

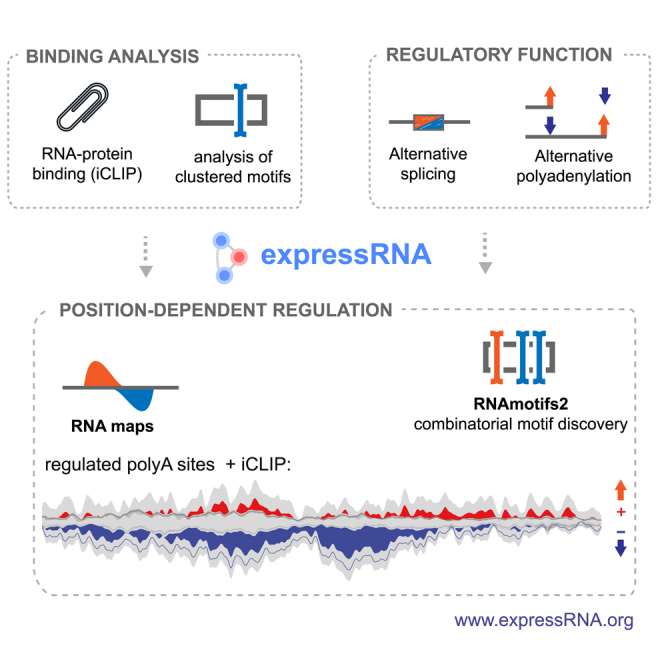
Summary
Many RNA-binding proteins (RBPs) regulate both alternative exons and poly(A) site selection. To understand their regulatory principles, we developed expressRNA, a web platform encompassing computational tools for integration of iCLIP and RNA motif analyses with RNA-seq and 3′ mRNA sequencing. This reveals at nucleotide resolution the “RNA maps” describing how the RNA binding positions of RBPs relate to their regulatory functions. We use this approach to examine how TDP-43, an RBP involved in several neurodegenerative diseases, binds around its regulated poly(A) sites. Binding close to the poly(A) site generally represses, whereas binding further downstream enhances use of the site, which is similar to TDP-43 binding around regulated exons. Our RNAmotifs2 software also identifies sequence motifs that cluster together with the binding motifs of TDP-43. We conclude that TDP-43 directly regulates diverse types of pre-mRNA processing according to common position-dependent principles.
Keywords: alternative polyadenylation, TDP-43, RNA map, positional regulatory principles, clustered sequence motifs, alternative splicing, expressRNA, RNAmotifs2, 3′ mRNA sequencing, iCLIP
Graphical Abstract
Highlights
-
•
TDP-43 regulates competing poly(A) sites in a highly position-dependent manner
-
•
expressRNA is a new platform for analysis of alternative polyadenylation and splicing
-
•
RNAmotifs2 is a cluster motif analysis platform integrated with expressRNA
-
•
Regulation of pre-mRNA processing might follow common position-dependent principles
Rot et al. investigate how TDP-43 regulates alternative polyadenylation in HEK293 cells. This defined position-dependent regulatory principles with high-resolution RNA maps. The authors provide an integrative computational platform for comprehensive analysis of alternative splicing and alternative polyadenylation (expressRNA), as well as software for positional analysis of clustered sequence motifs (RNAmotifs2).
Introduction
Biogenesis of most eukaryotic mRNAs involves splicing and cleavage and polyadenylation (3′ end processing) (Derti et al., 2012, Tian et al., 2005). Both mechanisms are required to produce functional mRNAs and are also important to regulate gene expression by producing alternative mRNA isoforms and for efficient transcription termination (Di Giammartino et al., 2011, Elkon et al., 2013, Shi, 2012). The alternative isoforms are produced by alternative splicing or by use of alternative polyadenylation (APA) sites. Both alternative splicing and APA are regulated by RNA-binding proteins (RBPs) (Di Giammartino et al., 2011, Elkon et al., 2013, Fu and Ares, 2014, Shi, 2012, Witten and Ule, 2011). However, few tools are available to study both processes in an integrated manner, and the overlap between their regulatory programs is poorly understood.
The regulatory function of many RBPs depends on their binding position in respect to the regulated exon (Witten and Ule, 2011). Such position-dependent regulatory principles have been visualized at high-resolution in the form of RNA maps (Ule et al., 2006), and were exploited to derive codes that can predict tissue-specific splicing patterns (Alipanahi et al., 2015, Barash et al., 2010). It is clear that the RBP binding on nascent RNA can also affect APA in a position-dependent manner (Batra et al., 2014, Li et al., 2015, Licatalosi et al., 2008, Masuda et al., 2015).
Three studies have used crosslinking and immunoprecipitation (CLIP) to define the RNA map of APA (Licatalosi et al., 2008, Masuda et al., 2015, Batra et al., 2014), but these studies have plotted the position of full CLIP reads around the regulated poly(A) sites, rather than the position of crosslink sites, and have not evaluated the statistical significance of identified enrichments. Therefore, the importance in binding position for guiding the repressing or enhancing effects of RBPs remains unclear. The nucleotide resolution that is obtained by the truncated cDNAs in iCLIP, and the quantitative nature gained by the analysis of unique molecular identifiers (UMIs), allowed us to examine the RNA maps of APA regulation in much greater detail.
Our study examines how the TAR (transactive response) DNA-binding protein 43 (TDP-43, also referred to as TARDBP) regulates splicing and APA via position-dependent principles. TDP-43 is an RBP involved in several neurodegenerative diseases, including frontotemporal lobar degeneration (FTLD-TDP) and amyotrophic lateral sclerosis (ALS) (Ratti and Buratti, 2016). TDP-43 regulates alternative splicing in a position-dependent manner by binding to intronic dinucleotide of uridine and guanosine (UG)-rich motifs, such that binding close to the splice sites represses splicing, whereas binding further downstream of the exon enhances splicing (Tollervey et al., 2011). The binding of TDP-43 is also enriched in 3′ UTRs, and it can regulate APA of its own transcript (Ratti and Buratti, 2016, Tollervey et al., 2011), indicating a possible role for regulating APA of other transcripts.
We identified poly(A) sites regulated by TDP-43 in HEK293 cells by 3′ mRNA sequencing and defined TDP-43 binding sites with individual nucleotide crosslinking and immunoprecipitation (iCLIP). To perform 3′ end data analysis, we developed expressRNA, a modular bioinformatics research platform that manages data and inter-connects recently developed (expressRNA.apa module, RNAmotifs2) and existing (STAR, DEXSeq) analysis software. The expressRNA.apa module identifies the sites where cleavage and polyadenylation takes place (poly(A) sites), marks and annotates the differentially regulated poly(A) sites, and visualizes binding patterns of RBPs around these sites. The resulting RNA maps defined the binding positions where TDP-43 can enhance or repress multiple types of poly(A) sites, and we validated its direct action with a minigene reporter. We demonstrated that the role of TDP-43 can be replaced in this minigene by TIA1, another known splicing regulator, if UG-rich motifs are replaced by UA-rich motifs. To identify enriched clusters of diverse regulatory motifs around the regulated exons or poly(A) sites, we extended and upgraded RNA motifs (Cereda et al., 2014) (now named RNAmotifs2). This identified enriched clusters composed of multiple types of motifs around tissue-specific exons or poly(A) sites, which can indicate a potential for combinatorial regulation of splicing and APA.
