

Abstract
Ubiquitylation controls protein function and degradation. Therefore, ubiquitin ligases need to be tightly controlled. We discovered an evolutionarily conserved allosteric restraint mechanism for Nedd4 ligases and demonstrated its function with diverse substrates: the yeast soluble proteins Rpn10 and Rvs167, and the human receptor tyrosine kinase FGFR1 and cardiac IKS potassium channel. We found that a potential trimerization interface is structurally blocked by the HECT domain α1‐helix, which further undergoes ubiquitylation on a conserved lysine residue. Genetic, bioinformatics, biochemical and biophysical data show that attraction between this α1‐conjugated ubiquitin and the HECT ubiquitin‐binding patch pulls the α1‐helix out of the interface, thereby promoting trimerization. Strikingly, trimerization renders the ligase inactive. Arginine substitution of the ubiquitylated lysine impairs this inactivation mechanism and results in unrestrained FGFR1 ubiquitylation in cells. Similarly, electrophysiological data and TIRF microscopy show that NEDD4 unrestrained mutant constitutively downregulates the IKS channel, thus confirming the functional importance of E3‐ligase autoinhibition.
Keywords: inactivation, Nedd4, oligomerization, Rsp5, ubiquitylation
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics
Introduction
Nedd4 E3 ubiquitin ligases play a pivotal role in cell growth and homoeostasis as they tag numerous proteins for functional or spatial modulation or destruction. Nine mammalian members of the Nedd4 family regulate a major part of cell signalling, membrane excitability and synaptic transmission by ubiquitylation‐mediated endocytosis and/or degradation of receptor tyrosine kinases (RTKs; Zeng et al, 2009; Persaud et al, 2011), Smads (Zhu et al, 1999), ion channels (Kamynina et al, 2001), post‐synaptic receptors (Lin et al, 2011), endocytic proteins (Boase & Kumar, 2015) and others. Consequently, mechanisms that precisely control Nedd4 activity in space, time and amplitude are essential. Accordingly, various malignancies correlate with abnormal expression of Nedd4 family members (Chen & Matesic, 2007; Zou et al, 2015). Moreover, spurious targeting or dysregulation of Nedd4 substrates has been linked to hypertension disorders (Staub et al, 1996), Parkinson's disease (Tofaris et al, 2011), epilepsy (Wu et al, 2015) and other pathologies.
Nedd4 family members typically contain a C2 membrane binding domain followed by several WW substrate‐interacting modules and a HECT (homologous to E6‐AP carboxyl terminus) catalytic domain (Hein et al, 1995; Harvey & Kumar, 1999; Fig 1A). Intra‐molecular interactions of the C2 or WW modules with the HECT domain of some Nedd4s inhibit the catalytic activity of these enzymes (Wiesner et al, 2007; Wang et al, 2010; Zhang et al, 2016). Over the years, several studies have identified numerous post‐translational modifications such as ubiquitylation and/or phosphorylation that positively or negatively regulate the activity of Nedd4 family members (Gallagher et al, 2006; Liu et al, 2014; Li et al, 2015a). For example, it was recently demonstrated how receptor tyrosine kinases (RTKs) open the self‐inhibited closed conformation of Nedd4 by c‐Src‐mediated phosphorylation of critical residues within the C2:HECT interface (Persaud et al, 2014).
Figure 1. Removal of α1 alters the oligomeric state of Rsp5.
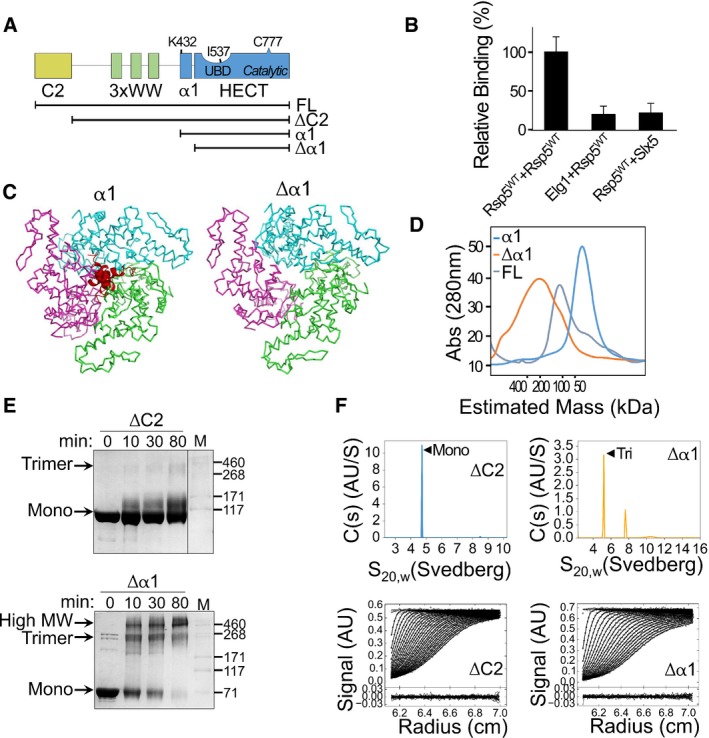
- Schematic representation of the Rsp5 domain architecture. Black strips below represent truncated proteins used in this study.
- Normalized, mean β‐galactosidase activity in a yeast two‐hybrid assay reporting association of Rsp5 with itself or with the negative controls, ELG1 or SLX5. Mean values and bars of standard deviation from triplicates are shown.
- Representation of a trimeric Rsp5 model in the presence (left) or absence (right) of α1 (red). The structure of Rsp5 (3OLM) was superimposed onto the E6AP trimeric structure (1C4Z), with its three HECT protomers depicted in green, pink and cyan. A clash of the α1 helices is apparent at the trimerization interface.
- Size‐exclusion chromatography of full‐length Rsp5 or of its HECT domain, with (α1) or without (Δα1) α1. Proteins were loaded on a Superdex 200 16/60 column, and elution was monitored by measuring absorption at 280 nm (A280).
- Time‐dependent cross‐linking of ΔC2 or Δα1 Rsp5 with 0.5 mM disuccinimidyl suberate (DSS). Samples were resolved by SDS–PAGE followed by Coomassie blue staining.
- Sedimentation velocity data, fits and residuals from analytical ultracentrifugation of ΔC2 or Δα1 Rsp5. Upper panels show continuous distributions analyses c(s) of the sedimenting species for ΔC2 (blue) or Δα1 (orange). Observed/calculated mass (oligomeric order), and distribution of species is presented in Table EV1.
Rsp5 is the only Nedd4 family member in yeast. Unlike some of its vertebrate orthologues, Rsp5 does not appear to be controlled by intra‐molecular inhibition (Wiesner et al, 2007). Interestingly, the semi‐dominant phenotype of the rsp5‐2 mutant cannot be rescued by the wild‐type protein, suggesting that Rsp5 undergoes oligomerization in vivo. Yeast two‐hybrid and pull‐down assays corroborated this conjecture (Dunn & Hicke, 2001). However, the purpose and the details of Rsp5 oligomerization remained unknown.
Here, we present a novel mechanism of Rsp5 regulation through auto‐ubiquitylation‐dependent oligomerization. We demonstrate, in vitro and in cells, molecular details of the conformational changes induced by ubiquitylation to promote oligomerization and consequent ligase inactivation. Interestingly, we also show that an Rsp5 point mutant (Tardiff et al, 2013) undergoes ubiquitylation‐independent oligomerization and inactivation.
The biological significance of this new regulatory mechanism was demonstrated in mammalian cells by following several cellular processes that are regulated by Nedd4. Specifically, we generated point mutations that impair the oligomerization mechanism of Nedd4, resulting in unrestrained activity and a subsequent increase in FGFR1 ubiquitylation. We also found that ubiquitylation‐dependent oligomerization and inactivation of human Nedd4 regulates the function of the cardiac IKS channel, thus affecting potassium current density. In conclusion, we present a novel molecular mechanism regulating Nedd4 activity with the broad implications for cell physiology.
Results
Rsp5 oligomerizes in yeast cells
It has been shown that ligand binding to RTKs signals for activation of their cognate Nedd4 ligases (Persaud et al, 2014). However, as yeast lack RTKs, we explored alternative mechanism that may regulate their only Nedd4 like protein, Rsp5. It was previously demonstrated that Rsp5 undergoes self‐assembly (Dunn & Hicke, 2001). Since oligomerization is a common mechanism for regulating protein activity, we first wanted to reproduce this observation. Specifically, we set up a yeast two‐hybrid (Y2H) binding assay to follow the self‐association of Rsp5 by monitoring the activity of the downstream reporter, β‐Galactosidase. We found that cells expressing both Gal4p‐DNA‐binding domain and Gal4p‐activating domain fused to Rsp5 showed a significantly higher β‐galactosidase activity compared to cells in which one of the Rsp5 copies was substituted with another unrelated ligase, such as Slx5 or with Elg1 (Fig 1B).
