

Abstract
Breast cancer (BC) continues to be the most frequently diagnosed cancer in American women, which disproportionately affects women of African-American (AA) descent. Previously, we reported greater serum levels of resistin in AA BC patients relative to Caucasian-American (CA) patients, and established its role in growth and aggressiveness of breast tumor cells. Here we have investigated the role of resistin in BC-chemoresistance. MDA-MB-231 and MDA-MB-468 BC cells of CA and AA origin, respectively, were incubated with resistin prior to doxorubicin treatment. Our data suggest that resistin conferred chemoresistance to both BC cell lines; however, the effect on AA cells was more profound. Furthermore, the resistin-induced doxorubicin-resistance was shown to occur due to suppression of apoptosis. Resistin treatment also affected the stemness of BC cells, as suggested by reduced cell surface expression of CD24, induced expression of CD44 and ALDH1, and increased capability of cells to form mammospheres. Mechanistic studies revealed that resistin-induced chemoresistance, apoptosis and stemness of BC cells were mediated through STAT3 activation. Taken together, our findings provide novel insight into the role of resistin in BC biology, and strengthen its role in racially disparate clinical outcomes.
Keywords: Racial disparity, breast cancer, resistin, chemoresistance, stemness, STAT3
1. Introduction
Breast cancer (BC) is the most frequently diagnosed cancer and the second leading cause of cancer-related deaths in women in the United States [1]. According to the American Cancer Society, approximately 252,710 women will be diagnosed with BC, and nearly 40,610 will die with this malignancy in 2017 [1]. More importantly, it disproportionately affects the women of African American (AA) descent with early onset and occurrence of more aggressive disease, poor therapeutic outcome, and a higher risk of recurrence, as compared to Caucasian-American (CA) women [2]. According to Surveillance, Epidemiology and End Results (SEER) 2013 data, despite low incidence rate of BC in AA women, compared to CA women (124.3 vs. 128.1, per 100,000, respectively), AA women face high death rates (28.19 vs. 20.26, per 100,000, respectively), contributing to BC disparity ratio of 39.14% [3]. These data suggest that BC is far more aggressive in AA women. Indeed, highly aggressive triple-negative breast cancer (TNBC), characterized by the absence of ER, progesterone receptor (PR) or HER2, is much more prevalent in AA women [4]. TNBC suffers from lack of targeted therapy and chemotherapy remains the only option for disease management [5–7]. Doxorubicin (adriamycin), a conventional anthracycline, has been an important mainstay in the treatment of BC [8]. However, its efficacy for BC treatment is hindered by inherent or acquired chemoresistance leading to poor clinical outcome [9]. Several mechanisms have been proposed for chemoresistance of BC, which may involve both intrinsic and extrinsic factors [10, 11]. Tumor microenvironment (TME) is comprised of a heterogeneous mixture of many different cells that produce and secrete a number of growth factors and cytokines [12, 13]. Interestingly, a recent study suggested the existence of disparate TME in AA vs. CA breast tumors [13–15]. Emerging data have provided convincing evidence for an important role of TME in cancer chemoresistance, which involves both direct and indirect mechanisms [16, 17]. TME may not only impact drug delivery to the tumor cells, but can also bestow tumor cells with chemoresistant phenotypes by altering their cell signaling [18, 19].
Resistin, initially described as an adipocyte-derived cytokine, has been studied extensively for its role in inflammation and obesity-related cancers [20, 21]. Several studies have shown that resistin is elevated in BC patients, and its elevated levels are associated with increased risk of BC [22–24]. In addition, the levels of resistin correlate with tumor stage, size and metastasis [25]. Importantly, expression of resistin was also found to affect the overall clinical outcome [25]. Further, we and others have reported increased levels of resistin in AA BC patients relative to those in Caucasian patients, and resistin is also shown to promote breast tumor growth and aggressiveness [13, 14, 26, 27]. Interestingly, elevated serum levels of resistin have also been reported in chemotherapy-treated BC patients [28]. In light of these observations, we attempted to investigate the role of resistin in chemoresistance of BC cells. TNBC cell lines of AA and CA origin (MDA-MB-468 and MDA-MB-231, respectively) were used to evaluate the effect of resistin on doxorubicin-sensitivity. Our data suggest that resistin promotes doxorubicin-resistance in BC cells by suppressing doxorubicin-induced apoptosis. Furthermore, resistin confers stem cell-like characteristics to BC cells. From the mechanistic standpoint, our data support an essential role of STAT3 activation in resistin-induced survival and stemness of BC cells. Altogether, our study suggests a novel role of resistin in BC chemoresistance, which could contribute, at least in part, toward a racially disparate therapeutic outcome in BC patients.
2. Material and methods
2.1 Cell culture
Human breast cancer (BC) cell lines, MDA-MB-231 and MDA-MB-468, were purchased from ATCC (Manassas, VA). Cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (GE Healthcare Life Sciences, Logan, Utah) supplemented with 10 % fetal bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA), penicillin (100 units/ml) and streptomycin (100 μg/ml) (Invitrogen, Carlsbad, CA) in a humidified atmosphere of 5 % CO2 at 37 °C. Cells were intermittently tested for mycoplasma contamination at our institutional flow cytometry core facility. Cells were routinely monitored for their typical morphology, and authenticated by in-house short-tandem repeats genotyping.
2.2 Antibodies and siRNAs
Antibodies used were: anti-cleaved caspase 7, -cleaved caspase 3 and -cleaved PARP1 (rabbit polyclonal), anti-BCL2, -BCL-xL, -KLF4, -Nanog (rabbit monoclonal) (Cell Signaling Technology, Danvers, MA); Anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA); Anti-human fluorochrome-conjugated antibodies against CD24 (Alexa Fluor-647 conjugated), CD44 (Brilliant violent-421 conjugated) (BioLegend, San Diego, CA) and β-actin (mouse monoclonal) antibody (Sigma-Aldrich, St. Louis, MO). Non-targeting scrambled siRNAs (NT-Scr), or STAT3 targeting siRNAs were purchased from GE Dharmacon (Lafayette, CO).
2.3 Treatments and transfection
MDA-MB-231 and MDA-MB-468 cells were seeded in 96 or 6 -well plates and treated with PBS or resistin (20 ng/ml) (Phoenix Pharma, Burlingame, CA) alone or in the presence of doxorubicin (0–2.0 μM), as mentioned in the pertinent figure legends. To examine the involvement of STAT3 in resistin-induced effects, BC cells were transfected with NT-Scr or STAT3-targeting siRNA (30 nM) and further treated with resistin and/or doxorubicin, and effects on cell viability, sphere formation, and associated proteins were examined.