Results
Identification and Validation of Poly(A) Sites with expressRNA
To study how TDP-43 regulates APA, RNA was isolated from six independent TDP-43 knockdown (KD) and control HEK293 cells. The 3′ ends of mRNAs were amplified with the QuantSeq Rev 3′ mRNA sequencing (mRNA-seq) library prep kit (Lexogen), which uses a poly(T) primer to reverse transcribe the mRNAs. The library was sequenced with HiSeq, producing 60-nt single-end reads and 10-nt index reads.
For data analysis, we developed the expressRNA web application and analysis platform (Figures 1 and S1B). The first step of expressRNA is to process and map the 3′ mRNA-seq data to the genome, classify the sites where cleavage and polyadenylation takes place (poly(A) sites), and identify the differentially regulated poly(A) sites. We first aligned the reads to the hg19 reference genome with STAR (Dobin et al., 2013), allowing soft clipping from both 5′ and 3′ end. Of the total 52.96 million reads, 54.83% aligned uniquely to the human genome (Table S1). To construct the database of identified poly(A) sites, we considered sequence data from all experiments as one dataset. Some of the aligned reads were soft-clipped due to the sequencing running into the poly(A) tail or imperfect primer annealing (Figure S2A).
Figure 1.

expressRNA Research Platform
The expressRNA platform performs analyses of alternative polyadenylation datasets and can include external alternative splicing datasets. The identified or provided regulated features (poly(A) sites, alternative exons) are then combined with RNA protein binding information (iCLIP). Motif analysis is performed with the RNAmotifs2 platform. The results are presented with RNA maps that elucidate position-dependent regulatory mechanisms.
Since the longest 3′ UTR isoforms are in some cases not fully annotated, we added 5 kb of the intergenic region downstream of each gene. If two genes are closer than 10 kb, only the region up to the middle was added. We found that alternative positions of cleavage and polyadenylation can be clustered in close proximity of a dominant poly(A) site, with most variation occurring within 5 nt of the dominant site, indicating that the cleavage position is not always precise and can vary by a few nucleotides (Figure S2B). Such cleavage positions occur downstream of a single polyadenylation signal (PAS), and therefore their variation likely reflects lack of cleavage precision by the cleavage and polyadenylation machinery. For quantification of poly(A) sites, we therefore summed up the counts of reads that identified cleavage up to 5 nt away from each dominant poly(A) site (Figure S2C). Published studies also indicate that auxiliary RNA motifs tend to be enriched in the region approximately up to 75 nt upstream (URE, upstream regulatory elements) and 50 nt downstream (DRE, downstream regulatory elements) of each cleavage site (Beaudoing et al., 2000, Shi, 2012). We wished to focus our study on fully independent cleavage sites that contain their own PAS and auxiliary motifs. Therefore, we identified the dominant poly(A) sites based on read count, such that all resulting sites were at least 125 nt apart. This produced an atlas of 30,213 putative poly(A) sites.
QuantSeq relies on annealing a poly(T) primer to the poly(A) tail of mRNAs to identify the 3′ end of the mRNAs. However, as has been shown previously, poly(T) primers often also anneal to internal A-rich sites in mRNAs (Derti et al., 2012). Therefore, poly(A) sites with A-tracts in the vicinity [–10..10] were first filtered out. The poly(A) sites were further classified into three classes by using the software poly(A)R (Akhtar et al., 2010), which evaluates the presence of preceding PAS. This identified 17,102 PAS-strong, 2,599 PAS-weak, and 10,513 PAS-less sites (Table S4). We examined the nucleotide composition, the efficiency of cleavage, and the level of overlap with two past studies (Derti et al., 2012, Gruber et al., 2012), which confirms that the PAS-strong and PAS-weak sites are the most reliable and efficiently used sites (Figures S2D and S2E–S2H).
Classification and broad identification of poly(A) sites in HEK293 cells has been achieved earlier by a comprehensive analysis of 3′ end targeted sequencing datasets (Gruber et al., 2016). In contrast, the aim of our study is not to further characterize newly identified sites, but rather to use a stringent filtering approach to examine the regulatory patterns at the functional poly(A) sites. We therefore focused only on the PAS-strong and PAS-weak sites, which identified a total of 19,701 poly(A) sites, 16,599 sites were annotated to genes (Ensembl v.74). To ensure robust analysis, poly(A) sites with less than ten read counts in either control or KD experiments were further filtered out, resulting in 16,221 poly(A) sites. We then identified the poly(A) site in each gene that contained the highest read count, which we consider as the “major poly(A) site.” To avoid poorly used sites, we then discarded those poly(A) sites that have less than 5% of reads compared to the number of reads present at the major poly(A) site in the same gene. 15,113 sites in 10,069 genes remained after these filtering steps (Figure S2D).
Validation of the Poly(A) Sites
Comparisons of our 15,113 sites to the published poly(A) database in human tissues (Derti et al., 2012) and HEK293 cells (Gruber et al., 2012) demonstrates that the majority of sites overlap at nucleotide precision (Figure 2D). Allowing for up to a 10-nt spacing, approximately 70% of our sites overlap with the sites in Derti et al. (2012) and 65% with the previously defined sites in HEK293 cells from Gruber et al. (2012) (Figure 2D). The overlapping sites have the characteristic nucleotide signature of A-rich PAS upstream of the cleavage site, which is followed by T-rich sequences, in agreement with past studies (Sheets et al., 1990) (Figure S2F). A larger number of poly(A) sites were identified in HEK293 cells by a previous study due to the use of milder filtering criteria (Gruber et al., 2012), but the sites that are not shared with our study have weaker nucleotide signature compared to the overlapping sites (Figure S2G). However, the sites identified in our study have similar nucleotide signature to the ones that overlap with the published sites, indicating that the stringent filtering criteria applied by our study are sufficient to ensure that most of the identified poly(A) sites are valid (Figure S2G). Moreover, de novo search of the most enriched hexamers in the region where the PAS is normally located ([–30..–18] upstream of the poly(A) site) found the expected consensus sequences (Beaudoing et al., 2000) (Figure S2H; Supplemental Experimental Procedures).
Figure 2.

Mapping Reads and Evaluating Poly(A) Site Loci and Expression
(A) Read alignment to the hg19 reference genome. Reads are soft clipped (red) due to poly(A) tail sequence at the 5′ end or imperfect primer annealing at the 3′ end.
(B) Classification of proximal and distal poly(A) site pairs (5′ s1 = 5′ splice site 1). The same-exon pairs are not limited to the last exons in the gene, 9% (201 of 2,202) of the same-exon pairs are annotated to a non-terminal exon.
(C) DEXSeq computes direction (log2 fold change) and significance (adjusted p value) of poly(A) sites in control versus TDP-43 KD. The proximal-distal site pair with highest fold change is selected among significantly changed poly(A) sites (adjusted p value <0.05). Nonsignificant changes are classified as control-enhanced and control repressed. Control distributions are drawn as black lines in all RNA maps.