The N‐terminal helix of Rsp5‐HECT domain blocks oligomerization
We sought to study whether Rsp5 self‐association in Y2H is a result of direct interaction or whether it is mediated by additional proteins. Intriguingly, early crystallographic data supported by later biophysical experiments revealed that the HECT domain of the Rsp5 relative, E6AP, forms trimers (Huang et al, 1999; Ronchi et al, 2014). However, all subsequent HECT structures, including that of Rsp5, were crystallized as monomers. Structural comparison reveals that all these monomeric E3s were crystallized in the presence of the N‐terminal helix (α1) of the HECT domain, which was omitted from the crystallized E6AP protein. To obtain a structural insight into the manner by which the α1 helix might affect complex formation, we superimposed the Rsp5 structure with or without α1 (red) onto the trimeric structure of E6AP (Fig 1C; close‐up in Appendix Fig S1).
The superimposed structures suggest two major hypotheses: (i) The Rsp5‐α1 helices (in red) inhibit self‐assembly by clashing at the oligomerization interface. (ii) Rsp5 may form trimers by adopting similar interfaces to E6AP. Energy minimization of the trimeric model showed nice fitting of the protomers with conserved residues at the interface (Appendix Fig S2). To test these hypotheses we generated several constructs, including full‐length Rsp5, and the HECT domain containing (α1) or lacking (Δα1) the α1 helix (Fig 1A), and evaluated their oligomeric state by several techniques. In a size‐exclusion chromatography (SEC) experiment, we found that the retention volume of full‐length Rsp5 corresponds to that of a monomer (Fig 1D). Interestingly, while α1‐containing HECT domain eluted as a monomer, Δα1 eluted in a volume corresponding to an oligomeric size, thus confirming that α1 removal is key to oligomerization. To assess the oligomeric order of Δα1, we performed a time‐dependent cross‐linking assay with His6‐MBP fused Δα1 or with the α1‐containing ΔC2 proteins in the presence of a mild cross‐linker, disuccinimidyl suberate (DSS). In agreement with our SEC results, the nearly full‐length control protein (ΔC2) remained predominantly monomeric, while Δα1 readily formed trimers and higher oligomers (Fig 1E). The minute amounts of ΔC2 trimers seen in the gel hint on the protein propensity to oligomerize.
To identify the oligomeric order with accuracy and quantify the distribution of the various Rsp5 species in solution, we performed analytical ultra‐centrifugation (AUC) sedimentation velocity (SV) experiments with ΔC2 or Δα1 following removal of the His6‐MBP tag. Sedimentation coefficient and molar mass of the sedimenting species were extracted by analysing the data in terms of continuous distribution analyses c(s), following conversion to c(M). Since molar mass calculation accounts for the fitted average frictional ratio (f/f 0), the obtained value is reliable only for the major peak of each experiment, as the minor peaks are underrepresented in the fit of f/f 0. Analysis of ΔC2 revealed that 81% of species sedimented in a monomeric peak (n = 0.97) with a sedimentation coefficient (S20,w) of 4.72S, and that 1.9% of the species were found in a peak with an S20,w of 8.48S (Fig 1F, top left with residuals presented below, Table EV1). Analysis of Δα1 showed distribution in two main population including 49.5% of species with an S20,w of 5.182S and the size of a trimer (n = 3.09), and 22.8% of the species with an S20,w of 7.62S3 (Fig 1F, top right, and Table EV1). Taken together, these results suggest that removal of α1 exposes oligomerization interfaces and promotes trimerization.
Interestingly, structural alignment showed a large number of conserved residues at the oligomerization interfaces (Appendix Fig S2). Moreover, the catalytic cysteines and the E2 binding sites are located outside of the trimeric structure (Appendix Fig S3). Together, these observations support the hypothesis that α1 blocks a natural trimerization interface.
Ubiquitylation on K432 induces Rsp5 oligomerization and inactivation
The contradiction between Rsp5 self‐assembly in cells (Dunn & Hicke, 2001; Fig 1B), and our in vitro results showing that α1 impedes oligomerization (Fig 1D–F), suggests that α1 must be displaced in a cellular context. As many E3 ligases are regulated by post‐translational modifications, we speculated that a post‐translational event might dislocate α1.
Proteomic studies in yeast revealed ubiquitylation of Rsp5 on K432 (Beltrao et al, 2012), a residue located at the N‐terminal tip of α1. To test whether Rsp5 ubiquitylates itself directly on K432, we co‐expressed MBP‐Rsp5232–809 along with E1, E2 and Ub in Escherichia coli (Keren‐Kaplan et al, 2012), purified ubiquitylated Rsp5 and mapped the modification sites by mass spectrometry (MS). Since E. coli K‐12 does not possess any ubiquitylation enzymes, the bacterial system provides a facile assay for testing E3 self‐ubiquitylation. Indeed, our MS data revealed Rsp5 self‐ubiquitylation on K432 along with additional ubiquitylation sites that concur with in vivo studies (Fig 2A).
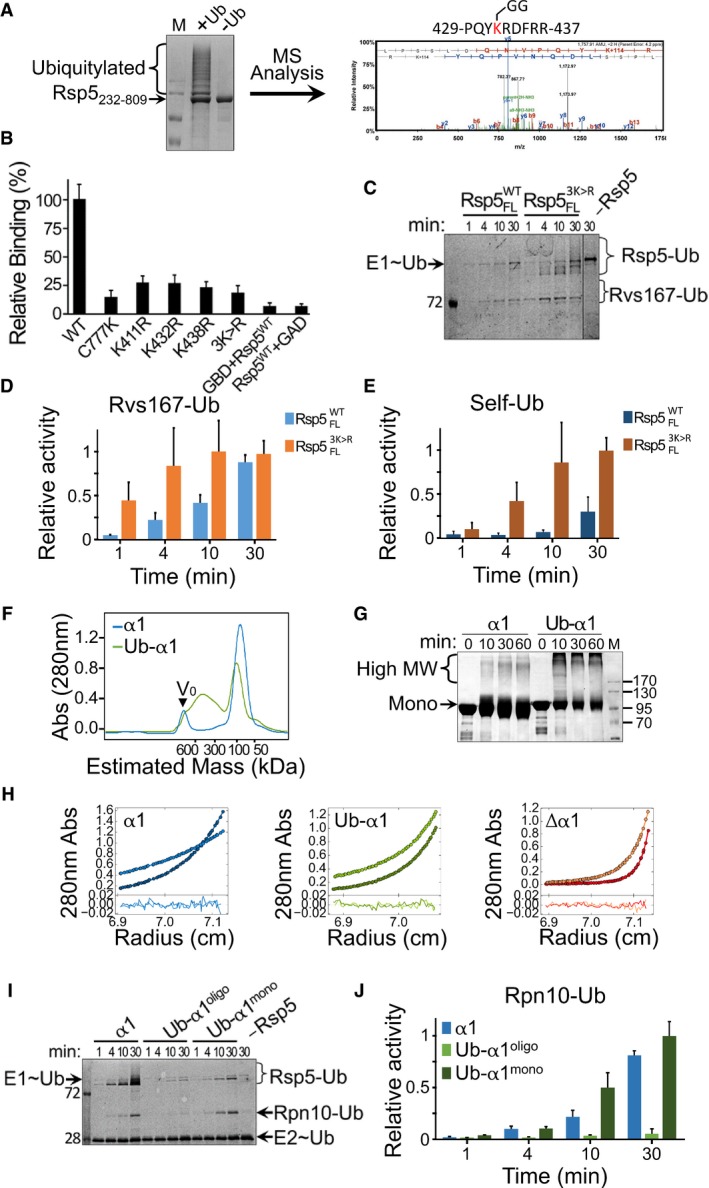
Figure 2. Ubiquitylation on α1 induces Rsp5 oligomerization and inactivation.
-
ARsp5232–809 was expressed in bacteria along with ubiquitin, E1 (UBA1) and E2 (Ubc5). The left panel shows Coomassie blue‐stained SDS–PAGE of Rsp5 purified from bacterial lysates. Identification of ubiquitylation sites was performed by mass spectrometry analysis following in‐gel trypsin digestion. A representative tandem mass spectrum of a peptide containing lysine 432 is shown (right panel).
-
BNormalized, mean β‐galactosidase activity reporting self‐association of wild‐type, triple or single K411R/K432R/K438R mutants of Rsp5. Mean values and standard deviation bars from triplicates are shown.
-
C–E(C) Representative gel of time‐dependent in vitro ubiquitylation of MBP‐Rvs167332–482 by Rsp5WT or Rsp53K>R in the presence of fluorescein‐labelled ubiquitin. Imaging of fluorescent ubiquitin conjugates was carried out with a Typhoon laser scanner at 488 nm. Gel quantification of (D) ubiquitylated Rvs167 or (E) self‐ubiquitylated Rsp5. Mean values and standard deviation from three experiment replicates are shown.
-
FSEC of apo HECT (α1) or Ub‐fused HECT (Ub‐α1) proteins tagged with His6‐MBP. The proteins were loaded on a Superdex 200 16/60 column, and elution was monitored by A280 detection.