2.4 Cell viability assay
Cells (1×104) were grown in 96-well plates and treated with PBS or resistin (20 ng/ml) for 12 h. After treatment, cells were further treated with doxorubicin (0–2.0 μM) in presence or absence of resistin for 72 h, and cell viability was calculated using WST-1 assay kit (Roche, Indianapolis, IN), as described earlier [29, 30].
2.5 Plating efficiency assay
Plating efficiency assay was performed to examine the effect of resistin on chemoresistance in the long term. For this, cells (5×102) were seeded in 6-well plates and cultured for 48 h. Subsequently, cells were treated with PBS or resistin (20 ng/ml) for 12 h before doxorubicin (0–1.0 μM) treatment and grown under normal culture conditions. Resistin was supplemented every 48 h during the culture. After two weeks, colonies were stained with crystal violet, photographed and counted using Image analysis software (Gene Tools, Syngene, Frederick, MD).
2.6 RNA isolation and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen), and reverse-transcribed with high capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA), as described earlier [31]. Subsequently, qRT-PCR was performed in 96-well plates using cDNA as a template and SYBR Green Master Mix on an iCycler system (Bio-Rad, Hercules, CA, USA) with specific primer pair sets. GAPDH served as an internal control. (Table S1). Following thermal conditions were used: cycle 1: 95 °C for 10 min, cycle 2 (40): 95 °C for 10 sec and 58 °C for 45 sec.
2.7 Immunoblot analysis
Immunoblotting was performed as previously described [13, 31]. Briefly, protein samples were resolved on 10 % polyacrylamide gels, transferred to PVDF membrane, and subjected to immunodetection using specific antibodies. Bands were visualized using ECL plus Western Blotting substrate kit (Thermo Scientific, Logan, UT) with an LAS-3000 image analyzer (Fuji Photo Film Co., Tokyo, Japan).
2.8 Measurement of apoptosis
Resistin or vehicle pre- treated (for 12 h) BC cells (1×106 cells/well) were exposed to 0.1 μM doxorubicin in presence or absence of resistin (20 ng/ml) for 72 h. After that, cells were stained with Annexin V APC and SYTOX Green (Invitrogen) and analyzed by flow cytometry, according to manufacturer’s protocol.
2.9 Detection of CD24+ and CD44+ cells
MDA-MB-231 and MDA-MB-468 cells were treated with PBS or resistin (20 ng/ml) for 48 h. Post-incubation, cells were washed and suspended in PBS supplemented with 2 % FBS. Subsequently, cells (1 × 106/ml) were incubated with fluorochrome-conjugated anti-CD24 (Alexa Fluor-647), and CD44 antibodies (Brilliant violent-421) for 15 min on ice. After incubation, cells were washed with PBS and subjected to flow cytometry analysis using FACS AriaII™ (BD Bioscience). 7-Aminoactinomycin D (7-AAD; BD Bioscience) was added to exclude non-viable cells before FACS analysis.
2.10 ALDEFLUOR assay
ALDEFLUOR assay was performed using ALDEFLOUR™ kit (Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. Briefly, cells (2.5 × 105) were incubated in ALDH1 assay buffer containing ALDH1 substrate (Bodipy Aminoacetaldehyde; BAAA) at 37 °C for 45 min with intermittent mixing. After incubation, cells were centrifuged at 250 g for 10 min, suspended in 0.5 ml of ALDH1 assay buffer and analyzed using FACS AriaII™. ALDH1 enzyme inhibitor [diethylaminobenzaldehyde (DEAB)] treated cells were used as a control. 7-AAD was added to the cell suspension before FACS analysis for the exclusion of dead cells.
2.11 Sphere formation assay
For sphere formation, MDA-MB-231 and MDA-MB-468 cells (1 × 103/well) were cultured in 6-well Ultra-Low attachment plates (Corning Incorporated, Corning, NY) in stem cell culture medium (DMEM:F-12K, 1:1; Life Technologies, Carlsbad, CA) containing penicillin (100 units/mL), streptomycin (100 μg/mL) and supplemented with basic fibroblast growth factor (bFGF; 20 ng/ml), epidermal growth factor (EGF; 20 ng/ml) (Life Technologies) and resistin (20 ng/ml) or vehicle every alternate day. After 15 days, the mammospheres were counted and photographed using phase contrast microscope, as described previously [32].
2.12 Statistical analysis
All the experiments were performed at least three times, and data are expressed as mean ± SD. Wherever suitable, the data were also subjected to unpaired two-tailed Student’s t-test and p< 0.05 was considered statistically significant.
3. Results
3.1 Resistin confers doxorubicin-resistance in breast cancer cells
Earlier, we have shown that elevated serum levels of resistin, particularly in AA BC patients, correlate with enhanced growth and aggressiveness of BC cell lines [13]. Also, recently, it was demonstrated that BC patients, who received chemotherapy, had significantly enhanced levels of serum resistin [28]. We, therefore, hypothesized that increased levels of resistin might confer resistance of BC cells against therapy-induced cell death. To test this, we pre-treated BC cell lines of AA (MDA-MB-468) and CA (MDA-MB-231) origin, with (20 ng/ml) resistin or vehicle (water) for 12 h, and then exposed them to increasing doses of doxorubicin (0–2 μM) in presence or absence of resistin for additional 72 h. Effect on cell viability was examined using WST-1 assay. Our data reveal that doxorubicin treatment resulted in a significant cell death in both MDA-MB-468 and MDA-MB-231 BC cells. Notably, resistin treatment significantly protected BC cells from doxorubicin-induced cell death (Fig. 1A). As indicated in Table 1, upon treatment with resistin, the IC50 values for doxorubicin significantly increased from 0.12 and 0.17 μM in MDA-MB-231 and MDA-MB-468 BC cells to 0.65 and 1.05 μM, respectively, indicating resistin-mediated resistance to doxorubicin.
Figure 1. Resistin promotes doxorubicin resistance in breast cancer cells.