(D) Overlap with Derti et al., 2012, Gruber et al., 2012, and Nam et al. (2014) poly(A) sites around the poly(A) sites defined by the present study.
We further tested the validity of our detected poly(A) sites by plotting the coverage of RNA sequencing (RNA-seq) experiments from published studies performed in HEK293 cells (Pham et al., 2016, Trakman et al., 2016) (Figures S2J–S2L). We computed the read coverage in the region −500..200 around proximal and distal poly(A) sites, defined by the present study, which were separated into the 85% that are shared with the previous study by Nam et al. (2014), and the 15% that are not. We find a peak of increased RNA-seq average coverage upstream of both types of poly(A) sites, and a drop to negligible levels of RNA-seq coverage downstream of the distal poly(A) sites (Figure S2J). This significant difference in read coverage just upstream and downstream of both the annotated and poly(A) sites that we have annotated additionally supports the validity of poly(A) sites uncovered here.
For further comparison, we also plotted heatmaps of RNA-seq read coverage around the 50 known and poly(A) sites uncovered by this analysis that contain most QuantSeq reads by displaying the QuantSeq 3′ end targeted sequencing coverage (data from our study) alongside the RNA-seq coverage data (Figures S2K and S2L). This shows that the RNA-seq read distribution at individual poly(A) sites is similar for known and sites uncovered by this analysis, since most sites contain enrichment of RNA-seq reads upstream, and lack the reads downstream of the poly(A) sites. Taken together, this indicates that the poly(A) sites are accurately identified by the expressRNA analysis of QuantSeq data in HEK293 cells.
Classification and Identification of the Regulated Poly(A) Sites
To define the regulatory principles with high fidelity, we focused our analyses on 3,291 genes where we can robustly annotate multiple poly(A) sites. This represents about 33% of detected genes, which is slightly lower compared to most past studies, which reported approximately ∼40% of protein coding genes with >1 poly(A) site (Ni et al., 2013). It is much lower than the report of ∼70% with multiple poly(A) sites that were detected with the cross-tissue analysis (Derti et al., 2012), but that is expected given that a single cell type has less RNA diversity compared to all the tissues. The lower proportion of genes with multiple poly(A) sites likely reflects also our stringent requirement for that each poly(A) site contains at least 5% of the reads that map to each gene.
Next, we wished to identify those genes with multiple poly(A) sites where the use of the poly(A) sites changes upon TDP-43 KD. Statistically significant changes in poly(A) site use between control and KD conditions were identified using DEXSeq (Anders et al., 2012) (Figure 2C). If more than two poly(A) sites were identified in a gene, then the two poly(A) sites most significantly changed (adjusted p value <0.05) were considered for further analyses of regulated sites. If only one site had an adjusted p value <0.05, then the second site was selected based on highest read count. If no site had p value <0.05, then both sites were selected based on highest read count.
Our approach identified 3,291 genes (poly(A) site pairs) that we then classified based on the position of the two poly(A) sites in the gene (Figure 2B). If both poly(A) sites are in the same exon, we classified them as same exon (the most common APA type, also referred as tandem in previous studies) (Elkon et al., 2013). If the proximal poly(A) site is part of a composite exon that contains an internal 5′ splice site, and the two poly(A) sites were generated by alternative 5′ splice site use, we classified them as a composite exon. Finally, if the proximal poly(A) site is in an exon that is fully skipped when the distal poly(A) site is used, we classified them as skipped exon (Figure 2B). The final filtered poly(A) data included 3,291 poly(A) site pairs, of which 2,202 belonged to the same-exon, 662 to the skipped-exon, and 427 to the composite-exon class.
To identify changes in APA between control and TDP-43 KD, we examined the changes in relative read counts at the proximal and distal poly(A) sites in each gene, which is referred to as fold change (reported by DEXSeq). Poly(A) sites with adjusted p value <0.05 are labeled as significantly regulated. The fold change is used to determine the direction of change as repressed (fold change <0) or enhanced (fold change >0). For genes with no significantly regulated poly(A) sites, two sites with highest read counts across both control and KD conditions were selected and classified into control enhanced and control repressed (Figure 2C).
iCLIP and Motif Analysis Demonstrate a Direct Role of TDP-43 in APA Regulation
To identify TDP-43 binding sites in HEK293 cells, we performed iCLIP of TDP-43 in two replicate experiments, which together generated 12,622,661 uniquely mapped iCLIP cDNAs. We report the number of iCLIP cDNAs around each regulated poly(A) site (Table S2). We then identified 415,238 significant crosslink clusters, 9.4% of which map to annotated 3′ UTRs (Figure S2I). We visualized the positions of these crosslink clusters around the same-exon class of regulated poly(A) sites, including both the proximal (Figures 3A–3C) and the distal poly(A) sites (Figure 3D). We also examined the UG-rich binding motifs (see Defining TDP-43 Binding Positions with iCLIP), which are known to bind TDP-43. Reassuringly, crosslink clusters and enriched UG-rich motifs are abundant at similar positions (Figures 3A and S3A–S3D), further indicating that these two independent approaches correctly define the binding sites of TDP-43.
Figure 3.

RNA Maps of TDP-43 around Proximal and Distal Same-Exon Poly(A) Sites with iCLIP Clusters
(A) Proximal poly(A) site RNA map of TDP-43 iCLIP at regulated genes. TDP-43 is bound around repressed poly(A) sites and to a lesser extent (sparsely) further upstream and downstream of enhanced poly(A) sites.
(B) Top 20 iCLIP mRNA targets contributing to proximal poly(A) site repression.
(C) Top 20 iCLIP mRNA targets contributing to proximal poly(A) site enhancement.
(D) Distal site RNA map of TDP-43 binding around regulated genes. The level of binding is lower compared to the proximal sites (Table S6).
To further assess the specificity of the RNA map, we redrew it by using just the 13,034 poly(A) sites that overlap with the ones identified also with the 3P-SEQ method in the previous study (Figure 2D). The resulting same-exon proximal RNA map is almost identical to the map that used all the sites (Figures 3A and S3E), which demonstrates that the map is robust and largely unaffected by the identification of additional poly(A) sites. Moreover, we compared iCLIP binding of TDP-43, TIAL1, and STAU1 around the same-exon proximal poly(A) sites regulated by TDP-43 (Figure S3F). We chose TIA1 and STAU1 as controls, since both of these RBPs also have enriched crosslinking to 3′ UTRs. We plotted the enrichment of crosslink clusters for each protein by comparing regulated versus control poly(A) sites, which demonstrated much stronger and position-specific enrichment for TDP-43 compared to the other control RBPs. Together, these analyses indicate the RNA map is robust and specific.
Since TDP-43 is a known regulator of splicing, we also examined whether use of the poly(A) sites might be indirectly affected due to co-regulation (binding) at nearby splice sites. This is possible in the case of composite-exon and skipped-exon poly(A) sites, which contain at least one splice between the regulated poly(A) sites (s1 and s2, as marked in Figure 2B). We found position-dependent TDP-43 crosslinking at the regulated composite-exon and skipped-exon poly(A) sites (Figures S4A–S4D) and much less at splice sites flanking these poly(A) sites (Figures S5A–S5F; Table S6). This indicates that TDP-43 rarely regulates APA indirectly via splicing but rather directly regulates both types of competing poly(A) sites.