-
GTime‐dependent cross‐linking of α1 or Ub‐α1 HECT domain in the presence of 0.5 mM DSS. Samples were resolved by SDS–PAGE followed by Coomassie blue staining.
-
HSedimentation equilibrium (SE) data, fit and residuals of α1, Δα1 or Ub‐α1 HECT domain. A280 data were collected at 8,000 rpm (lighter colours) and at 12,000 rpm (darker colours). The best fit for α1 HECT was obtained with a model of a single ideal solute with the size with a monomer. Alternatively, the best fits for Δα1 and Ub‐α1 HECT were obtained with a model of multiple ideal solutes with the size of monomers, trimers and hexamers (see also Table EV1).
-
ITime‐dependent in vitro ubiquitylation of Rpn10 by apo (α1), oligomeric or monomeric Ub‐fused HECT (Ub‐α1mono and Ub‐α1oligo, respectively), in the presence of fluorescein‐labelled ubiquitin. Reaction set‐up and detection were performed as described in Fig 2C.
-
JHistogram plot showing mean values and standard deviation bars of triplicate measurements.
To test in a cellular context whether Rsp5 self‐assembly depends on self‐ubiquitylation on its α1 helix, we performed mutational analysis using the yeast two‐hybrid assay presented in Fig 1B. Specifically, we tested Rsp5 self‐assembly following mutations that either abolish its catalytic activity or that substitute target lysine residues in and around α1. In line with our hypothesis, self‐assembly of the catalytically inactive Rsp5C777K is impaired (Fig 2B). Moreover, a catalytically active Rsp5 where K432, K438 and K411 were substituted by arginine residues (Rsp53K>R) significantly reduced Rsp5 self‐assembly. Interestingly, mutating each lysine individually reduced self‐association to almost similar extent as the triple mutant. These results confirm that self‐assembly of Rsp5 relies upon self‐ubiquitylation on α1 in the cell.
To investigate the functional implications of ubiquitylation‐dependent oligomerization, we set up an in vitro ubiquitylation assay using purified proteins of the ubiquitylation cascade, including a fluorescein‐labelled version of ubiquitin. Specifically, we measured time‐dependent ubiquitylation of the PPXY‐containing substrate‐Rvs167, by an untagged full‐length wild‐type (Rsp5WT) or triple lysine mutant (Rsp53K>R) ligase. Strikingly, while the mutant ligase presented immediate substrate ubiquitylation, the wild‐type protein showed reduced and delayed activity (Fig 2C and D). Interestingly, enhanced ubiquitylation was even greater when Rsp5 self‐ubiquitylation was analysed (Fig 2C and E).
To further characterize the molecular details of the mechanism underlying Rsp5 oligomerization and inactivation by self‐ubiquitylation, it is necessary to produce and purify a homogenous population of the protein modified on α1, for downstream in vitro biophysical/biochemical assays. This could prove challenging in the light of our MS data. Likewise, directing ubiquitylation to K432, K438 or K411 by mutating all the other target lysine residues could compromise the natural fold of the HECT domain. To overcome this hurdle, we employed a well‐established approach used in studies of endocytosed proteins (Prag et al, 2003; Dunn et al, 2004; Léon et al, 2008; Lee et al, 2009; Li et al, 2015b), namely mimicking ubiquitylation by direct fusion of ubiquitin to the target site. Here, we fused the C‐terminus of Ub to the N‐terminus of α1, thus mimicking ubiquitylation on K432 (proximity of the mimicry site to the target lysine is evident in Fig 3). The oligomerization states of apo HECT (α1) or Ub‐HECT (Ub‐α1) proteins fused to His6‐MBP were tested by several methods. Analysis by SEC revealed that while α1‐containing HECT exhibited a uniform population of monomeric size, the Ub‐α1 HECT distributed between a monomeric and an oligomeric peak (Fig 2F). Time‐dependent cross‐linking with DSS corroborated this finding (Fig 2G). For a more accurate insight into the order of oligomerization, we performed multispeed AUC sedimentation equilibrium (SE) experiments with each protein (Fig 2H and Table EV1). Sedimenting species analysis of apo HECT (α1) was consistent with a single ideal solute of monomeric size (90,662 Da compared to calculated 90,264 Da). However, the average size obtained for Δα1‐HECT and Ub‐HECT (Ub‐α1) was larger than their calculated monomeric size, that is 394,824 Da (versus calculated 86,244 Da) and 175,207 Da (versus calculated 97,913 Da), respectively. To identify the oligomerization order of Δα1, we tested various self‐association models by substituting the molar masses with full integers of the monomeric size in a model of multiple sedimenting species. Analysis of Δα1 in terms of mono‐tri‐hexa self‐association retrieved the best result with an A280 ratio of 0.062: 0.298: 0.640 and a local RMSD of 0.004832. Alternative models, namely mono‐di‐tetra and mono‐tri‐nona, resulted in poor fits with high local RMSD values of 0.0285 and 0.0098, respectively (Fig EV1). Consistent with this, the best fit for Ub‐HECT was obtained in a model of mono‐tri‐hexa with an A280 ratio of 0.0525: 0.27: 0.205. Altogether, it appears that ubiquitylation on K432 promotes self‐assembly in the same oligomeric order as Δα1‐HECT.
Figure 3. Structural model of ubiquitylation‐dependent oligomerization.
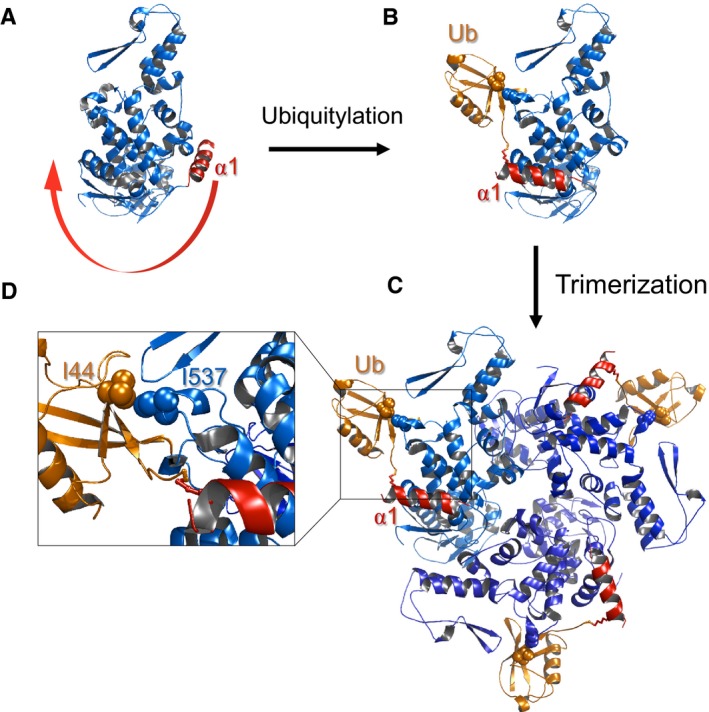
- In the apo form, α1 (red) blocks the oligomerization interface.
- Upon ubiquitylation, the conjugated ubiquitin binds to the HECT‐UBD, and by thus pulls α1 out of the interface and enables oligomerization.
- Model of trimeric Ub‐HECT. Three HECT molecules are depicted in various shades of blue, and ubiquitin moieties are represented in orange.
- Enlarged view of the HECT:Ub interface including key residues chosen for experimental assessment of the model.
Figure EV1. Comparing alternative models for Δα1 self‐association in SE analysis.