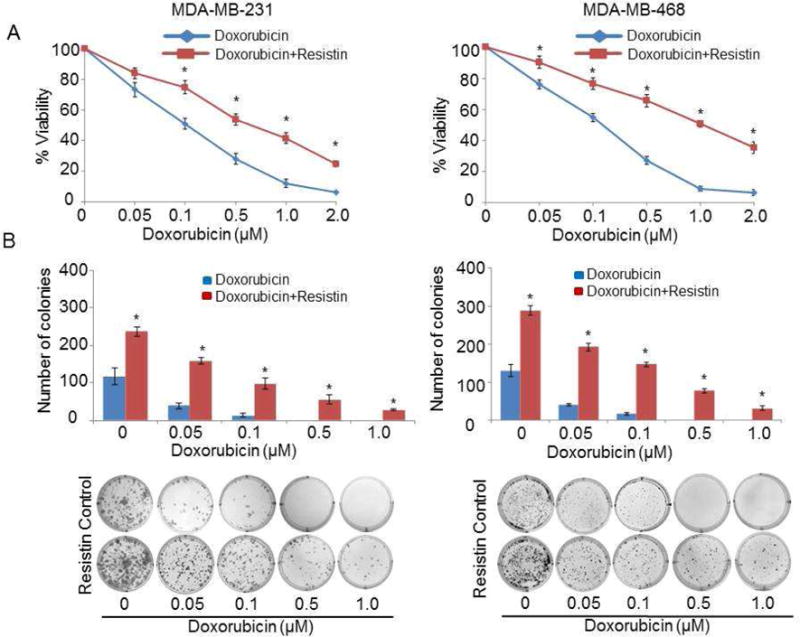
(A) MDA-MB-231 and MDA-MB-468 cells pretreated with resistin (20 ng/ml) for 12 h were further treated with indicated doses of doxorubicin (0–2.0 μM) for 72 h in the presence of resistin, and cell viability was determined by WST-1 assay. Cells untreated with resistin served as control. OD value of untreated cells was taken as 100 % viable. Data represent the mean ± SD; n = 3; *p < 0.01. (B) Cells were seeded (500 cells/well) in 6 well plates and allowed to adhere and establish for 48 h. Subsequently, cells were treated with resistin (20 ng/ml) for 12 h and then with varying doses of doxorubicin. Resistin was then supplemented to the media after every 48 h. After two weeks, colonies were stained with crystal violet and visualized and photographed using imaging system. Bars represent mean ± S.D. n=3, *p < 0.01. Representative images of colonies, formed in 2 weeks after treatment with increasing concentrations of doxorubicin in presence or absence of resistin, are shown at the bottom.
Table 1.
Doxorubicin IC50 values for MDA-MB-231 and MDA-MB-468 breast cancer cells.
| Cell lines | IC50 (in μM) of Doxorubicin at 72 h | |
|---|---|---|
| Vehicle | Resistin | |
| MDA-MB-231 | 0.12 | 0.65 |
| MDA-MB-468 | 0.17 | 1.05 |
Further, we analyzed the effect of resistin on doxorubicin resistance in BC cells by plating efficiency (an ideal test to monitor growth in long-term). Our data show a significant chemoprotective effect of resistin on both the cell lines (Fig. 1B). Importantly, no visible colony was detected in vehicle-treated MDA-MB-231 and MDA-MB-468 cells at 0.5 and 1.0 μM doxorubicin doses. However, a remarkably higher number of colonies were observed in both the BC cell lines with resistin and doxorubicin (0.5 and 1.0 μM dose) (Fig. 1B). Interestingly, a considerably greater resistin-induced doxorubicin resistance was observed in the cell line of AA origin (MDA-MB-468), as compared to MDA-MB-231 cells (CA origin). Altogether, our data thus suggest that enhanced resistin level may be one of the key mechanisms underlying high chemoresistance of this tumor type, especially in patients of AA origin.
3.2 Resistin-protects breast cancer cells from doxorubicin-induced apoptosis
Since induction of apoptotic cell death is a major mechanism of chemotherapeutic drugs [33, 34], we were interested to see if resistin has any effect on doxorubicin-induced apoptosis. For this, resistin pre-treated MDA-MB-231 and MDA-MB-468 cells were treated with doxorubicin alone or in combination with resistin and, the extent of apoptosis was monitored by Annexin V staining. We observed a lower apoptotic index (Annexin V positive/SYTOX negative cells) in BC cells treated with a combination of resistin and doxorubicin, as compared with doxorubicin treatment alone. Data shows 79.9 % and 43.6 % apoptotic cells in MDA-MB-231 and MDA-MB-468 cells, respectively, that were treated with doxorubicin only (Fig. 2A). Importantly, the doxorubicin-induced apoptosis was significantly abolished following co-treatment with resistin, as only 33.0 % (MDA-MB-231 cells) and 29.2 % (MDA-MB-468 cells) apoptosis was observed in the BC cells treated with doxorubicin along with resistin (Fig. 2A).
Figure 2. Resistin abrogates doxorubicin-induced apoptosis in BC cells.
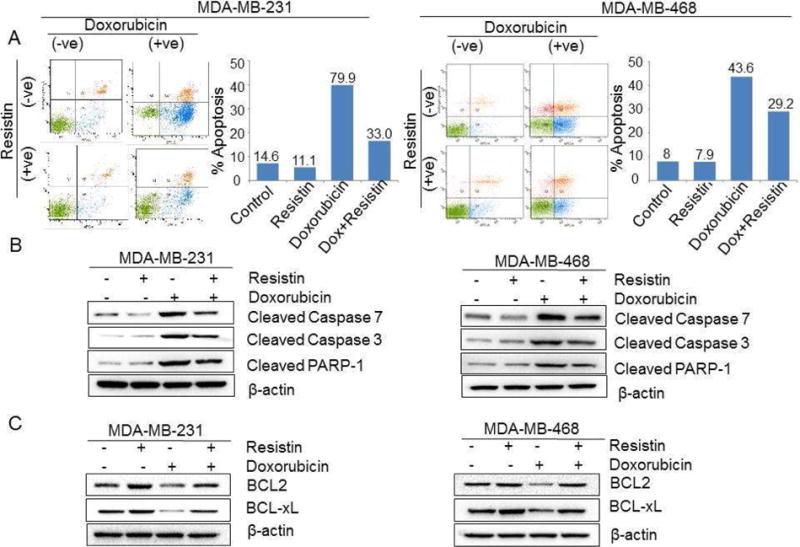
(A) Resistin-pretreated breast cancer cells were treated with doxorubicin (0.1 μM) in the presence of resistin for 72 h. Following treatment, the number of apoptotic cells was determined by Annexin V-binding assay, using flow cytometry. (B and C) Resistin-pretreated breast cancer cells were exposed to doxorubicin (0.1 μM) in presence and absence of resistin for 48 h. After that, total protein was isolated, and immunoblot analysis was performed to analyze the expression of (B) cleaved-caspase 7, caspase 3 and PARP1 and (C) BCL2 and BCL-xL. β-actin was used as loading control.