To demonstrate in a simple way that TDP-43 binds at different positions to repress or enhance the poly(A) sites, we defined the 40-nt window around each type of regulated poly(A) sites that had the strongest enrichment of crosslink clusters compared to controls (repressed/enhanced versus control genes) (Table S6). To determine the number of genes that contain a specific class of alternative poly(A) site that is directly regulated by TDP-43, we counted the number of iCLIP crosslink clusters at this 40-nt window, which allowed us to calculate the bound regulated genes score (BRG) (see Experimental Procedures and Table S6). This shows that TDP-43 most often binds next to the proximal same-exon poly(A) sites (−97..–57 relative to proximal poly(A) site, BRG = 33, p value 1E-6, Figure 3), while enriched binding is also seen further downstream of the enhanced sites (72..112 relative to proximal poly(A) site, BRG = 19, p value 3E-5, Figure 3). This pattern is reminiscent of the RNA maps of splicing regulation, where TDP-43 binds directly upstream or within the exon to repress and further downstream of the exon to enhance splicing (Tollervey et al., 2011).
RNAmotifs2 Allows Unbiased Discovery of the Regulatory RNA Maps
To understand whether the RNA map of APA regulation by TDP-43 can be discovered without any knowledge of its binding specificity, we upgraded our RNAmotifs software (Cereda et al., 2014) so that it could assess regulated poly(A) sites, in addition to alternative exons. Moreover, while the original RNAmotifs could only identify clusters of highly similar motifs around alternative exons, the version (named RNAmotifs2) can identify clusters of diverse motifs enriched around groups of exons or poly(A) sites. This can assess the potential for multiple RBPs to combinatorially regulate pre-mRNA processing. We applied RNAmotifs2 to analysis of the same-exon class of poly(A) sites regulated by TDP-43. The enrichment of the detected significant motif clusters (Fisher’s exact test, p < 0.01) is plotted in blue at the repressed and in red at enhanced poly(A) sites (Figure 4). UG-rich motif clusters were enriched mainly around the proximal regulated sites at similar positions as the iCLIP crosslink sites (Figures 3A and S3A; Table S6). This further confirms that TDP-43 can directly either repress or enhance poly(A) sites. Interestingly, the distal sites mainly had enrichment of U-rich and YA-rich motifs (Figure 4B), indicating a potential for regulation of competing poly(A) sites by different RBPs.
Figure 4.

Motif Analysis around TDP-43-Regulated Same-Exon Poly(A) Sites
Up to two of the most significant motif clusters are shown for each search region (R1, R2, R3).
(A) Motif analysis around proximal poly(A) sites show significant UG-rich clusters in R1 [–100..–40] and R2 [–40..20] around repressed proximal poly(A) sites. The repressive effect concords with TDP-43 iCLIP binding analysis (Figure 3). More distal binding in R3 [20..80] results in enhancement.
(B) Motif analysis around distal poly(A) sites reveals less pronounced regulatory effects, mostly enhancement guided by UC and UA-rich clusters.
Binding Site Swap Validates the Direct APA Regulation by TDP-43 or TIA1/L1
To study whether the binding of TDP-43 alone is sufficient to regulate APA, we produced a minigene reporter with the 3′ UTR of structural maintenance of chromosomes 1A (SMC1A) gene, which contains regulated poly(A) sites from the same-exon class (Figure 5A). The 3′ UTR of SMC1A gene was cloned downstream of the firefly luciferase ORF in a modified pcDNA3 plasmid, which does not contain any additional poly(A) sites. qRT-PCR was then used to quantify the use of distal poly(A) sites, which was normalized by the total amount of mRNA that was produced. To distinguish the minigene expression from the endogenous SMC1A gene, we quantified the total mRNA with a forward primer that recognizes part of the luciferase coding sequence. To monitor the distal poly(A) site use, the forward sequence was designed across the artificial junction that was introduced with production of the shortened SMC1A 3′ UTR. This confirmed the significant increase in the use of the proximal poly(A) site upon TDP-43 KD (Figure 5B).
Figure 5.

SMC1A Mini-Gene
(A) iCLIP TDP-43 binding along SMC1A 3′-UTR and zoomed in binding upstream of proximal poly(A) site. The intact SMCA1 sequence around the proximal polyA site is shown in the lower part (wild-type [WT] SMCA1), which includes the region upstream of the polyA signal (in bold) that contains a UG-rich region (in bold) that crosslinks to TDP-43 (shown above the sequence with blue bars). We introduced mutations (MUT) or deletion of TG dimers (DEL) to prevent TDP-43 binding to the UG-rich region in the RNA. The final minigene was designed with mutations that replaced G in TG dimers into T or A to convert the sequence into a binding site for TIA proteins (TIA). The mutant nucleotides are marked in red.
(B) Ratio in the use of distal vs. proximal polyA site in control cells, TDP-43 KD or the double KD of TIA1 and TIAL1 (TIA KD).
TDP-43 also affects mRNA stability (Volkening et al., 2009), and thus it is theoretically possible that it binds to the longer mRNA isoform and stabilizes this isoform, which could explain why the shorter mRNA isoform is increased upon TDP-43 KD. To rule out this possibility, we mutated or deleted a TDP-43 binding site that is located upstream of the proximal poly(A) site (Figure 5A). This binding site is present in both short and long isoforms, and it is unlikely that this site could lead to differential stability of the two isoforms. In contrast, the site is positioned upstream of the repressed poly(A) site, thus representing the pattern where the RNA map detects most binding enrichment (Figure 3A). TDP-43 binding in this region is ideally positioned to repress the nearby poly(A) site by blocking the recruitment of cleavage and polyadenylation factors. Indeed, disruption and deletion of the TDP-43 binding site in the minigene caused a strong increase in proximal poly(A) site use under control conditions, which is comparable with the derepression of the site that is seen upon TDP-43 KD in the wild-type minigene (Figure 5B). TDP-43 KD caused no further effect on the mutated or deleted minigene, confirming that these mutations abolished the capacity of TDP-43 to regulate the proximal poly(A) site.
Another protein, TIA1/TIAL1, can also bind to U-rich sequences and has been shown to bind to the 3′ UTR (Wang et al., 2010). To test whether the position-dependent activity of TDP-43 is shared by other RBPs, we therefore replaced the UG-rich motifs with a UA-rich sequence that is designed to promote binding of cytotoxic granule-associated RNA binding (TIA) proteins based on its similarity to the TIA-binding sites (Figure 5A) identified previously by iCLIP (Wang et al., 2010). Addition of this site restored repression of the proximal poly(A) site, and KD of TIA1/TIAL1 proteins confirmed that this repression is caused by binding of TIA proteins rather than TDP-43 (Figure 5B). This demonstrates that diverse RBPs can bind in the vicinity of the poly(A) site to block its use. Both TDP-43 and TIA proteins are also important regulators of splicing, thus indicating that splicing and 3′ end processing could often be regulated by the same proteins.