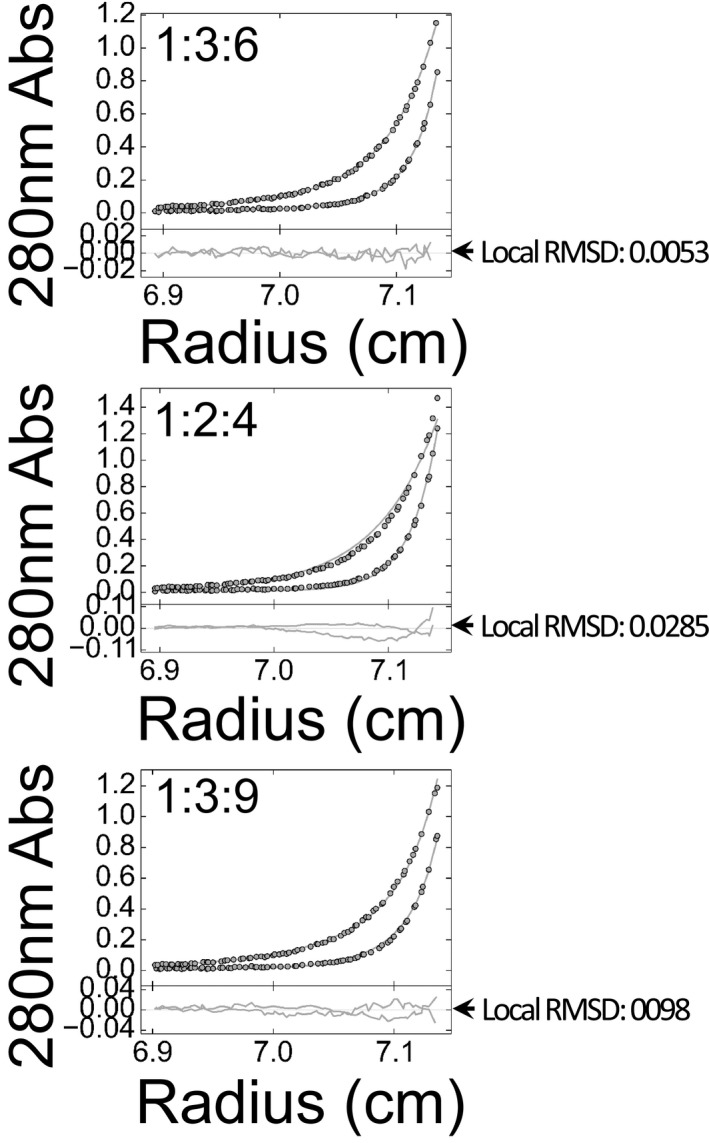
Interpretation of SE data from analytical ultracentrifugation of Δα1 in terms of self‐assembly with orders of 1:3:6, 1:2:4 or 1:3:9. A280 was collected at 8,000 and 12,000 rpm. Data points, best fits, residuals and local RMSD values of each analysis are shown.
To make further use of the Ub‐HECT chimeras for the investigation of ubiquitylation‐dependent oligomerization, we tested whether their substrate ubiquitylation activity corresponds to that of authentically ubiquitylated Rsp5 presented in Fig 2C and E. Rsp5 employs several substrate recognition mechanisms, including interactions between its WW domains and PPXY motif‐containing substrates (as shown in Fig 2C and D), or interaction between a conjugated ubiquitin on the ligase and a substrate containing a Ub‐interacting motif (UIM; Harvey & Kumar, 1999; Springael et al, 1999; Polo et al, 2002; Woelk et al, 2006). As the recombinant proteins lack the WW domains, we chose the UIM‐containing proteasome subunit Rpn10, a well‐known substrate of Rsp5 (Lu et al, 2008; Isasa et al, 2010; Udeshi et al, 2012). We kinetically monitored ubiquitylation of Rpn10 by apo HECT (α1), oligomeric Ub‐HECT (Ub‐α1oligo) or monomeric Ub‐HECT (Ub‐α1mono) isolated by SEC (see chromatogram in Fig 2F). In agreement with our results from the assay with the full‐length proteins (Fig 2C and D), oligomeric Ub‐HECT presents a significantly low rate of Rpn10 ubiquitylation as compared to apo HECT (Fig 2I and J). Strikingly, the rate of Rpn10 ubiquitylation by monomeric Ub‐HECT exceeds the rate with apo HECT, implying that oligomerization rather than ubiquitylation inactivates the ligase. As previously observed in Fig 2C and E, ubiquitylation‐dependent inactivation is also evident in the view of Rsp5 self‐ubiquitylation (Fig 2I). Together, these results indicate that oligomerization derived from α1 ubiquitylation inactivates the ligase.
Structural model for ubiquitylation‐dependent Rsp5 oligomerization
Fusing of MBP (alone) instead of MBP‐Ub directly onto α1 did not promote Rsp5 self‐assembly (Fig 2F–H). This suggests that the effect of ubiquitin is caused by its unique characteristic rather than by simple conjugation of any globular protein onto α1. Previous studies revealed the existence of a conserved ubiquitin‐binding domain (UBD) within the Rsp5 HECT domain (French et al, 2009; Kim et al, 2011; Maspero et al, 2011). We hypothesized that the attraction between the α1‐ubiquitin conjugate and the HECT‐UBD pulls α1 out of the oligomerization interface, and thereby promotes oligomerization. To examine whether distances and orientations support this hypothesis, we generated a structure‐based molecular model of trimeric Ub‐Rsp5. Our model was constructed by superimposing the structure of the Rsp5‐HECT:Ub non‐covalent complex onto the trimeric structure of E6AP (Huang et al, 1999; Kim et al, 2011). In the apo form, α1 (red) blocks the oligomerization interface of the HECT domain (Fig 3A). Upon ubiquitylation on K432, α1 is displaced from the oligomerization interface with the energy that derives from the UBD:Ub interaction (Fig 3B), and thus facilitates Rsp5 HECT domain trimerization (Fig 3C). Insight into the HECT‐UBD:Ub interface shows key residues selected for downstream experimental validation of the model, namely ubiquitin I44 and Rsp5 I537 (Fig 3D). This structural model shows that the length of α1 and the following loop perfectly match the distance needed to juxtapose the conjugated ubiquitin with the UBD.
The HECT‐UBD displace Ub‐α1 from the oligomerization interface
To test whether the UBD:Ub interaction provides the driving force for α1 displacement and consequent oligomerization, we mutated key residues at the UBD:Ub interface, namely Ub I44E and HECT I537D, which were previously shown to disrupt the HECT‐UBD:Ub interaction (French et al, 2009; Kim et al, 2011). SEC analysis of the His6‐MBP fused proteins revealed that the wild‐type Ub‐HECT (Ub‐α1) distributed into monomeric and oligomeric peaks. However, both UbI44E‐HECT (UbI44E‐α1) and Ub‐HECTI537D (Ub‐α1HECT I537D) mutants eluted as monomers only (Fig 4A). In agreement with this, cross‐linking experiments revealed that the strong shift of wild‐type Ub‐HECT towards high MW species was significantly reduced in UbI44E‐HECT and almost abolished in Ub‐HECTI537D (Fig 4B). SE experiments with both UbI44E‐HECT and Ub‐HECTI537D revealed average molar masses of nearly monomeric size, that is 106,239 Da and 105,208 Da, respectively (versus calculated MW of 97,927 and 97,913 Da, respectively). Analysis of self‐association exhibited mono‐tri‐hexa A280 ratios of 0.95: 0.05: 0 and 0.94: 0.06: 0 for UbI44E‐HECT and Ub‐HECTI537D, respectively (Fig 4C). To assess the exchange between monomers and oligomers in the oligomeric peak of Ub‐HECT obtained in SEC (Figs 2F and 4A), we measured self‐association of the wild‐type or I44E Ub‐HECT proteins by microscale thermophoresis (MST). One of the principles of this technique relies on the exchange between unlabelled and fluorescently labelled proteins, a tool that facilitates assessment of homomeric complexes. Here, we titrated labelled Rsp5 with an increasing concentration of identical unlabelled protein and measured the exchange. In line with our previous results, while the wild‐type Ub‐HECT showed dynamic exchange with an apparent K d of 123 ± 8 nM in a 1:1 model (currently the only applicable model for this new technique), the I44E mutation completely abolished self‐association (Fig 4D).
Figure 4. Oligomerization depends on binding of α1‐conjugated Ub to the HECT‐UBD. The following experiments were conducted with His6‐MBP‐tagged proteins.
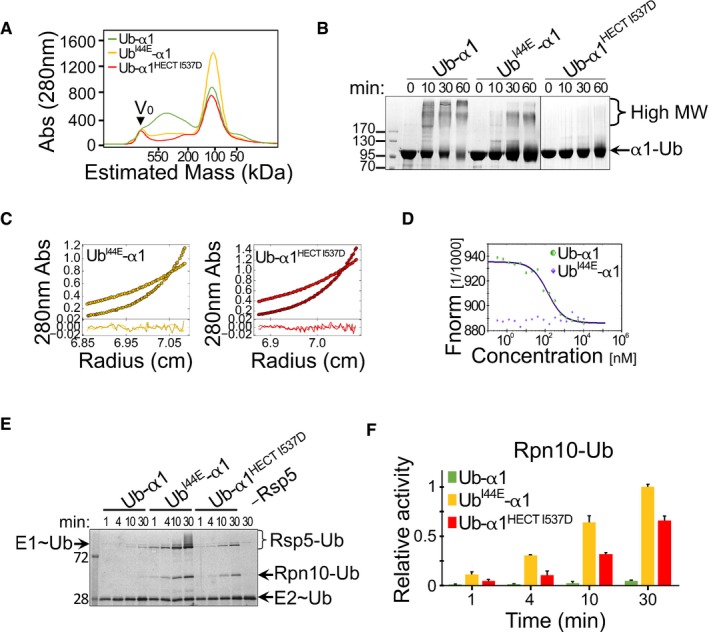
-
ASize‐exclusion chromatography of wild‐type and mutant Ub‐HECT proteins. Ub‐α1, Ub‐α1HECT‐I537D or UbI44E‐α1 HECT proteins were loaded on a Superdex 200 16/60 column, and the elution profile was monitored by A280 detection.
