

Abstract
Sandhoff disease is a neurodegenerative disease caused due to deficiency of hexosaminidase (HEX) A and B. A 1-year-old male child presented with regression of milestones, exaggerated startle response, decreased vision, and seizures from 6 months of age. The child had coarse facies without hepatosplenomegaly. Serum levels of β hexosaminidase total (A + B) were low. Genetic testing for Sandhoff disease revealed a homozygous missense variant on HEXB gene. The case is presented to highlight that the absence of hepatosplenomegaly should not restrain in suspecting Sandhoff disease.
KEYWORDS: Cherry red spot, hepatosplenomegaly, hexosaminidase, hexosaminidase B gene, Sandhoff disease
INTRODUCTION
Sandhoff disease is a rare but severe autosomal recessive lysosomal storage disorder. It is caused by a deficiency of both hexosaminidase (HEX) A and B, resulting in accumulation of glycosphingolipids and oligosaccharides in the brain. It has three clinical subtypes (infantile, juvenile, and adult forms) and represents around 7% of cases among all the lysosomal storage disorders.[1] The infantile form presents in the first 6–18 months of age with regression of milestones, developmental delay, startle response, hypotonia, cherry red spots, and convulsions.[2] We report this case as the infant presented with regression without hepatosplenomegaly and confirmed by gene testing.
CASE REPORT
A 1-year-old second born male child, born to a third-degree consanguineously married couple with uneventful perinatal history, was brought with regression of milestones and seizures. The onset of clinical symptoms began at the age of 6 months with gradual loss of the milestones. The child had achieved response to sounds by 2 months; head holding, social smile, and recognition of mother by 3 months of age. Initially, he lost social smile and recognition of mother followed by control of neck. He lost all the milestones by 8–9 months of age. The child developed exacerbated startle response since 8 months of age and multiple episodes of right-sided focal seizures from 11 months of age. There was a history of sibling death at the age of 18 months with similar complaints.
On examination, head circumference was 44 cm (between 3rd and 15th centile) with coarse facies. The tone was increased in all the limbs with power of 3/5 (Medical Research Council grade). Deep tendon reflexes were exaggerated with extensor plantar response. Fundus examination showed cherry-red spot in the macula, and there was no hepatosplenomegaly. Magnetic resonance imaging (MRI) shows hyperintensity of the basal ganglia (long white arrow) and hypointensity of the ventral thalami (open arrow) on T2-weighted sequences [Figure 1]. Based on above findings suspected to be a case of Sandhoff disease, the enzyme assay in leukocytes for β hexosaminidase total (A + B) revealed 61 nmhol/h/mg (normal, 905–2878). Gene testing was positive for homogenous missense substitution p. Cys534Tyr, c1601G>A, chr5:74016560G>A in exon 13 of HEXB gene.
Figure 1.
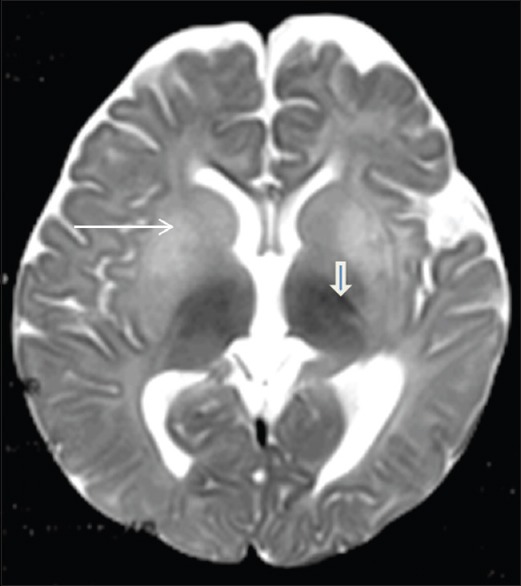
Axial T2-weighted imaging showing hyperintensity of the basal ganglia (long white arrow) and hypointensity of the ventral thalami (open arrow)
DISCUSSION
Our patient had presented with regression of milestones, exaggerated startle response, decreased vision, and seizures. The child had coarse facies, cherry red spot, and normocephaly without hepatosplenomegaly. In a study conducted among 18 GM2 gangliosidosis patients by Karimzadeh et al., seven patients had macrocephaly, three patients had microcephaly, and eight patients had a normal head circumference.[3]
