D.; de La Rochefoucauld E.; Etcheberry A. Effects of Controlled Anodic Treatments on Electrochemical Behaviour of Boron Doped Diamond. Diamond Relat. Mater. 2007, 16, 888–891. 10.1016/j.diamond.2006.12.002. [DOI] [Google Scholar]
- Shenderova O. A.; Zhirnov V. V.; Brenner D. W. Carbon Nanostructures. Crit. Rev. Solid State Mater. Sci. 2002, 27, 227–356. 10.1080/10408430208500497. [DOI] [Google Scholar]
- Ryl J.; Burczyk L.; Bogdanowicz R.; Sobaszek M.; Darowicki K. Study on Surface Termination of Boron-Doped Diamond Electrodes under Anodic Polarization in H 2 SO 4 by Means of Dynamic Impedance Technique. Carbon 2016, 96, 1093–1105. 10.1016/j.carbon.2015.10.064. [DOI] [Google Scholar]
- Szunerits S.; Boukherroub R. Different Strategies for Functionalization of Diamond Surfaces. J. Solid State Electrochem. 2008, 12, 1205–1218. 10.1007/s10008-007-0473-3. [DOI] [Google Scholar]
- Salazar-Banda G. R.; Andrade L. S.; Nascente P. A. P.; Pizani P. S.; Rocha-Filho R. C.; Avaca L. A. On the Changing Electrochemical Behaviour of Boron-Doped Diamond Surfaces with Time after Cathodic Pre-Treatments. Electrochim. Acta 2006, 51, 4612–4619. 10.1016/j.electacta.2005.12.039. [DOI] [Google Scholar]
- Chaplin B. P.; Hubler D. K.; Farrell J. Understanding Anodic Wear at Boron Doped Diamond Film Electrodes. Electrochim. Acta 2013, 89, 122–131. 10.1016/j.electacta.2012.10.166. [DOI] [Google Scholar]
- Wang M.; Simon N.; Decorse-Pascanut C.; Bouttemy M.; Etcheberry A.; Li M.; Boukherroub R.; Szunerits S. Comparison of the Chemical Composition of Boron-Doped Diamond Surfaces upon Different Oxidation Processes. Electrochim. Acta 2009, 54, 5818–5824. 10.1016/j.electacta.2009.05.037. [DOI] [Google Scholar]
- Simon N.; Girard H.; Manesse M.; Ballutaud D.; Etcheberry A. Electrochemical Preconditioning of Moderately Boron Doped Diamond Electrodes: Effect of Annealing. Diamond Relat. Mater. 2008, 17, 1371–1375. 10.1016/j.diamond.2008.03.003. [DOI] [Google Scholar]
- Chaplin B. P. Critical Review of Electrochemical Advanced Oxidation Processes for Water Treatment Applications. Environ. Sci. Process. Impacts 2014, 16, 1182–1203. 10.1039/c3em00679d. [DOI] [PubMed] [Google Scholar]
- Yano T.; Popa E.; Tryk D. A.; Hashimoto K.; Fuishima A. Electrochemical Behavior of Highly Conductive Boron-Doped Diamond Electrodes for Oxygen Reduction in Acid Solution. J. Electrochem. Soc. 1999, 146, 1081. 10.1149/1.1391724. [DOI] [Google Scholar]
- Kikuchi K.; Ioka A.; Oku T.; Tanaka Y.; Saihara Y.; Ogumi Z. Concentration Determination of Oxygen Nanobubbles in Electrolyzed Water. J. Colloid Interface Sci. 2009, 329, 306–309. 10.1016/j.jcis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Pan G.; Yang B. Effect of Surface Hydrophobicity on the Formation and Stability of Oxygen Nanobubbles. ChemPhysChem 2012, 13, 2205–2212. 10.1002/cphc.201100714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.