-
BTime‐dependent cross‐linking of Ub‐α1, UbI44E‐α1 or Ub‐α1HECT‐I537D Ub‐HECT proteins in the presence of 0.5 mM DSS. Samples were resolved by SDS–PAGE followed by Coomassie blue staining.
-
CSedimentation equilibrium (SE) data, fits and residuals of UbI44E‐α1 and Ub‐α1HECT‐I537D mutants. A280 was collected at 8,000 rpm (lighter colours) and at 12,000 rpm (darker colours). The best fits of both mutants were obtained in a model of non‐interacting ideal solutes with mostly monomeric size and < 5% trimers (see also Table EV1).
-
DMicroscale thermophoresis (MST) measurements of Ub‐α1 (green) or UbI44E‐α1 (blue). Concentration of red fluorescent NT‐647‐NHS‐labelled Rsp5 was kept constant and titrated with 0.2–1,000 nM of identical unlabelled protein. The y‐axis shows the fluorescence change ratio × 1,000.
-
E, F(E) Representative gel and (F) quantification of in vitro ubiquitylation of Rpn10 by α1, UbI44E‐α1 or Ub‐α1HECT I537D Rsp5, in the presence of fluorescein‐labelled ubiquitin. Mean values and standard deviation bars of three replicates are shown. Reaction set‐up and detection were performed as described in Fig 2C.
To test whether the disruption of the Ub:HECT‐UBD interface would also restore ligase activity, we tested Rpn10 ubiquitylation, by using the in vitro system presented in Fig 2. As hypothesized, Ub I44E or I537D mutations restored the catalytic activity of Ub‐HECT (Fig 4E and F). Interestingly, with regard to Rsp5 self‐ubiquitylation, I537D restored mono‐ but not poly‐ubiquitylation activity. This finding agrees with previous studies on the role of the HECT‐UBD in poly‐ubiquitin chain elongation (French et al, 2009; Kim et al, 2011). Collectively, the biochemical and biophysical data support our structural model, showing that the non‐covalent interaction between the ubiquitin that is conjugated to α1 and the ubiquitin‐binding patch within the HECT domain promotes Rsp5 oligomerization and consequent inactivation.
G747E mutation induces ubiquitylation‐independent Rsp5 oligomerization
To further validate our model in which oligomerization per se restrains the ligase activity, we sought conditions where oligomerization occurs in an ubiquitylation‐independent manner. Lindquist and co‐workers discovered that N‐aryl‐benzimidazole2 (NAB2) as a potential drug for Parkinson's disease (PD; Tardiff et al, 2013). Using yeast genetics, they found that Rsp5 is a central node of a gene network affected by NAB2. Moreover, they found that the rsp5 G747E mutant prevented the protective effect of NAB2 and presented with a defect in membrane protein trafficking. Examining the Rsp5 structure (3OLM), we found that G747 induces a turn‐like structure, stabilized by a hydrogen bond between G747 and V745 (Fig 5A). Interestingly, residues V745 and N746 of this turn‐like structure hold α1 in a closed conformation by forming hydrogen bonds with the K432 and R436 residues, located on α1. We hypothesized that the G747E mutation would destabilize the turn‐like structure and consequently disrupt its interactions with α1. Furthermore, we speculated that such a disruption might affect oligomerization in an ubiquitylation‐independent manner. To test this hypothesis, we loaded purified WT or G747E proteins of Rsp5232–809 onto a SEC column. Indeed, whereas the WT protein construct eluted as a monomer, the mutant eluted as an oligomer (Fig 5B). Furthermore, migration in a native polyacrylamide gel revealed higher dimerization and trimerization by the mutant protein (Fig 5C). In agreement with these data, AUC‐SE experiments revealed that while the WT protein behaved as an ideal solute with the size of a monomer, the G747E mutant exhibited mixed populations of monomers, trimers, and hexamers (Fig 5D and Table EV1).
Figure 5. Ubiquitylation‐independent oligomerization of the Rsp5 G747E mutant.
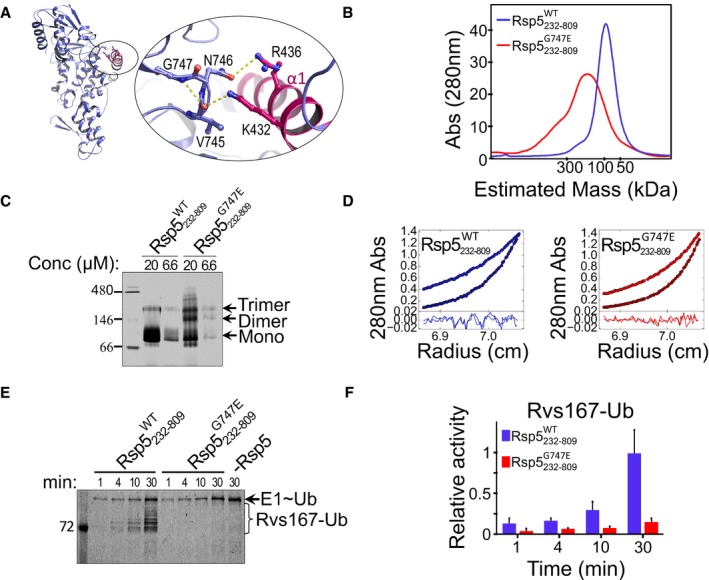
-
AModel showing stabilizing hydrogen bonds within the G747 turn‐like structure, as well as hydrogen bonds between the loop (purple) and the α1 (pink) residues in the Rsp5‐HECT structure (3OLM).
-
BSize‐exclusion chromatography profile of Rsp5232–809 WT or Rsp5232–809 G747E, run on a Superdex 200 16/60 column.
-
CMigration of 6.6 μM and 20 μM of Rsp5232–809 WT or Rsp5232–809 G747E in a native polyacrylamide gel.
-
DAUC‐SE analysis in terms of non‐interacting ideal solutes for Rsp5232‐809 WT or Rsp5232‐809 G747E fused to His6‐MBP at 8,000 and 12,000 rpm (see also Table EV1).
-
E, F(E) Representative gel and (F) quantification (mean and standard deviation from three replicates) of in vitro ubiquitylation of Rvs167 by Rsp5232–809 WT or Rsp5232–809 G747E, in the presence of fluorescein‐labelled ubiquitin. Reaction set‐up and detection were performed as described in Fig 2C.
We then asked whether Ub‐independent oligomerization of the G747E mutant restrains ligase activity similarly to Ub‐dependent oligomerization of the WT protein. Indeed, an Rvs167 ubiquitylation assay by untagged full‐length Rsp5 showed that the G747E mutant was inactive (Fig 5E and F). Accordingly, the model suggests that the G747E defective phenotype in trafficking that was demonstrated by Lindquist and co‐workers, resulted from a constitutive oligomerization‐dependent ligase inactivation.
Conservation and cellular outcome of ubiquitylation‐dependent oligomerization: Implication for a receptor tyrosine kinase and a potassium channel
Rsp5 K432 is conserved in eukaryotic Smurf2 proteins. However, K438, which was previously detected as a target residue in higher eukaryotes, is conserved across the whole Nedd4 family (Gao et al, 2012; Udeshi et al, 2012; Fig EV2). Moreover, in our yeast two‐hybrid assay we showed that ubiquitylation on K438R promotes self‐association of Rsp5 in yeast cells. Since α1 is connected to the HECT domain by a long loop, it may afford the structural flexibility to accommodate the Ub:HECT‐UBD interaction even if ubiquitin is conjugated further down the helix. We thus tested whether ubiquitylation of this conserved lysine on α1 promotes oligomerization and inactivation of human Nedd4. For this end, we employed our approach undertaken with Rsp5 and mimicked ubiquitylation on the conserved K525 of human Nedd4 by fusing the C‐terminus of Ub to this site. Remarkably, SEC experiments showed that while apo HECT (α1Nedd4) remained a monomer, Ub fusion onto α1 promoted oligomerization of the entire Ub‐HECT (Ub‐α1Nedd4) population of Nedd4 (Fig 6A). As observed in Rsp5, mutating the conserved Ub I44 residue abolished oligomerization of the Ub‐HECT Nedd4 (Fig EV3).
Figure EV2. Sequence and structural conservation of the α1 helix.