Motifs Known to Regulate Brain-Specific Processing Cluster next to UG-Rich Sequences
To provide insight into shared mechanisms of splicing and 3′ end processing, we used RNAmotifs2 to further examine the poly(A) sites and exons that are differentially used in the brain compared to other tissues. We first examined the poly(A) sites that are differentially used between brain and universal human reference (Derti et al., 2012). This detected several types of motifs, including enrichment of TCAT immediately downstream of the proximal poly(A) sites that are repressed in the brain (Figure S4E), which agrees with the previous finding that NOVA (Neuro-Oncological Ventral Antigen) proteins generally repress poly(A) sites when binding in close vicinity (Licatalosi et al., 2008) and indicates that NOVA proteins promote formation of mRNA isoforms with longer 3′ UTRs, which are known to be more common in neurons (Derti et al., 2012, Miura et al., 2013).
Finally, we analyzed motifs at alternative exons that are differentially spliced between brain and heart as defined by splicing microarray (ArrayExpress EMTAB-1911). As in our previous study, this detected TC-rich motifs and TCAT-related motifs, which correspond to the TC-rich and TCAT-related preferences of polypyrimidine tract-binding protein (PTBP) and NOVA proteins, respectively (Cereda et al., 2014) (Figure 6). The positional enrichment agreed with the known RNA maps of the two proteins, since PTBP proteins mainly bind upstream of brain-specific exons to repress them, while NOVA proteins bind upstream and within the exons that are repressed, and downstream of exons that are enhanced in the brain (Ule et al., 2006, Witten and Ule, 2011). By detecting the NOVA-binding motifs both at the poly(A) sites and exons that are differentially regulated in the brain, RNAmotifs2 confirmed the important role of NOVA proteins in both splicing and 3′ end processing.
Figure 6.

Motif Clusters Regulating Alternative Splicing in the Human Brain
Significant motif clusters involved in alternative splicing (comparing brain and heart tissue) obtained with RNAmotifs2. Both PTBP and NOVA proteins major effect is silencing of alternative exons. Alternative exons flanked by upstream TC and T-rich clusters (PTBP targets) are enhanced in the brain, where PTBP expression is low compared to other tissue. Contrary, NOVA proteins are highly expressed in the brain, and exons flanked by upstream TCAT-rich clusters (NOVA targets) are therefore repressed in this tissue. For previous analysis of data, see Cereda et al. (2014).
Our upgraded RNAmotifs2 can discover clusters of diverse motifs, and therefore it can provide insights into potential combinatorial regulation of poly(A) sites or exons. Indeed, it showed that UG-rich motifs tend to cluster close to both UC-rich and UCAU-rich motifs around the regulated exons (Figure 6) and regulated poly(A) sites (Figure S4E) in the brain. To further examine this clustering, we plotted the co-occurrence of UG and UC-rich motif clusters at 3′ splice sites of silenced alternative exons (Figure S4F) in the brain. Moreover, analysis of the alternative exons regulated by PTB, NOVA, and TDP-43 proteins discloses that UG and UCAU-rich motifs tend to cluster together around the exons regulated by NOVA proteins (Figure S6). These results indicate that proteins such as TDP-43 might cooperate with tissue-specific RBPs to regulate pre-mRNA processing; however, this hypothesis will need further experimental validation.
Discussion
In this study, we developed several tools to examine how cis-acting elements recruit RBPs to regulate splicing and APA in a transcriptome-wide manner (Figure S7). We developed expressRNA, a platform that processes 3′ mRNA sequencing data, classifies the sites where cleavage and polyadenylation takes place (poly(A) sites), and identifies the differentially regulated poly(A) sites. We also integrated the expressRNA platform with an upgraded RNAmotifs2 software to identify enrichment of combinatorial motif clusters around regulated exons or poly(A) sites. We used the motifs and the iCLIP data to define high-resolution RNA maps of APA regulation by TDP-43. This showed that TDP-43 can directly regulate both proximal and distal poly(A) sites of same-exon or skipped classes (Figure 7, Table S6, p value <0.01, BRG >10). When binding close to the poly(A) signal or the poly(A) site, TDP-43 represses use of the site, whereas it can enhance the site when binding further downstream. Similarly, TDP-43 generally represses splicing when binding close to 3′ splice site or the exon, while enhancing when binding further downstream of the regulated alternative exon (Tollervey et al., 2011). This indicates common position-dependent regulatory principles of both mechanisms.
Figure 7.

Summary of the Main Position-Dependent Modes of APA Regulation by TDP-43
(A) The binding patterns that are most enriched for the same-exon type of APA, as shown in Figures 3A–3D and S3B-E.
(B) The binding patterns that are most enriched for the skipped-exon type of APA, as shown in Figures S4C and S4D; the arrow marks the position of the regulated poly(A) site, and the circle marks the main position of TDP-43 binding. The blue color denotes the repressive, and the red the enhancing effect of TDP-43, and the positions of the regulatory patterns can be found in the Table S6 (p value <0.01, BRG >10).
Our findings are consistent with previous studies of RNA maps for APA regulation by several other RBPs (Batra et al., 2014, Licatalosi et al., 2008, Liu et al., 2013, Masuda et al., 2015). We extend these findings by assessing the regulatory principles at high resolution and in a quantitative manner, which shows the strongest direct role of TDP-43 is in repression of proximal poly(A) sites, especially by binding in close proximity upstream and downstream of the poly(A) site. While PAS is the primary element recruiting the cleavage and polyadenylation specificity factor (CPSF), other auxiliary sequences tend to be UG or U-rich in mammalian transcripts (Yang and Doublié, 2011). These include the TGTA motif upstream of the PAS that recruits the cleavage factor I(m) (CFIm) and the downstream UG-rich motifs that recruit the cleavage stimulation factor (CstF) (Hoque et al., 2013, Martin et al., 2012, Yao et al., 2012). TDP-43 thus appears to act as a competitor to displace CFI and CstF from the UG-rich sites on pre-mRNAs. Interestingly, we also find that TDP-43 binding is pronounced further downstream of the enhanced poly(A) sites, in which cases it might be able to stabilize the binding of processing factors at the nearby poly(A) site. The RNA maps of splicing suggest that TDP-43 might compete with binding of U2AF and other splicing factors when binding close to 3′ splice sites or recruit U1 small nuclear ribonucleoprotein (snRNP) when binding downstream of 5′ splice sites. It remains to be seen how TDP-43 manages to block or enhance pre-mRNA binding of such a variety of factors.
The use of RNAmotifs2 confirms that APA is, like alternative splicing, often regulated by RBPs that bind clusters of closely spaced short motifs on pre-mRNAs. It is clear that the precise position of UG-rich motifs defines the function of TDP-43 in repressing or enhancing either alternative exons or poly(A) sites (Tollervey et al., 2011). Mutation of the UG to UA-rich motifs replaced the function of TDP-43 with TIA proteins, which are also otherwise major regulators of splicing. This indicates that many RBPs may have pleiotropic functions in splicing and APA, with the position of their RNA-binding site being the primary determinant of their function.