Activation of caspases (caspase 3 and 7) and subsequent activation of PARP-1, a known cellular substrate of caspases, is considered a hallmark of apoptotic cell-death [35, 36]. We, therefore, examined the effect of resistin and/or doxorubicin on these apoptosis effector molecules. In concordance to our flow-cytometry data, doxorubicin treatment resulted in the enhanced cleavage of caspases-3 and -7, and PARP-1, in both BC cells (Fig. 2B), an effect that was abrogated upon resistin-treatment (Fig. 2B). To study the underlying molecular mechanism, we further analyzed the expression of survival related proteins (BCL2 and BCL-xL) in the above mentioned treatment groups, by qRT-PCR and immunoblot assay. We observed a drastic reduction in the expression of anti-apoptotic proteins BCL2 and BCL-xL, following doxorubicin treatment, at both mRNA (data not shown) and protein levels (Fig. 2C). Interestingly, doxorubicin-mediated downregulation of BCL2 and BCL-xL was abolished in BC cells upon resistin treatment (Fig. 2C). Our findings thus clearly suggest a significant protection of BC cells by of resistin against doxorubicin-induced apoptosis.
3.3 Resistin induces stemness in breast cancer cells
The role of cancer stem cells in chemoresistance is well established [37]. Therefore, we investigated the significance of resistin in stemness potential of BC cells. CD24 (low) and CD44 and ALDH1 (high) expression/activities are well-characterized markers of BC stem cells (BCSCs). We, therefore, first examined the effect of resistin on the expression of stemness-associated markers in BC cells by quantitative RT-PCR and flow-cytometry. Our data reveal that resistin treatment enhanced the expression of CD44 and ALDH1 in MDA-MB-231 and MDA-MB-468 cells at mRNA level, with a concomitant decrease in the CD24 expression, as compared to respective vehicle-treated cells (Fig. 3A). Further, we stained the BC cells for CD24 and CD44, using Alexa Fluor-647 and Brilliant violent-421 antibodies, respectively, and performed flow-cytometric analysis. Our data indicate that the individual geometric mean fluorescence intensity (MFI) of CD44 was increased in resistin-treated MDA-MB-231 (1.1 fold) and MDA-MB-468 (3.2 fold) cells, as compared to vehicle-treated cells (Fig. 3B). In contrast, reduction in MFI for CD24 (1.1 and 2.0 folds, respectively) was observed upon treatment with resistin in both MDA-MB-231 and MDA-MB-468 cells. BCSCs are enriched with ALDH1 [38], and our data show that the number of ALDH1 positive cells increased from 29.8 % to 37.4 % in the MDA-MB-468 cells upon resistin treatment (Fig. 3C), while opposite effect on ALDH1 activity was seen in MDA-MB-231 cells upon resistin treatment.
Figure 3. Effects of resistin on the breast cancer-specific stemness-markers.
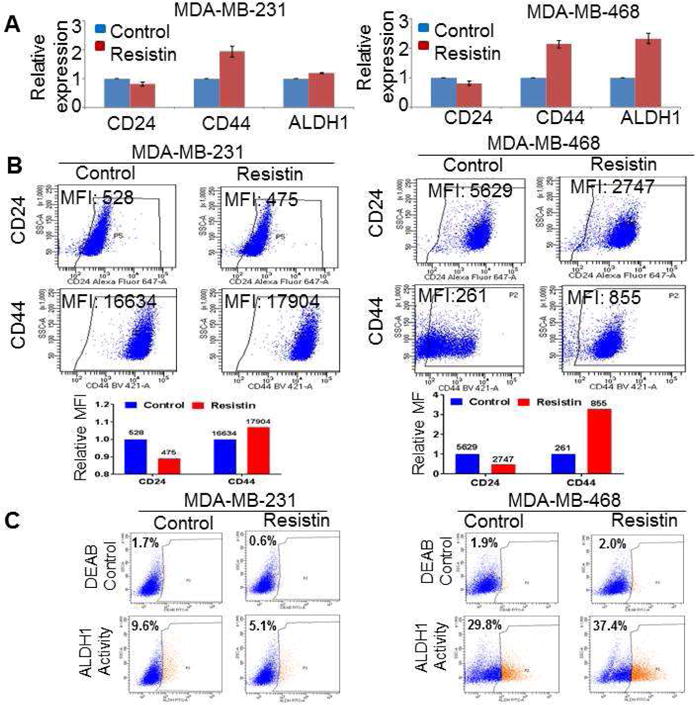
(A) Breast cancer cells were treated with resistin for 48 h, total RNA was isolated, c-DNA prepared and expression of stemness-associated markers (CD24, CD44, and ALDH1) examined by real-time quantitative RT-PCR assay. GAPDH was used as an internal control. (B and C) Resistin-treated BC cells were incubated with fluorochrome-conjugated (B) anti– CD44, –CD24 and (C) –ALDH1 antibodies for 15 min on ice and analyzed by flow cytometry. The data obtained from FACS analysis is presented as a geometric mean of fluorescence intensity (MFI) or percentage of cells.
Further, to identify if these changes in surface markers also translate into functional alterations, we tested the ability of these BC cell lines to generate mammospheres upon resistin treatment. For this, cells were seeded at low density in ultra-low attachment plate and supplemented with resistin and growth factors every alternate day. The cells were allowed to grow in stem cell culture medium for 15 days, and the number of spheres formed was counted. Data demonstrate greater sphere forming ability (~2.0 fold) in MDA-MB-231 cells treated with resistin, as compared to the control cells (Fig. 4A). Notably, the effect of resistin on the sphere formation was more potent (~2.8 fold) in BC cells of AA origin i.e. MDA-MB-468 (Fig. 4A). Furthermore, we also examined the effect of resistin on the expression of stemness-associated transcription factors (Nanog and KLF4) at mRNA and protein levels. Our data exhibit significant induction in the expression of both Nanog and KLF4 in MDA-MB-231 as well as MDA-MB-468 cells, at both mRNA (Fig. 4B) and protein levels (Fig. 4C). Taken together, our data show that resistin potentiates stemness in BC cells.
Figure 4. Resistin enhances stemness potential in breast cancer cells.