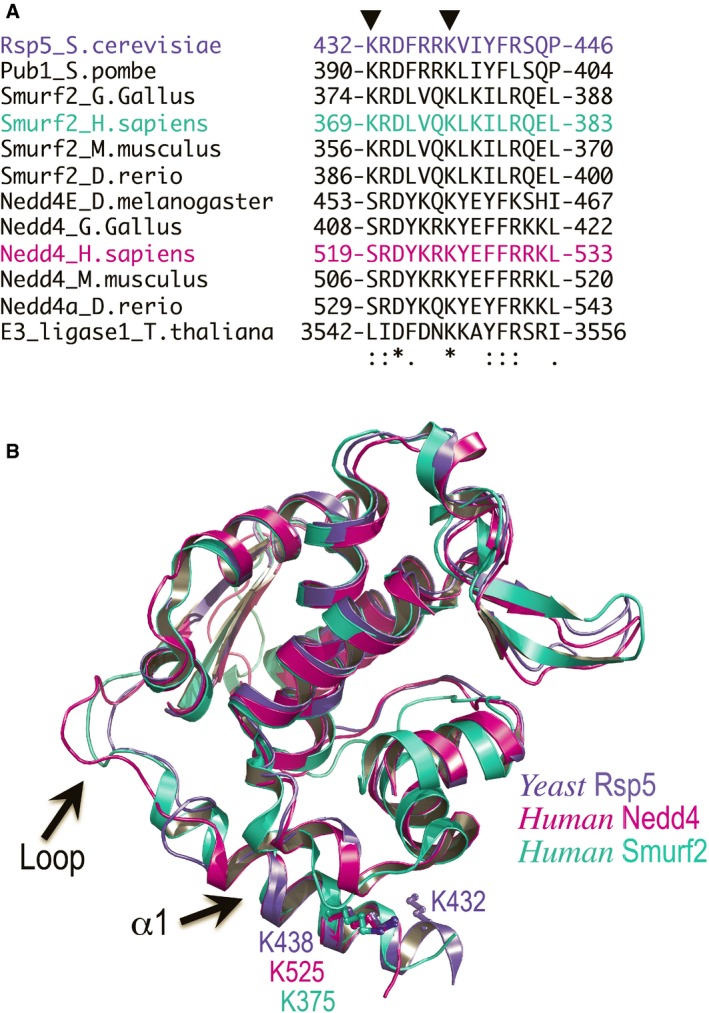
- Sequence alignment of α1 showing that K525 of human Nedd4 is fully conserved in the indicated organisms.
- The conserved Rsp5 K432 and the Nedd4 K525 are shown as ball and sticks. The loop connecting a1 to the HECT domain is highlighted. The significantly high B‐factor values of the loop atoms and the different conformations assumed in each of the structures indicate that the loop is highly flexible.
Figure 6. Conservation of ubiquitylation‐dependent oligomerization in human Nedd4 and implications in targeting of FGFR1.
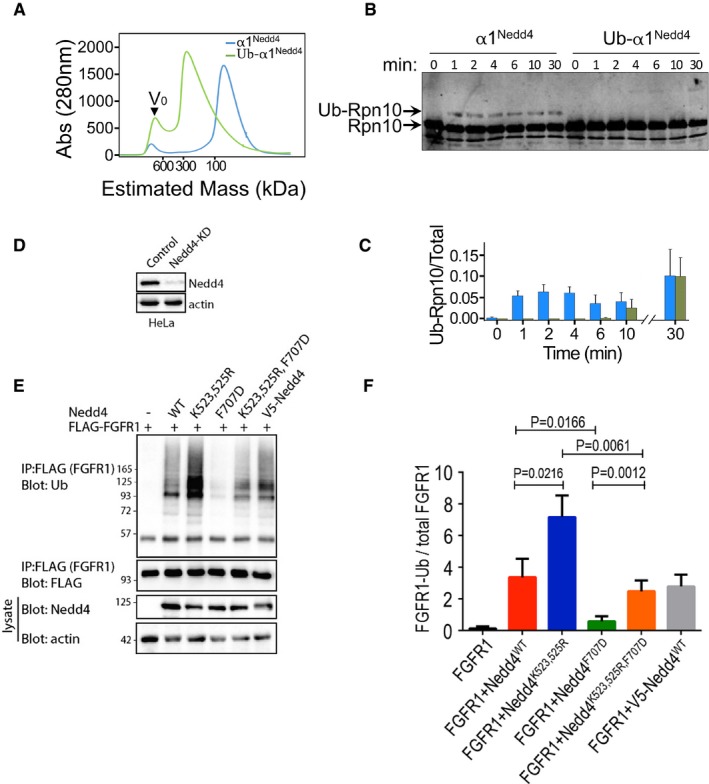
- Elution profile from size‐exclusion chromatography of apo HECT (α1Nedd4) or Ub‐fused HECT (α1Nedd4‐Ub) of Nedd4. Proteins were loaded on a Superdex 200 16/60 column, and elution was monitored by A280 measurements.
- In vitro ubiquitylation of Rpn10 by apo HECT (α1Nedd4) or Ub‐fused HECT (α1Nedd4‐Ub) of Nedd4. Reaction products were resolved by SDS–PAGE and blotted against a primary rabbit α‐Rpn10 antibody, followed by secondary IR labelled mouse α‐rabbit. Odyssey infrared imaging system was used for IR detection.
- Quantified ubiquitylated/total Rpn10 ratio (mean values and standard deviation bars from three replicates).
- Documentation of Nedd4 knockdown by shRNA in HeLa cells.
- Representative immunoblot of FGFR1 ubiquitylation (in the presence of serum) upon transfection of the indicated wild‐type (WT) and mutant human Nedd4 constructs. V5‐Nedd4 was used as an additional control.
- Quantified (mean ± SEM) ubiquitylated/total FGFR1 ratio from three separate experiments. P‐values are from Student's t‐test.
Figure EV3. Size‐exclusion chromatography of His6‐MBP‐Nedd4 fusion proteins.
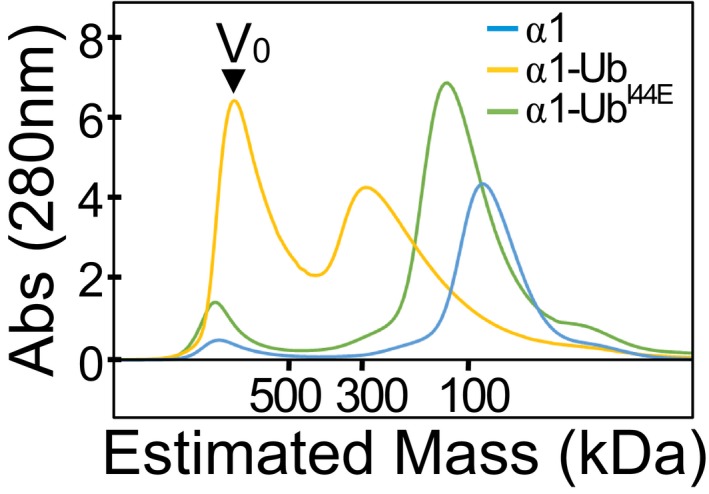
Full‐length protein and WT or I44E mutant of Ub‐fused HECT were separated on a Superdex 200 16/60 column, and elution was monitored by A280 detection.
Subsequently, we tested the effect of ubiquitylation‐dependent oligomerization on Nedd4 activity in vitro and in cells. In vitro ubiquitylation of Rpn10, followed by SDS–PAGE and anti‐Rpn10 immunoblot revealed perfect agreement with the behaviour of Rsp5 presented in Fig 2H and I (Fig 6B and C). While the apo HECT rapidly ubiquitylated Rpn10, the Ub‐HECT Nedd4 was largely inactive during the first 10 min. To investigate the significance of our in vitro results in cells, we examined the ubiquitylation of FGFR1, a well‐known substrate of Nedd4 (Persaud et al, 2011). HeLa cells stably depleted of Nedd4 by shRNA targeting its 3′UTR (Fig 6D) were transfected with wild‐type or K523,525R Nedd4. Strikingly, Western immunoblot analyses revealed that Nedd4K523,525R exhibited a significantly higher activity than Nedd4WT (Fig 6E and F). Moreover, K523,525R restored the reduced activity of the F707D mutant, which was previously shown to decrease ligase activity (Maspero et al, 2013). This suggests that K>R mutations on the Nedd4‐α1 prevented oligomerization and resulted in an unrestrained ligase.