Our preliminary analyses indicate that an integrative analysis of tissue-specific splicing and 3′ end processing could uncover additional RBPs that can regulate both processes. For example, we detected the known function of the cis-acting element TCAT in the brain-specific splicing and APA. Moreover, we find that UG-rich motifs are present within clusters of UC motifs at the tissue-specific exons, indicating a potential for crosstalk between proteins binding these motifs, such as TDP-43, CELF, and PTBP proteins. Enrichment of UG-rich motifs was recently also identified next to binding sites of Rbfox proteins (Damianov et al., 2016). Since our software can detect clusters of diverse motifs, it is well suited for identification of such sites of overlapping binding motifs for multiple RBPs, which might allow cooperative or competitive regulation of alternative splicing or alternative polyadenylation across various tissues and conditions.
Our findings are available online through an interactive web application at http://expressRNA.org/paper. The platform can examine published poly(A) sites and exons as well as discover additional ones. We apply state-of-the-art statistical methodology to identify differentially polyadenylated genes (DEXSeq). In this context, the ability of successfully identifying the regulatory RNA maps could also be an estimator (benchmark) of the validity and success of the presented approaches. Therefore, expressRNA provides a flexible (modular) data integrative research platform, making computational analysis highly reproducible and allowing user-friendly visualization with sharing of data and results.
Experimental Procedures
Experimental Setup in HEK293 Cells and Poly(A) Sequencing
HEK293 FlpIn T-Rex cells were maintained in DMEM with 10% fetal bovine serum (FBS), supplemented with 3 μg/mL blasticidine and 50 μg/mL zeocin.
For the small interfering RNA (siRNA)-induced TDP-43 KD, 20 nM of TARDBP stealth siRNA (Invitrogen, A-012394-14, 5′-GGCUCAAGCAUGGAUUCUA-3′) was mixed with 10 μL of RNAiMAX following the manufacturer’s reverse transfection protocol and added to a 10-cm dish of HEK293 FlpIn cells. For the non-targeting siRNA (Stealth RNAi siRNA negative control med GC content, Invitrogen 12935-300), 20 nM was also used to distinguish off-target effects from biologically relevant ones. After the first 24 hr of transfection, the medium was replaced with DMEM with 10% FBS, and, after an additional 24 hr, the cells were collected for analysis.
RNA was extracted using the Direct-zol RNA kit, and an in-column DNase digestion step was performed at room temperature for 15 min. The poly(A)seq libraries for samples that were transfected with either non-targeting siRNA or TARDBP siRNA were generated using the reverse QuantSeq 3′ mRNA-seq kit. Libraries were prepared from 500 ng of total RNA. In the protocol, one fragment per transcript was generated, which resulted in extremely accurate gene expression values. For the initial step of this kit, oligodT priming, including Illumina-compatible linker sequences, was carried out. The second strand synthesis was followed by purification with magnetic beads. Barcodes were introduced during the PCR amplification step as standard external barcodes.
Single-end sequencing (60 nt) was performed on a Illumina GA-2 with a Rapid Run flow-cell. This kit makes use of a custom sequencing primer that anneals to a linker sequence previously introduced in the oligo(dT) priming step for reverse transcription. The obtained reads are strand specific.
expressRNA
We developed two independent research platforms, expressRNA for computational analysis of post-transcriptional modifications including analysis of 3′ end sequence data (expressRNA.apa) and RNAmotifs2 for clustered motif analysis (see RNAmotifs2). The web application part of expressRNA (jQuery and JavaScript) allows the exploration of combined analysis results over an interactive web interface (Figure 1).
expressRNA supports several open-source and commercial 3′ end sequencing protocols (Lexogen forward/reverse, 3P-seq, pA-seq, PolyAseq). The platform is modular and scalable and runs on desktop computers for small sequence samples and on multi-core machines for large datasets. It provides a complete analytical framework incorporating several tools for reads alignment, genome annotation, calling of differentially polyadenylated genes, and integration of RNA-protein binding (iCLIP) datasets (Figure S1). We established an online server (http://expressrna.org/paper) to provide interactive browsing of results presented in this publication.
Processing the 3′ End Sequencing Data
We processed each of the 12 experimental datasets (Table S1) by aligning the reads to the reference human genome (hg19) using STAR aligner (Dobin et al., 2013) with default parameters. Tagging only one position per alignment (the first 5′ aligned nucleotide), we constructed the database of genome-wide polyadenylation events (Figure 2A).
Since internal priming (annealing to the genomic sequence instead of the poly(A) tail) is a major problem in 3′ end sequencing protocols, we checked the genomic sequence in the region [–10..10] surrounding the polyadenylation events and filtered out alignments containing stretches of six consecutive A or with 70% A coverage in any 10-nt sub-window in this region. Published studies indicate that auxiliary RNA motifs tend to be enriched in the region approximately up to 75 nt upstream (URE) and 50 nt downstream (DRE) of each cleavage site (Beaudoing et al., 2000, Shi, 2012). We wished to focus our study on fully independent cleavage sites that contain their own PAS and auxiliary motifs. Therefore, we identified the dominant poly(A) sites based on read count, such that all resulting sites were at least 125 nt apart. For this purpose, we ranked the polyadenylation events by read count in descending order and considered only the high-ranking events that are more than 125 nt apart. This resulted in the overall poly(A) site database.
It has been shown that cleavage is not an exact process (Pauws et al., 2001), and we find that cleavage can occur within a small window of positions around the dominant cleavage site (Figure S2B). To allow for the variation in cleavage precision (as seen in Figure S2B), we computed the per-experiment expression of each poly(A) site by summing the read counts that identify any position in the region [–5..5].
Identifying the Differentially Polyadenylated Genes with DEXSeq
To identify regulated poly(A) sites in genes, we input the read counts of all poly(A) sites remaining after filtering into DEXSeq. The analogy to alternative exons counts usually input to DEXSeq are read counts at poly(A) sites. We input count values for all replicates for control and TDP-43 KD 3′ end sequencing experiments. DEXSeq returns fold change (log2) and adjusted p value for each site. The genes where no poly(A) site reaches significance (p < 0.05) are classified as controls.
In genes with more than one poly(A) site, only two poly(A) sites are selected for further analysis. In control genes, the two poly(A) sites with highest read count are considered for further analysis. In regulated genes, two significantly changed sites (adjusted p value <0.05) with highest difference in fold change are selected for each gene, additionally requiring that fold changes are of opposite signs.
If the gene was marked as control, the proximal and distal control poly(A) sites are further labeled as control-down and control-up (dependent on their fold change). For regulated sites, if a proximal site has fold change <0, the site is marked as repressed, and if fold change >0, marked as enhanced (the reverse holds for distal sites). Gene site pairs are then used for further analyses, including RNA maps, RNAmotifs2, and gene ontology (GO) term analysis.
Classifying Poly(A) Site Pairs
After we select the proximal and distal site for each gene, we classify the pair (and consequently the gene) into same exon, composite exon, and skipped exon. For this classification, we use the gene level annotation that is computed by linearizing the Ensembl gene annotation by merging the transcript annotation. When no annotated splice site is present between the selected poly(A) sites, then the gene is assigned to the same-exon category (Figure 2B). If there is a splice site present between the selected poly(A) sites, further classification depends on the type of splice site preceding the proximal poly(A) site. If there is a 5′ splice site immediately upstream of the proximal site, the gene is assigned to the composite-exon category, and, if there is a 3′ splice site immediately upstream of the proximal site, the gene is assigned to the skipped-exon category (Figure 2B).