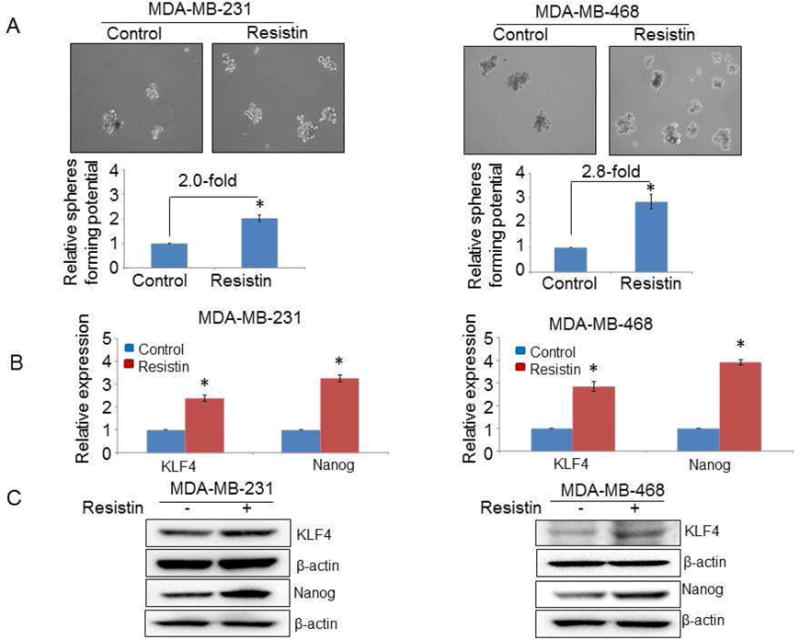
(A) MDA-MB-231 and MDA-MB-468 cells (1 × 103) were seeded in ultra-low attachment plate and treated with resistin or vehicle, and allowed to grow for 15 days in stem cell culture medium. Resistin (20 ng/ml) was added every alternate day during incubation. Images were taken under a light microscope at 200x magnification, and the number of spheres (size > 50 μm) was counted. Error bars represent the mean ± SD; n = 3; *p < 0.05. (B and C) Breast cancer cells were treated with resistin and the expression of stemness-associated markers was examined (B) at mRNA level by quantitative RT-PCR (C) and protein level by immunoblot assay. GAPDH (for mRNA) and β-actin (for protein) were used as an internal control.
3.4 STAT3 signaling mediates resistin-induced drug resistance and stemness in breast cancer cells
In our earlier study [13], we demonstrated that resistin enhances the expression and phosphorylation of STAT3 that mediates resistin-induced growth and aggressiveness of BC cells. Furthermore, studies document the critical role of STAT3 in proliferation and self-renewal of CSCs and the promotion of therapy-resistant phenotype in several cancer types, including BC [39]. Therefore, we sought to examine whether STAT3 is mechanistically involved in resistin-induced drug resistance and stemness. For this, BC cells were transfected with non-target scrambled (NT-Scr) or STAT3-specific siRNA, and silencing was confirmed by immunoblot assay. STAT3 siRNAs efficiently silenced its expression (basal as well as resistin-induced) by 24 h, an effect that sustained at least up to 72 h post-transfection (data not shown). STAT3-silenced BC cells were pre-treated with resistin or vehicle for 12 h and further treated with increasing doses of doxorubicin, and the effect on cell viability was examined. Data show that resistin protects BC cells, that were transfected with NT-Scr, from doxorubicin-induced cytotoxicity (Fig. 5A and Table 2), while no chemoprotective effect of resistin is seen in both MDA-MB-231 and MDA-MB-468 cells transfected with STAT3-specific siRNAs (Fig. 5A and Table 2).
Figure 5. Silencing of STAT3 abrogates resistin-induced effects in breast cancer cells.
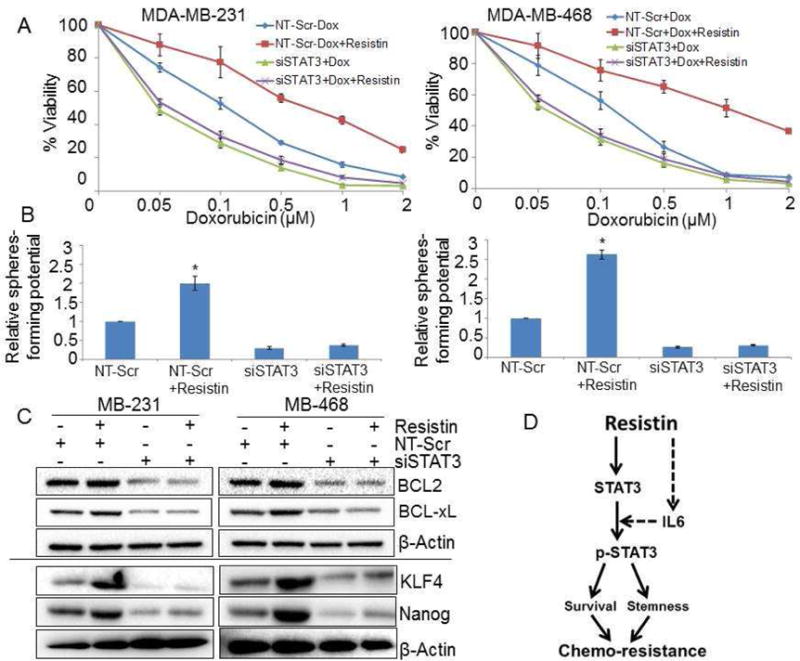
(A) Cells were seeded in 96 well plates and transfected with non-targeting (NT)-Scr or siSTAT3. 24 h after transfection, cells were treated with resistin (20 ng/ml) for 12 h, followed by incubation with various concentration of doxorubicin (0.05-2 μM) in presence or absence of resistin for 72 h and cell viability was measured by WST-1 assay. Bars represent mean ± S.D (n=3). (B) To examine the role of STAT3 in resistin-induced stemness, BC cells were transfected with NT-Scr or siSTAT3 and sphere formation assay was performed in presence or absence of resistin as described earlier. Sphere >50 μm in diameter was counted at 200× magnification. Error bars represent the mean ± SD; n = 3; *p < 0.05. (C) MDA-MB-231 and MDA-MB-468 cells were transfected with NT-Scr or STAT3-targeting siRNAs for 24 h, post transfection, cells were treated with resistin (20 ng/ml) for next 48 h and expression of survival (BCL2 and BCL-xL) and stemness (KLF4 and Nanog) associated genes were observed by immunoblot assay. (D) A schematic diagram showing the molecular basis of resistin induced-chemoresistance in breast cancer cells. Resistin not only regulates the expression of STAT3 but also enhances its activation. Phosphorylation of resistin-induced STAT3 is mediated through IL-6. Further, activation of STAT3 results in enhanced stemness potential, survival and chemoresistance. Arrow dash indicates earlier published findings [13]
Table 2.
Doxorubicin IC50 values for control Vs. STAT3 silenced MDA-MB-231 and MDA-MB-468 breast cancer cells
| Cell lines | IC50 (in μM) of Doxorubicin at 72 h | |||
|---|---|---|---|---|
| NT-Scr | siSTAT3 | |||
| Vehicle | Resistin | Vehicle | Resistin | |
| MDA-MB-231 | 0.15 | 0.72 | 0.05 | 0.06 |
| MDA-MB-468 | 0.19 | 1.1 | 0.06 | 0.07 |
Moreover, we also analyzed the role of STAT3 in resistin-induced stemness potential of BC cells. Data demonstrate that silencing of STAT3 significantly diminished the sphere forming ability of BC cells (Fig. 5B). Notably, when STAT3-silenced BC cells were treated with resistin, no induction in the sphere formation was observed (Fig. 5B). Also, the effect of STAT3 silencing on the resistin-induced expression of survival (BCL2 and BCL-xL) and stemness (Nanog and KLF4)-associated proteins was examined (Fig. 5C). Our immunoblot data showed that silencing of STAT3 efficiently blocks the resistin-induced expression of anti-apoptotic (BCL2 and BCL-xL) (Fig. 5C, upper panel), and stemness-associated transcription factors (Nanog and KLF4) (Fig. 5C, lower panel) in both the BC cell lines. Taken together, our data suggest that STAT3 mediates resistin-induced chemoresistance and stemness in the BC cells by regulating the expression of associated proteins.