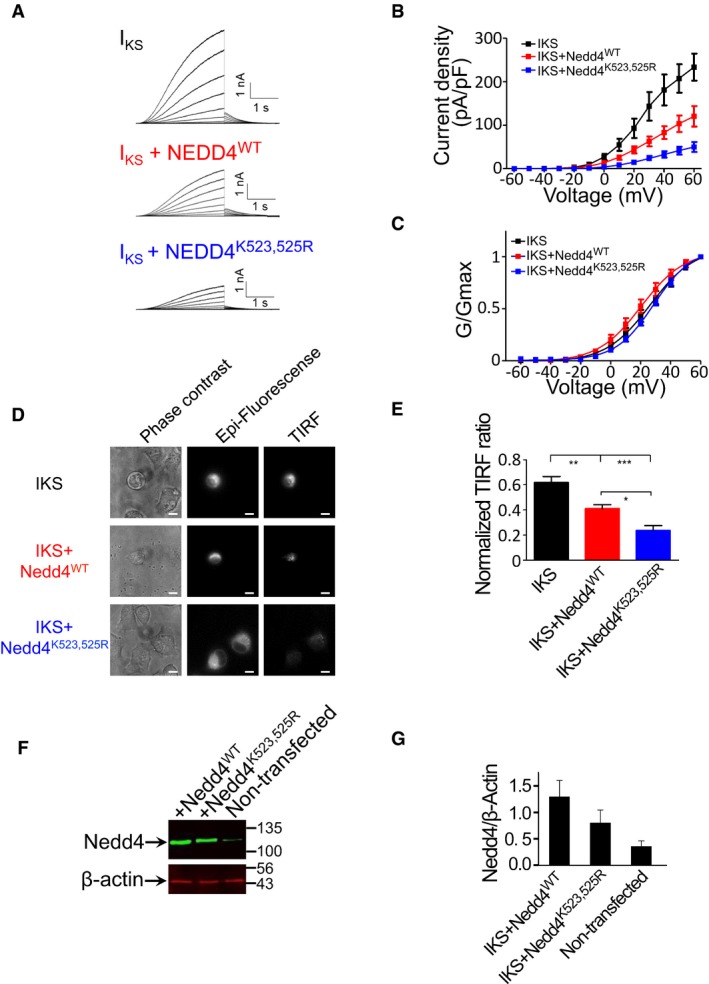
To investigate whether the physiological significance of ubiquitylation‐dependent oligomerization extends beyond RTKs, we tested the effect of this Nedd4 regulatory mechanism on the cardiac IKS potassium channel. The IKS channel is formed by the co‐assembly of KCNQ1 and KCNE1 α and β subunits and is crucial for cardiac action potential repolarization (Barhanin et al, 1996; Sanguinetti et al, 1996). Mutations in KCNQ1 and KCNE1 genes cause severe cardiac arrhythmias such as long QT syndrome and atrial fibrillation (Dvir et al, 2014a). Nedd4 and Nedd4‐like proteins bind, ubiquitylate and thereby modulate the internalization of certain ion channels bearing PY motifs in the intracellular C‐terminus of their subunits (Abriel & Staub, 2005; Staub & Rotin, 2006; Bongiorno et al, 2014). Nedd4‐dependent degradation is likely to be important for the regulation of the cell surface density of IKS potassium channels in cardiomyocytes. We therefore recorded the currents from human IKS channels as a downstream reporter for Nedd4 activity. Specifically, we co‐transfected Chinese hamster ovary (CHO) cells with a vector driving the functional expression of the tandem chimera of KCNE1 and KCNQ1 (Wang et al, 1998) in the absence or presence of WT Nedd4 ligase and recorded IKS currents by the whole‐cell patch‐clamp technique (Fig 7A and B). In line with previous work (Jespersen et al, 2007), co‐expression of WT Nedd4 significantly reduced the IKS current density. At +60 mV, the current densities were 233 ± 31 pA/pF and 120 ± 23 pA/pF for IKS and IKS + WT Nedd4, respectively (Fig 7A and B; n = 11–12; P < 0.0001). When the IKS channel subunits (KCNE1‐KCNE1) were co‐expressed with the Nedd4 K523,525R double mutant, which cannot undergo ubiquitylation‐dependent oligomerization, the IKS current density was significantly lower compared to that obtained by the co‐expression with WT Nedd4. At +60 mV, the current densities were 50 ± 11 pA/pF and 120 ± 23 pA/pF for IKS + Nedd4K523,525R and IKS + WT Nedd4, respectively (Fig 7A and B; n = 10–11; one‐way ANOVA: F(2, 32) = 15.43, P < 0.0001). Post hoc analysis confirmed significant differences between the current densities of IKS and IKS + Nedd4WT, IKS and IKS + Nedd4K523,525R and between IKS + Nedd4WT and IKS + Nedd4K523,525R (P < 0.0003). No significant differences were found in the voltage dependence of IKS channel activation in the presence of Nedd4WT or Nedd4K523,525R (Fig 7C).
Figure 7. Ubiquitylation‐dependent oligomerization mechanism regulates the cardiac IKS potassium current.
-
ARepresentative current traces from CHO cells expressing either IKS (KCNQ1 + KCNE1), IKS + Nedd4WT or IKS + Nedd4K523,525R. Cells were held at −90 mV. Membrane voltage was stepped for 3 s from −60 mV to +60 mV in 10 mV increments followed by repolarization to −60 mV for 1.5 s.
-
B, C(B) Current‐voltage (mean ± SEM) and (C) conductance‐voltage (mean ± SEM) relationships of the recorded cells (n = 10–11). Normalized conductance curves were fitted to a single Boltzmann function.
-
DPhase contrast (left panels), epi‐fluorescence (central panels) and TIRF fluorescence (right panels) images of CHO cells expressing either IKS, IKS + Nedd4WT or IKS + Nedd4K523,525R. Scale bars: 10 μm.
-
EQuantification (mean ± SEM) of the TIRF signals was normalized by the TIRF/epi‐fluorescence intensity ratio, using Fiji NIH viewer. n = 10–17; one‐way ANOVA and Bonferroni's multiple comparison test; *P < 0.05, **P < 0.01, ***P < 0.001.
-
FRepresentative immunoblot with anti‐Nedd4 of total CHO cell lysates from non‐transfected cells and cells transfected with wild‐type or mutant Nedd4. Lysates were also blotted with antibodies against endogenous β‐actin.
-
GQuantified (mean ± SEM) Nedd4/β‐actin ratio (n = 3).
To evaluate whether the decreased IKS current densities recorded from Nedd4WT or Nedd4K523,525R transfected cells corresponded to increased endocytosis, we co‐transfected CHO cells with Nedd4WT or Nedd4K523,525R in the presence of YFP tagged IKS channel. We then imaged the cells using total internal reflection fluorescence (TIRF) microscopy. In TIRF, fluorophores located within ~300 nm of the plasma membrane are selectively excited but not cytoplasmic fluorophores (Steyer & Almers, 2001; Dvir et al, 2014b). We quantified the KCNQ1‐YFP signal using a normalized TIRF value measured for a given cell as the fluorescence intensity ratio between the TIRF and the epifluorescence illumination. The lower the channel density on the cell surface, the lower the ratio. We found that the normalized TIRF ratio was significantly lower for KCNQ1‐EYFP + KCNE1 co‐expressed with Nedd4K523,525R (0.236 ± 0.045) when compared to that of KCNQ1‐EYFP + KCNE1 co‐expressed with Nedd4WT (0.411 ± 0.029). The latter ratio was also significantly lower than that of KCNQ1‐EYFP + KCNE1 expressed alone (Fig 7D and E; 0.618 ± 0.048, n = 10–17; one‐way ANOVA: F(2, 36) = 23.332, P < 0.0001). Post hoc analysis confirmed significant differences between the TIRF ratios of IKS and IKS + WT Nedd4, IKS and IKS + Nedd4K523,525R and between IKS + Nedd4WT and IKS + Nedd4K523,525R (P < 0.003). Western immunoblot analysis confirms similar cellular level of the transfected Nedd4 variants (Fig 7F and G).
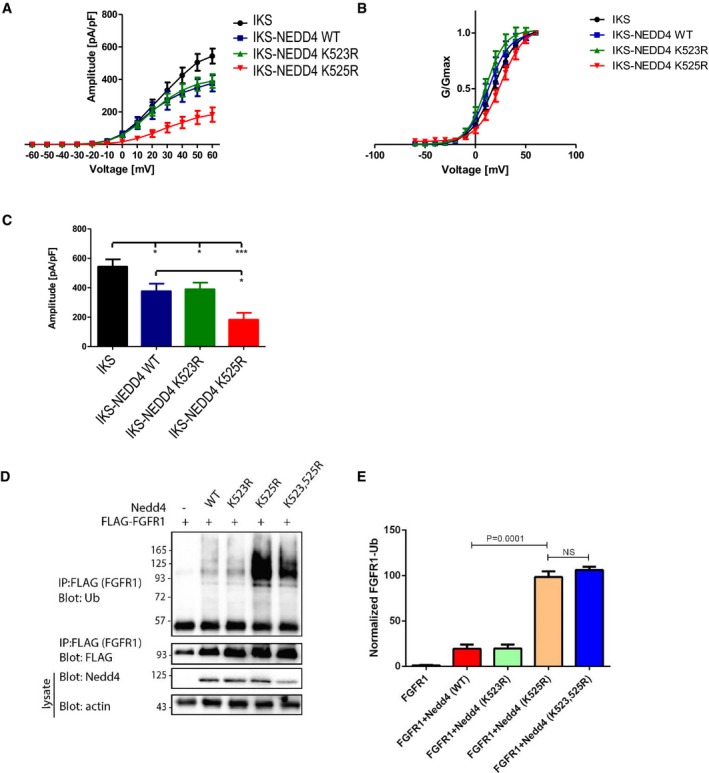
In order to pinpoint the principal lysine residue partaking in the regulation mechanism of human Nedd4, we repeated our cell biochemistry assay and electrophysiological measurements with single residue Nedd4 mutants, that is K523R or K525R. In accord with the degree of conservation (Fig EV2), it is K525, but not K523 that is entirely accountable for inactivation of the ligase. Specifically, mutating K525 and (but not K523) resulted in a dramatic increase of FGFR1 ubiquitylation, and in reduction of IKS current density (Fig EV4).