A distinguishing feature in standoff's disease from other GM2 gangliosidosis is presence of hepatosplenomegaly and N-acetylglucosamine-containing oligosaccharides in urine.[4] However, hepatosplenomegaly was not present in our patient. Likewise, Karimzadeh et al. also noted, and only two patients of Sandhoff disease out of their nine cases had hepatosplenomegaly.[3] Similar observation was made by Ozkara et al. in their study of 18 cases affected by Sandhoff disease, and hepatosplenomegaly was not found in 11 out of 18 infantile Sandhoff disease patients, while the remaining seven had mild hepatosplenomegaly.[5] Closest differential diagnosis of Sandhoff disease is Tay–Sachs disease, which is characterized by the absence of coarse facies, hepatosplenomegaly, skeletal deformities, and signs of peripheral nerve involvement.[4]
Axial T2-weighted imaging on MRI in our patient revealed hyperintensity of the basal ganglia and hypointensity of the ventral thalami. Beker-Acay et al. also reported as bilateral thalamic hyperdensity on computed tomography and hypointensity on T2-weighted MRI images as the earliest diagnostic markers of Sandhoff disease.[6]
The gold standard method for diagnosis of GM2 gangliosidosis is the measurement of β-HEX activity in plasma, serum, and/or fibroblasts.[7] Molecular characterization of the HEXA and HEXB mutations can also be performed for confirmation by direct sequencing of the entire coding region and intron/exon boundaries using genomic DNA.[8] The same mutation was reported previously in a Japanese case which showed hepatosplenomegaly unlike our case.[8] Prenatal diagnosis advised for next pregnancy. Substrate reduction therapy is currently under investigation.[4] Sandhoff disease should be suspected in addition to Tay–Sachs disease if any child presenting with regression of milestones and cherry red spot without hepatosplenomegaly. Genetic testing should be considered for early diagnosis and genetic counseling.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Sheth J, Mistri M, Sheth F, Shah R, Bavdekar A, Godbole K, et al. Burden of lysosomal storage disorders in India: Experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014;12:51–63. doi: 10.1007/8904_2013_244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yun YM, Lee SN. A case refort of Sandhoff disease. Korean J Ophthalmol. 2005;19:68–72. doi: 10.3341/kjo.2005.19.1.68. [DOI] [PubMed] [Google Scholar]
- 3.Karimzadeh P, Jafari N, Nejad Biglari H, Jabbeh Dari S, Ahmad Abadi F, Alaee MR, et al. GM2-gangliosidosis (Sandhoff and Tay Sachs disease): Diagnosis and neuroimaging findings (an Iranian pediatric case series) Iran J Child Neurol. 2014;8:55–60. [PMC free article] [PubMed] [Google Scholar]
- 4.Pastores GM. Lysosomal storage diseases. In: Swaiman KF, editor. Swaiman's Pediatric Neurology. 5th ed. Philadelphia: Elsevier Saunders; 2012. pp. 403–51. [Google Scholar]
- 5.Ozkara HA, Topçu M, Renda Y. Sandhoff disease in the Turkish population. Brain Dev. 1997;19:469–72. doi: 10.1016/s0387-7604(97)00061-2. [DOI] [PubMed] [Google Scholar]
- 6.Beker-Acay M, Elmas M, Koken R, Unlu E, Bukulmez A. Infantile type Sandhoff disease with striking brain MRI findings and a novel mutation. Pol J Radiol. 2016;81:86–9. doi: 10.12659/PJR.895911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maegawa GH, Stockley T, Tropak M, Banwell B, Blaser S, Kok F, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics. 2006;118:e1550–62. doi: 10.1542/peds.2006-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuroki Y, Itoh K, Nadaoka Y, Tanaka T, Sakuraba H. A novel missense mutation (C522Y) is present in the beta-hexosaminidase beta-subunit gene of a Japanese patient with infantile Sandhoff disease. Biochem Biophys Res Commun. 1995;212:564–71. doi: 10.1006/bbrc.1995.2007. [DOI] [PubMed] [Google Scholar]