4. Discussion
Elevated levels of resistin have been reported in several types of malignancies, including BC [13, 40, 41]. Further, the levels of resistin correlate with breast tumor size, stage and lymph node metastasis [25]. Expression of resistin also affects disease-free as well as overall survival rates in BC patients [25]. Studies also report a role of resistin in the neovascularization process [42]. Our earlier work demonstrated a significantly disparate overexpression of resistin in AA BC serum, as compared to CA patients, in addition to its role in growth and aggressiveness of BC cell lines [13]. Interestingly, treatment of BC patients with chemotherapeutic drugs has been shown to result in increased expression of resistin [28]. One of the known side-effects of anthracycline-based therapy, such as doxorubicin, is the associated cardiotoxicity. Interestingly, a role of resistin in doxorubicin-induced cardiotoxicity has been reported in a transgenic mouse model [43]. Doxorubicin treatment was shown to result in four-fold induction of resistin in mice. The study also evaluated resistin levels in BC patients undergoing anthracycline-containing chemotherapy, and found significantly elevated resistin levels in such patients [43]. These studies and our own findings, presented herein, establish resistin, as a key determinant of BC therapeutic response. While our study provides first evidence for a role of resistin in cancer chemoresistance, its role in insulin resistance is well characterized [44]. Incidentally, insulin resistance is also associated with high risk of human cancers [45], including BC [46].
Development of drug-resistance often compromises the clinical efficacy of chemotherapeutic drugs/regimens. A number of possible mechanisms have been suggested for both primary and acquired chemoresistance, and these include enhanced DNA repair, overexpression of drug efflux pump related genes and induction of survival pathways [47]. Cancer cells escape apoptosis by activating several signaling pathways and stemness-associated factors, resulting in reduced sensitivity to doxorubicin. Here, we report the effects of resistin on doxorubicin resistance, protection against doxorubicin-induced apoptosis and enhanced stemness potential of BC cells. Apoptosis is a tightly regulated and highly complex process of cell death that has a critical role in cell growth and tissue homeostasis [48]. Tumor cells have inherent ability to resist apoptosis, which has also been associated with chemoresistance [49–51]. In this study, we observed that resistin conferred doxorubicin resistance through suppression of apoptosis. Another interesting finding from our study was that we see a significantly greater effect of resistin in imparting chemoresistance in the MDA-MB-468 BC cells, which are of AA racial background as compared to MDA-MB-231 cells of CA origin. These findings are significant, and are supported by our earlier observation that BC cells of AA origin exhibit greater expression of CAP1 (receptor for resistin) [13]. Therefore, it appears that differential expression of CAP1 by tumor cells, along with disparate levels of resistin, may serve as determinant of racially disparate therapeutic outcome.
Chemoresistance in BC is also suggested to be due to stem cells phenotype acquired by the tumor cells [52–54]. Calcagno et al. reported that long-term continuous culture of MCF7 cells resulted in doxorubicin resistant MCF7/ADR cells that exhibited cancer stem cell-like population and contained a larger CD44+/CD24− population. Moreover, these cells were more invasive than MCF7 cells in vivo [55]. Furthermore, it has been suggested that BC cells with CD44high/CD24low/ALDHhigh phenotype exhibit greater self-renewal capacity and tumorigenicity, compared to cells that are CD44low/CD24high/ALDHlow [56–58]. Wang et al. characterized the CD44+CD24− stem cells derived from BC cells, which rapidly formed mammospheres and had potent tumorigenicity in vivo [59]. Therefore, therapeutic targeting of factors that are associated with BCSCs and therapy resistance would have profound clinical implications for better management of this disease. A direct induction of stemness associated markers (CD44high and CD24low) by resistin, as reported here, provides a rationale for targeting of resistin in an attempt to control BCSCs. It is known that the therapeutic resistance of cancer cells is largely attributed to self-renewing subpopulations of CSCs present in the bulk tumor, and enhanced sphere-forming potential is one of the important characteristics of CSCs. Data from our study demonstrate that resistin enhanced sphere forming capacity of BC cells.
In our earlier study, we have established that STAT3 is a critical mediator of resistin-induced growth and aggressiveness of BC cells. STAT3 is constitutively activated in numerous cancer types, including BC [60, 61]. Further, BC cells expressing activated STAT3 exhibit poorer therapeutic response to neo-adjuvant chemotherapy [61], and a role of STAT3 in the maintenance of BC stemness has also been well explored [39, 62]. Guha et al. demonstrated a critical role of STAT3 in apoptosis resistance and development and maintenance of CSC characteristics in breast cancer cells [63]. In other cancers, such as pancreatic cancer, STAT3 has been shown to be important in maintaining stem cell phenotypes [32]. In addition to maintaining the stemness phenotypes, activation of STAT3 is also linked to the conversion of non-CSCs to CSCs [64]. Our data from the present study clearly demonstrates STAT3 to be a crucial mediator of resistin induced chemoresistance and stemness potential (Fig. 5D).
In conclusion, our study establishes a role of resistin in doxorubicin resistance and BC stemness, an effect that is mediated through STAT3 activation. Thus, selective targeting of resistin could be of great significance in BC therapy.
Supplementary Material
Highlights.
Resistin confers doxorubicin-resistance in breast cancer cells
Effect of resistin on doxorubicin-resistance is due to suppression of apoptosis
Resistin promotes stemness in breast cancer cells
STAT3 mediates resistin-induced drug resistance and stemness in breast cancer cells
Acknowledgments
We would like to thank Mr. Steven McClellan, Manager, Flow cytometry core at the USA Mitchell Cancer Institute for his assistance with flow cytometry.