Visualizing Position-Dependent Regulation with RNA Maps
RBPs play a significant role not only in the regulation of alternative splicing but also in the regulation of APA. We designed experiments with intact cells as controls and KD samples as test experiments. After identifying alternatively polyadenylated genes, we defined three sets of genes (repressed, enhanced, and controls) and cumulatively plotted the iCLIP data around poly(A) sites of these three categories. The position of the polyA site (also referred to as the cleavage site) is marked with a red line at the center of the RNA maps (Figures 3 and S3–S5). The approach for visualizing an RNA map of APA is analogous to any other region of interest, such as, for instance, the intron-exon boundaries in the context of alternative splicing (Ule et al., 2006).
To display the variability of positional binding enrichment detected by RNA maps, we performed 10,000 bootstraps (sampling with replacement) of each gene class to obtain the SD (gray). The colored areas (red, blue) on RNA maps are drawn between the signal (repressed, enhanced) and control regions, excluding SDs, which conveys a picture of the most reliable enrichment of candidate regulatory binding sites (Figure 3A).
To determine the number of regulated genes that contain TDP-43 binding within a specific set of positions, we calculated the BRG, where the binding sites are defined either by the significant iCLIP crosslink clusters or RNA motif clusters (Table S6). BRG corresponds to the number of genes that contain at least one binding site in a specific window within the RNA map at a specific class of poly(A) sites. The BRG allows a comparative analysis of RNA maps, for example, showing the BRG in the entire map (reported BRG400, since the entire RNA map window is 400 nt).
Furthermore, to determine positions on the RNA map that have the strongest regulatory significance (fold change between repressed/enhanced and control genes), we performed 1M bootstraps (sampling with replacement) on the entire dataset. We then used a 40-nt sliding window and computed the p value by considering data versus bootstrap fold changes. For same-exon proximal poly(A) sites, we found that the most significant 40-nt window is between the −97th and −57th nucleotide relative to the repressed proximal poly(A) sites (p value = 1E-6, log2 fold change = 2.04, BRG = 33). The positions of the 40-nt most significant windows with respective BRGs and fold changes are available for each type of poly(A) site (Table S6).
We additionally plotted heatmaps of the repressed and enhanced top 20 bound genes, which demonstrate positional contributions of individual sites (Figures 3B, 3C, S3B, and S3C; Table S2).
Defining TDP-43 Binding Positions with iCLIP
iCLIP data were processed as described previously by iCount web server (http://icount.biolab.si) (Wang et al., 2010). The crosslink clusters were identified considering all crosslink sites that were significant with a false discovery rate (FDR) <0.05 at a maximum spacing of 20 nt between crosslink sites. Each binding site position was then defined by the exact position of the “bound GU-rich motifs” (GTGTG, TGTGT, TGCGT, TGTGC, CGTGT, ATGTG, GTATG, GTGTA, GCGTG, GTGCG, TGTGA, TGTAT, GTGTT, CTGTG, TATGT, TTGTG, TGAGT, GGTGT, GAGTG, GTGTC, TGTGG, AGTGT, GTTTG) that were present in iCLIP crosslink clusters.
RNAmotifs2
RNAmotifs2 is an upgraded version of the previously published RNAmotifs software (Cereda et al., 2014). We extended the motif search from alternative splicing regions to three regions surrounding poly(A) sites and modified the search algorithm with an iterative clustering method. The approach finds the motif with the strongest signal that differentiates up/downregulated sequences to control sequences first. Next, it removes sequences where the motif signal is strongest and compensates the loss of signal by clustering the strongest motif with additional motifs, iteratively constructing a cluster consisting of up to four different motifs.
RNAmotifs2 is a standalone application written in Python. The input of RNAmotifs2 is a list of genomic positions, either alternative exons or alternative poly(A) sites. Each input needs to be assigned to one of the three classes: control, enhanced, or repressed. After computing cluster motif analysis, the results are reported either independently or integrated with the expressRNA web application.
Minigene Analysis
The region used for the SMC1A minigene is composed of two sequences surrounding both poly(A) sites. The first part of SMC1A minigene contains an 812-bp-long sequence surrounding the proximal poly(A) site (positions 53406176..53406987), which is coupled to the 537-nt-long 3′ UTR sequence close to the distal poly(A) site (positions 53400970..53401506). The intervening part of the 3′ UTR was not incorporated in the minigene reporter due to the limiting size of the plasmid. The minigene (0.3 μg) was co-transfected into HeLa cells together with siRNA against TDP-43 or control or TIA, and the cells were harvested after 48 hr. Reverse transcription was performed using RevertAid (Fermentas), and qPCR was performed to assess the level of different poly(A) site usage using SYBRgreen according to manufacturer’s protocol. The expression of 3′ UTR of minigene was assessed using forward primer in the luciferase gene and reverse primer in the short 3′ UTR. The use of distal poly(A) site was assessed by using forward primer across the junction between proximal poly(A) site and the downstream sequence surrounding the distal poly(A) site, and reverse primer around the distal poly(A) site. Ratio between the distal and proximal poly(A) site use in each condition was normalized to the ratio in the control cells.
Author Contributions
G.R. developed expressRNA analysis software. I.H. and Z.W. carried out TDP-43 HEK293 experiments. T.L., M.M., and Z.W. carried out mini-gene experiments. M.H. carried out TDP-43 iCLIP experiments. N.H. performed iCLIP UG-rich cluster analysis. G.R. and J.U. developed the extended RNAmotifs2 software. G.R., T.C., and J.U. carried out APA bioinformatics. J.U., T.C., and C.v.M. supervised the project. J.U., G.R., Z.W., and C.v.M. prepared the manuscript.
Acknowledgments
We thank João F. Matias Rodrigues and Sebastian Schmidt for discussions on data analysis, Jan Attig for assistance with the minigene experiments, Dalia Daujotyte for assistance with the QuantSeq experiments, and von Mering and Ule lab members for comments on the manuscript. This work was supported by the European Research Council (206726-CLIP and 617837-Translate to J.U.) and the Slovenian Research Agency (J7-5460 to J.U. and T.C.); G.R. and C.v.M. were funded by the Swiss National Science Foundation (grant 31003A_160095).
Published: May 2, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.04.028.
Contributor Information
Gregor Rot, Email: gregor.rot@uzh.ch.
Jernej Ule, Email: jernej.ule@crick.ac.uk.
Accession Numbers
The accession number for the 3′ end (Lexogen QuantSeq Rev) and iCLIP data reported in this paper are ArrayExpress: E-MTAB-4732 and E-MTAB-4733.
Supplemental Information
Category specifies the type and loci of the regulation (enhanced, repressed and proximal, distal).