Funding
This work is supported by National Institutes of Health/National Cancer Institute [CA204801 (to SS) and CA185490 (to APS)] and USAMCI. SKS has an SBIR contract funding [HHSN261201600039C] from National Institutes of Health/National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
SS and APS are co-founders and serve on executive management team of Tatva Biosciences LLC, which is involved in the development of tools and models for cancer health disparity research. SKS serves as the Director of Cell Biology and Genetics at Tatva Biosciences LLC.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Deshmukh SK, Azim S, Ahmad A, Zubair H, Tyagi N, Srivastava SK, Bhardwaj A, Singh S, Rocconi RP, Singh AP. Biological basis of cancer health disparities: resources and challenges for research. Am J Cancer Res. 2017;7:1–12. [PMC free article] [PubMed] [Google Scholar]
- 3.DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI, Jemal A. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin. 2016;66:290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 4.Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol. 2012;23(Suppl 6):vi7–12. doi: 10.1093/annonc/mds187. [DOI] [PubMed] [Google Scholar]
- 5.Collignon J, Lousberg L, Schroeder H, Jerusalem G. Triple-negative breast cancer: treatment challenges and solutions. Breast Cancer (Dove Med Press) 2016;8:93–107. doi: 10.2147/BCTT.S69488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13:674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munzone E, Colleoni M. Metronomics in the neoadjuvant and adjuvant treatment of breast cancer. Cancer Lett. 2017 doi: 10.1016/j.canlet.2016.12.041. [DOI] [PubMed] [Google Scholar]
- 8.Aas T, Borresen AL, Geisler S, Smith-Sorensen B, Johnsen H, Varhaug JE, Akslen LA, Lonning PE. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996;2:811–814. doi: 10.1038/nm0796-811. [DOI] [PubMed] [Google Scholar]
- 9.Smith L, Watson MB, O’Kane SL, Drew PJ, Lind MJ, Cawkwell L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol Cancer Ther. 2006;5:2115–2120. doi: 10.1158/1535-7163.MCT-06-0190. [DOI] [PubMed] [Google Scholar]
- 10.Majidinia M, Yousefi B. Breast tumor stroma: A driving force in the development of resistance to therapies. Chem Biol Drug Des. 2017 doi: 10.1111/cbdd.12893. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Yang M, Li Y, Han B. The Role of MicroRNAs in the Chemoresistance of Breast Cancer. Drug Dev Res. 2015;76:368–374. doi: 10.1002/ddr.21275. [DOI] [PubMed] [Google Scholar]
- 12.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 13.Deshmukh SK, Srivastava SK, Bhardwaj A, Singh AP, Tyagi N, Marimuthu S, Dyess DL, Dal Zotto V, Carter JE, Singh S. Resistin and interleukin-6 exhibit racially-disparate expression in breast cancer patients, display molecular association and promote growth and aggressiveness of tumor cells through STAT3 activation. Oncotarget. 2015;6:11231–11241. doi: 10.18632/oncotarget.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart PA, Luks J, Roycik MD, Sang QX, Zhang J. Differentially expressed transcripts and dysregulated signaling pathways and networks in African American breast cancer. PLoS One. 2013;8:e82460. doi: 10.1371/journal.pone.0082460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin DN, Boersma BJ, Yi M, Reimers M, Howe TM, Yfantis HG, Tsai YC, Williams EH, Lee DH, Stephens RM, Weissman AM, Ambs S. Differences in the tumor microenvironment between African-American and European-American breast cancer patients. PLoS One. 2009;4:e4531. doi: 10.1371/journal.pone.0004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velaei K, Samadi N, Barazvan B, Soleimani Rad J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast. 2016;30:92–100. doi: 10.1016/j.breast.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25:2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aoudjit F, Vuori K. Integrin signaling in cancer cell survival and chemoresistance. Chemother Res Pract. 2012;2012:283181. doi: 10.1155/2012/283181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang ZM, Wu JF, Luo QC, Liu QF, Wu QW, Ye GD, She HQ, Li BA. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/beta-catenin pathway. Oncogene. 2016;35:4787–4797. doi: 10.1038/onc.2016.10. [DOI] [PubMed] [Google Scholar]
- 20.Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174:5789–5795. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- 21.Gong WJ, Zheng W, Xiao L, Tan LM, Song J, Li XP, Xiao D, Cui JJ, Li X, Zhou HH, Yin JY, Liu ZQ. Circulating resistin levels and obesity-related cancer risk: A meta-analysis. Oncotarget. 2016;7:57694–57704. doi: 10.18632/oncotarget.11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou WK, Xu YX, Yu T, Zhang L, Zhang WW, Fu CL, Sun Y, Wu Q, Chen L. Adipocytokines and breast cancer risk. Chin Med J (Engl) 2007;120:1592–1596. [PubMed] [Google Scholar]
- 23.Kang JH, Yu BY, Youn DS. Relationship of serum adiponectin and resistin levels with breast cancer risk. J Korean Med Sci. 2007;22:117–121. doi: 10.3346/jkms.2007.22.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun CA, Wu MH, Chu CH, Chou YC, Hsu GC, Yang T, Chou WY, Yu CP, Yu JC. Adipocytokine resistin and breast cancer risk. Breast Cancer Res Treat. 2010;123:869–876. doi: 10.1007/s10549-010-0792-4. [DOI] [PubMed] [Google Scholar]
- 25.Lee YC, Chen YJ, Wu CC, Lo S, Hou MF, Yuan SS. Resistin expression in breast cancer tissue as a marker of prognosis and hormone therapy stratification. Gynecol Oncol. 2012;125:742–750. doi: 10.1016/j.ygyno.2012.02.032. [DOI] [PubMed] [Google Scholar]
- 26.Vallega KA, Liu N, Myers JS, Yu K, Sang QX. Elevated Resistin Gene Expression in African American Estrogen and Progesterone Receptor Negative Breast Cancer. PLoS One. 2016;11:e0157741. doi: 10.1371/journal.pone.0157741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JO, Kim N, Lee HJ, Lee YW, Kim SJ, Park SH, Kim HS. Resistin, a fat-derived secretory factor, promotes metastasis of MDA-MB-231 human breast cancer cells through ERM activation. Sci Rep. 2016;6:18923. doi: 10.1038/srep18923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coskun T, Kosova F, Ari Z, Sakarya A, Kaya Y. Effect of oncological treatment on serum adipocytokine levels in patients with stage II–III breast cancer. Mol Clin Oncol. 2016;4:893–897. doi: 10.3892/mco.2016.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan MA, Srivastava SK, Bhardwaj A, Singh S, Arora S, Zubair H, Carter JE, Singh AP. Gemcitabine triggers angiogenesis-promoting molecular signals in pancreatic cancer cells: Therapeutic implications. Oncotarget. 2015;6:39140–39150. doi: 10.18632/oncotarget.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel GK, Khan MA, Bhardwaj A, Srivastava SK, Zubair H, Patton MC, Singh S, Khushman M, Singh AP. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br J Cancer. 2017 doi: 10.1038/bjc.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zubair H, Azim S, Srivastava SK, Ahmad A, Bhardwaj A, Khan MA, Patel GK, Arora S, Carter JE, Singh S, Singh AP. Glucose Metabolism Reprogrammed by Overexpression of IKKepsilon Promotes Pancreatic Tumor Growth. Cancer Res. 2016;76:7254–7264. doi: 10.1158/0008-5472.CAN-16-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyagi N, Marimuthu S, Bhardwaj A, Deshmukh SK, Srivastava SK, Singh AP, McClellan S, Carter JE, Singh S. p-21 activated kinase 4 (PAK4) maintains stem cell-like phenotypes in pancreatic cancer cells through activation of STAT3 signaling. Cancer Lett. 2016;370:260–267. doi: 10.1016/j.canlet.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makin G, Hickman JA. Apoptosis and cancer chemotherapy. Cell Tissue Res. 2000;301:143–152. doi: 10.1007/s004419900160. [DOI] [PubMed] [Google Scholar]
- 34.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 35.Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;18:1441–1449. doi: 10.1038/cdd.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiandalo MV, Kyprianou N. Caspase control: protagonists of cancer cell apoptosis. Exp Oncol. 2012;34:165–175. [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol Ther. 2016;160:145–158. doi: 10.1016/j.pharmthera.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA, Polyak K. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. The Journal of clinical investigation. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang G, Fan W, Luo B, Xu Z, Wang P, Tang S, Xu P, Yu M. Circulating Resistin Levels and Risk of Colorectal Cancer: A Meta-Analysis. Biomed Res Int. 2016;2016:7367485. doi: 10.1155/2016/7367485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Assiri AM, Kamel HF. Evaluation of diagnostic and predictive value of serum adipokines: Leptin, resistin and visfatin in postmenopausal breast cancer. Obes Res Clin Pract. 2016;10:442–453. doi: 10.1016/j.orcp.2015.08.017. [DOI] [PubMed] [Google Scholar]
- 42.Pang L, Zhang Y, Yu Y, Zhang S. Resistin promotes the expression of vascular endothelial growth factor in ovary carcinoma cells. Int J Mol Sci. 2013;14:9751–9766. doi: 10.3390/ijms14059751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartz DR, Briggs ER, Qatanani M, Sawaya H, Sebag IA, Picard MH, Scherrer-Crosbie M, Lazar MA. Human resistin in chemotherapy-induced heart failure in humanized male mice and in women treated for breast cancer. Endocrinology. 2013;154:4206–4214. doi: 10.1210/en.2013-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang X, Yang Z. Resistin’s, obesity and insulin resistance: the continuing disconnect between rodents and humans. J Endocrinol Invest. 2016;39:607–615. doi: 10.1007/s40618-015-0408-2. [DOI] [PubMed] [Google Scholar]
- 45.Elsayed EY, Mosalam NA, Mohamed NR. Resistin and Insulin Resistance: A Link Between Inflammation and Hepatocarcinogenesis. Asian Pac J Cancer Prev. 2015;16:7139–7142. doi: 10.7314/apjcp.2015.16.16.7139. [DOI] [PubMed] [Google Scholar]
- 46.Dumais V, Lumingu J, Bedard M, Paquet L, Verma S, Fontaine-Bisson B. Prevalence of Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes in Canadian Women at High Risk for Breast Cancer. Breast J. 2017 doi: 10.1111/tbj.12772. [DOI] [PubMed] [Google Scholar]
- 47.Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2:3. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simstein R, Burow M, Parker A, Weldon C, Beckman B. Apoptosis, chemoresistance, and breast cancer: insights from the MCF-7 cell model system. Exp Biol Med (Maywood) 2003;228:995–1003. doi: 10.1177/153537020322800903. [DOI] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 51.Mani J, Vallo S, Rakel S, Antonietti P, Gessler F, Blaheta R, Bartsch G, Michaelis M, Cinatl J, Haferkamp A, Kogel D. Chemoresistance is associated with increased cytoprotective autophagy and diminished apoptosis in bladder cancer cells treated with the BH3 mimetic (−)-Gossypol (AT-101) BMC Cancer. 2015;15:224. doi: 10.1186/s12885-015-1239-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity — Implications for chemoresistance. Cancer Lett. 2013;341:56–62. doi: 10.1016/j.canlet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 53.O’Brien CS, Farnie G, Howell SJ, Clarke RB. Breast cancer stem cells and their role in resistance to endocrine therapy. Horm Cancer. 2011;2:91–103. doi: 10.1007/s12672-011-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Brien CS, Howell SJ, Farnie G, Clarke RB. Resistance to endocrine therapy: are breast cancer stem cells the culprits? J Mammary Gland Biol Neoplasia. 2009;14:45–54. doi: 10.1007/s10911-009-9115-y. [DOI] [PubMed] [Google Scholar]
- 55.Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD, Gottesman MM, Varticovski L, Ambudkar SV. Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J Natl Cancer Inst. 2010;102:1637–1652. doi: 10.1093/jnci/djq361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mannello F. Understanding breast cancer stem cell heterogeneity: time to move on to a new research paradigm. BMC Med. 2013;11:169. doi: 10.1186/1741-7015-11-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ricardo S, Vieira AF, Gerhard R, Leitao D, Pinto R, Cameselle-Teijeiro JF, Milanezi F, Schmitt F, Paredes J. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64:937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 58.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Lu P, Zhang H, Luo M, Zhang X, Wei X, Gao J, Zhao Z, Liu C. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget. 2014;5:10803–10815. doi: 10.18632/oncotarget.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 61.Diaz N, Minton S, Cox C, Bowman T, Gritsko T, Garcia R, Eweis I, Wloch M, Livingston S, Seijo E, Cantor A, Lee JH, Beam CA, Sullivan D, Jove R, Muro-Cacho CA. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin Cancer Res. 2006;12:20–28. doi: 10.1158/1078-0432.CCR-04-1749. [DOI] [PubMed] [Google Scholar]
- 62.Wei W, Tweardy DJ, Zhang M, Zhang X, Landua J, Petrovic I, Bu W, Roarty K, Hilsenbeck SG, Rosen JM, Lewis MT. STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells. 2014;32:2571–2582. doi: 10.1002/stem.1752. [DOI] [PubMed] [Google Scholar]
- 63.Guha P, Bandyopadhyaya G, Polumuri SK, Chumsri S, Gade P, Kalvakolanu DV, Ahmed H. Nicotine promotes apoptosis resistance of breast cancer cells and enrichment of side population cells with cancer stem cell-like properties via a signaling cascade involving galectin-3, alpha9 nicotinic acetylcholine receptor and STAT3. Breast Cancer Res Treat. 2014;145:5–22. doi: 10.1007/s10549-014-2912-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, Lee YJ. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stemlike cells. Cell Signal. 2013;25:961–969. doi: 10.1016/j.cellsig.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.