Detailed information with additional % mapped (unique alignments only) and % of internally primed (reads that were filtered out due to A-richness in the surrounding -5..5 nt region on the reference genome) of Rot et al. and Gruber et al. datasets, Related to Figure S2
References
- Akhtar M.N., Bukhari S.A., Fazal Z., Qamar R., Shahmuradov I.A. POLYAR, a new computer program for prediction of poly(A) sites in human sequences. BMC Genomics. 2010;11:646. doi: 10.1186/1471-2164-11-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipanahi B., Delong A., Weirauch M.T., Frey B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015;33:831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- Anders S., Reyes A., Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J. Deciphering the splicing code. Nature. 2010;465:53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- Batra R., Charizanis K., Manchanda M., Mohan A., Li M., Finn D.J., Goodwin M., Zhang C., Sobczak K., Thornton C.A., Swanson M.S. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol. Cell. 2014;56:311–322. doi: 10.1016/j.molcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoing E., Freier S., Wyatt J.R., Claverie J.M., Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10:1001–1010. doi: 10.1101/gr.10.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereda M., Pozzoli U., Rot G., Juvan P., Schweitzer A., Clark T., Ule J. RNAmotifs: Prediction of multivalent RNA motifs that control alternative splicing. Genome Biol. 2014;15:R20. doi: 10.1186/gb-2014-15-1-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damianov A., Ying Y., Lin C.-H., Lee J.-A., Tran D., Vashisht A.A., Bahrami-Samani E., Xing Y., Martin K.C., Wohlschlegel J.A., Black D.L. Rbfox Proteins Regulate Splicing as Part of a Large Multiprotein Complex LASR. Cell. 2016;165:606–619. doi: 10.1016/j.cell.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A., Garrett-Engele P., Macisaac K.D., Stevens R.C., Sriram S., Chen R., Rohl C.A., Johnson J.M., Babak T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino D.C., Nishida K., Manley J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell. 2011;43:853–866. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R., Ugalde A.P., Agami R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- Fu X.-D., Ares M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A.R., Martin G., Keller W., Zavolan M. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 2012;9:1405–1412. doi: 10.4161/rna.22570. [DOI] [PubMed] [Google Scholar]
- Gruber A.J., Schmidt R., Gruber A.R., Martin G., Ghosh S., Belmadani M., Keller W., Zavolan M. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016;26:1145–1159. doi: 10.1101/gr.202432.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M., Ji Z., Zheng D., Luo W., Li W., You B., Park J.Y., Yehia G., Tian B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods. 2013;10:133–139. doi: 10.1038/nmeth.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., You B., Hoque M., Zheng D., Luo W., Ji Z., Park J.Y., Gunderson S.I., Kalsotra A., Manley J.L., Tian B. Systematic Profiling of Poly(A)+ Transcripts Modulated by Core 3’ End Processing and Splicing Factors Reveals Regulatory Rules of Alternative Cleavage and Polyadenylation. PLoS Genet. 2015;11:1–28. doi: 10.1371/journal.pgen.1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi D.D., Mele A., Fak J.J., Ule J., Kayikci M., Chi S.W., Clark T.A., Schweitzer A.C., Blume J.E., Wang X. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Hu W., Murakawa Y., Yin J., Wang G., Landthaler M., Yan J. Cold-induced RNA-binding proteins regulate circadian gene expression by controlling alternative polyadenylation. Sci. Rep. 2013;3:2054. doi: 10.1038/srep02054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G., Gruber A.R., Keller W., Zavolan M. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 2012;1:753–763. doi: 10.1016/j.celrep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Masuda A., Takeda J., Okuno T., Okamoto T., Ohkawara B., Ito M., Ishigaki S., Sobue G., Ohno K. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev. 2015;29:1045–1057. doi: 10.1101/gad.255737.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P., Shenker S., Andreu-Agullo C., Westholm J.O., Lai E.C., Miura P., Shenker S., Andreu-Agullo C., Westholm J.O., Lai E.C. Widespread and extensive lengthening of 3′ UTRs in the mammalian brain. Genome Res. 2013;23:812–825. doi: 10.1101/gr.146886.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J.W., Rissland O.S., Koppstein D., Abreu-Goodger C., Jan C.H., Agarwal V., Yildirim M.A., Rodriguez A., Bartel D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell. 2014;53:1031–1043. doi: 10.1016/j.molcel.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni T., Yang Y., Hafez D., Yang W., Kiesewetter K., Wakabayashi Y., Ohler U., Peng W., Zhu J. Distinct polyadenylation landscapes of diverse human tissues revealed by a modified PA-seq strategy. BMC Genomics. 2013;14:615. doi: 10.1186/1471-2164-14-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauws E., van Kampen A.H., van de Graaf S.A., de Vijlder J.J., Ris-Stalpers C. Heterogeneity in polyadenylation cleavage sites in mammalian mRNA sequences: Implications for SAGE analysis. Nucleic Acids Res. 2001;29:1690–1694. doi: 10.1093/nar/29.8.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham, X., Song, G., Lao, S., Goff, L., Zhu, H., Valle, D., Avramopoulos, D., 2016. The DPYSL2 gene connects mTOR and schizophrenia. Published online November 1, 2016. http://dx.doi.org/10.1038/tp.2016.204. [DOI] [PMC free article] [PubMed]
- Ratti A., Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138:95–111. doi: 10.1111/jnc.13625. [DOI] [PubMed] [Google Scholar]
- Sheets M.D., Ogg S.C., Wickens M.P. Point mutations in AAUAAA and the poly (A) addition site: Effects on the accuracy and efficiency of cleavage and polyadenylation in vitro. Nucleic Acids Res. 1990;18:5799–5805. doi: 10.1093/nar/18.19.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Alternative polyadenylation: New insights from global analyses. RNA. 2012;18:2105–2117. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B., Hu J., Zhang H., Lutz C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33:201–212. doi: 10.1093/nar/gki158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey J.R., Curk T., Rogelj B., Briese M., Cereda M., Kayikci M., König J., Hortobágyi T., Nishimura A.L., Župunski V. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakman L., Hewson C., Burdach J., Morris K.V. RNA directed modulation of phenotypic plasticity in human cells. PLoS ONE. 2016;11:e0152424. doi: 10.1371/journal.pone.0152424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J., Stefani G., Mele A., Ruggiu M., Wang X., Taneri B., Gaasterland T., Blencowe B.J., Darnell R.B. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Volkening K., Leystra-Lantz C., Yang W., Jaffee H., Strong M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS) Brain Res. 2009;1305:168–182. doi: 10.1016/j.brainres.2009.09.105. [DOI] [PubMed] [Google Scholar]
- Wang Z., Kayikci M., Briese M., Zarnack K., Luscombe N.M., Rot G., Zupan B., Curk T., Ule J. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol. 2010;8:e1000530. doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten J.T., Ule J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011;27:89–97. doi: 10.1016/j.tig.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Doublié S. Structural biology of poly(A) site definition. Wiley Interdiscip. Rev. RNA. 2011;2:732–747. doi: 10.1002/wrna.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C., Biesinger J., Wan J., Weng L., Xing Y., Xie X., Shi Y. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc. Natl. Acad. Sci. USA. 2012;109:18773–18778. doi: 10.1073/pnas.1211101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Category specifies the type and loci of the regulation (enhanced, repressed and proximal, distal).
Detailed information with additional % mapped (unique alignments only) and % of internally primed (reads that were filtered out due to A-richness in the surrounding -5..5 nt region on the reference genome) of Rot et al. and Gruber et al. datasets, Related to Figure S2