Figure EV4. Dissecting the role of individual lysine residues in Nedd4 regulation.
-
A, BQuantification (mean ± SEM) of (A) current‐voltage and (B) conductance‐voltage relationships of the recorded cells (n = 8–12), following co‐expression of IKS with Nedd4WT or mutants bearing individual lysine mutations, that is Nedd4K523R or Nedd4K525R. Cells were held at −90 mV. Membrane voltage was stepped for 3 s from −60 mV to +60 mV in 10 mV increments followed by repolarization to −60 mV for 1.5 s. Normalized conductance curves were fitted to a single Boltzmann function.
-
CQuantified (mean ± SEM) amplitude at −60 mV, for each cell type (n = 8–12). One‐way ANOVA; and Bonferroni's multiple comparison test; *P < 0.05, ***P < 0.001.
-
DRepresentative immunoblot of FGFR1 ubiquitylation (in the presence of serum) upon transfection of the indicated wild‐type and mutant human Nedd4 constructs.
-
EQuantified (mean ± SEM) ubiquitylated/total FGFR1 ratio from three separate experiments. P‐values are from Student's t‐test.
Discussion
Nedd4 family ubiquitin ligases target a large variety of membrane as well as soluble proteins. As such, it is little surprising that these ligases exhibit stringent mechanisms of control and that dysregulation of these enzymes has been implicated in various pathologies. For example, it was recently shown that amyloid‐β induces Nedd4‐dependent proteolytic targeting of AMPA receptors, leading to reduction in dendritic spine density and synaptic weakening in Parkinson's disease (Rodrigues et al, 2016). Our study reveals a novel auto‐ubiquitylation‐mediated regulatory mechanism that is conserved from yeast to humans. The described mechanism relies on both ubiquitylation of specific lysine residues located in the α1 helix and on the presence of a UBD within the HECT domain.
Involvement of the HECT‐UBD in Ub chain elongation has been demonstrated for Rsp5 and Nedd4 (French et al, 2009; Kim et al, 2011; Maspero et al, 2011). In this study, we used size‐exclusion chromatography, cross‐linking assays, sedimentation equilibrium, and functional assays, to unravel an additional role for the UBD in attracting the Ub‐conjugated α1 thus leading to a conformational change and to oligomerization‐dependent inactivation. The self‐ubiquitylation pattern of the Ub‐HECTI537D mutant shown in this study strengthens the notion of the dual role of the HECT‐UBD: for one, Ub‐HECTI537D restores Rpn10 and self‐mono‐ubiquitylation by impairing the UBD:Ub interface and reinstating the monomeric form of the ligase. At the same time, it restricts chain elongation as shown in previous studies (French et al, 2009; Kim et al, 2011; Maspero et al, 2011). The UbI44E‐HECT mutation separates these two roles by restoring the monomeric active state (like Ub‐HECTI537D) while maintaining the chain elongation activity as clearly seen for self‐ubiquitylation in Fig 4E. Ubiquitin chain elongation is enabled by interaction of the HECT‐UBD with the conjugated Ub on the substrate (Kee & Huibregtse, 2007; French et al, 2009; Maspero et al, 2011). In the crude classification of the ubiquitin “language” into “writers” (ligases), “editors” (DUBs) and “readers” (Ub‐receptors, which decode Ub signals into cellular responses), it has been suggested that ubiquitylation of Ub‐receptors (“readers”) renders them in a closed inactive conformation (Hoeller et al, 2006; Hurley et al, 2006). Within this framework, HECT domain ligases such as Rsp5 and Nedd4 would appear unique in that they share features common to both “writers” as well as “readers.”
Activity assays using monomeric and oligomeric species of Ub‐HECT Rsp5 strongly suggest that it is ubiquitylation‐induced oligomerization, rather than ubiquitylation per se, which restrains the activity of Rsp5 and Nedd4. This conclusion is strengthened by the reduced activity of the Rsp5G747E mutant, which undergoes ubiquitylation‐independent oligomerization.
The results presented in this study raise several intriguing questions regarding the control of Nedd4‐related E3 enzymes that remain to be addressed: (i) How does oligomerization inactivate these enzymes? Schulman and co‐workers highlighted the need for flexibility between the N‐lobe and the C‐lobe of the HECT domain for catalytic activity (Kamadurai et al, 2013). We thus speculate that in the oligomeric state, the tight interaction between the protomers constrains mobility between the two lobes that appears to be vital for enzyme activity. (ii) What is the basal steady state of these ligases: active monomer or inactive trimer? Furthermore, does this decision depend on the spatial and temporal context? An intriguing possibility is an “on demand” based model according to which these E3s would be kept dormant as ubiquitylated trimers until their activity is prompted by cognate substrate. Alternatively, substrate binding may be insufficient, and ligase activation may be triggered by deubiquitylation through a specific DUB enzyme. Indeed, Rsp5 was shown to be in constant interaction with Ubp2 through the intermediator protein, Rup1 (Kee et al, 2005). It will be interesting to distinguish between these possibilities in future work. (iii) Since Nedd4 regulation involves at least two mechanisms—ubiquitylation‐mediated oligomerization as shown here and inhibitory interactions between the C2 and the HECT domain that is relieved by phosphorylation—further studies are required to decipher potential crosstalk between these regulatory mechanisms. The specific challenge will be to understand whether these mechanisms are complementary, redundant or mutually exclusive.
Several Nedd4 ligases ubiquitylate multiple proteins. However, deubiquitylation, degradation and redundancy in many cases impede the ability to assign a particular Nedd4 to its cognate targets. The latter is often achieved by looking for mistargeting on a background of damaged ligase. Our discovery of a Nedd4 hyper‐activating mutation provides an alternative strategy by identification of new potential substrates through monitoring the excessive accumulation of their ubiquitylated form.
In the broader context of the extended HECT ligase family, we speculate that phosphorylation serves as an alternative mechanism for ubiquitylation‐dependent oligomerization. For example, it was shown that threonine (T485) phosphorylation of E6AP/UBE3A on α1 downregulates its activity. Disruption of this phosphorylation site (T485A) causes excessive dendritic spine development in the brain and autism (Yi et al, 2015). Alignment of human HECT proteins shows that the identified lysine (K525 in NEDD4) is conserved beyond the Nedd4 subfamily (Fig EV5). Moreover, the threonine (T485 in UBE3A) is also conserved and is sometimes substituted by serine. We speculate that the restraint mechanism is therefore conserved in other HECT proteins [possibly with some exceptions such as Huwe1 (Pandya et al, 2010; Ronchi et al, 2014)] and functions either by ubiquitylation that pulls α1 from the oligomerization interface through the attraction of Ub to the UBD or by phosphorylation that repels α1 from the oligomerization interface (probably in a similar manner to the G747E mutation in Rsp5). Along this view, we speculate that this highly conserved mechanism regulates the ubiquitylation of numerous targets and thus has a pivotal role in homoeostasis.
Figure EV5. Multiple sequence alignment of α1 sequences and post‐translation modification sites.

The α1 sequences of human Nedd4, Rsp5 and other HECT ligases were aligned with ClustalW. The conserved lysine residue of the Nedd4 family and the threonine reside of E6AP that undergo ubiquitylation and phosphorylation are indicated.
Materials and Methods
DNA cloning and mutagenesis
Rsp5 or Nedd4 and their derivatives were subcloned in‐frame with His6‐MBP into AvrII and AscI restriction endonuclease sites of a modified pCDF‐duet vector (Keren‐Kaplan et al, 2012). Ubiquitin‐Rsp5 chimeras were generated by PCR amplification of overlapping DNA fragments and plasmid reconstruction according to the Gibson protocol (Gibson et al, 2009). Point mutations were introduced using the ExSite protocol (Stratagene).
The sequence of all constructs was verified by DNA sequencing at our home facilities to confirm that we obtained the desired clones with no unintended mutations. Cloning of Rpn10 in pCDF‐His6‐MBP and of the pGEN plasmids used for ubiquitylation in bacteria was previously described in (Keren‐Kaplan et al, 2012). Plasmids are listed in Table EV3.
β‐galactosidase Y2H assays
Saccharomyces cerevisiae strain PJ694 (trp1‐901 leu2‐3,112, ura3‐52, his3Δ200, gal4Δ, gal80Δ, prGAL2:ADE2 lys2::prGAL1::HIS3 met2::prGAL7‐LacZ) was transformed with plasmids expressing wild‐type or mutant GBD‐Rsp5 and GAD‐Rsp5 by standard lithium acetate methods (Knop et al, 1999). β‐galactosidase assays were performed using cells in log‐phase (–Leu –Ura medium with 2% glucose). Cell concentration was determined by reading 80 μl of cells at 595 nm. 40 μl of cells was added to the β‐galactosidase reaction mix (40 μl YPER (Pierce 78990), 80 μl Z‐buffer (120 mM Na2HPO4, 80 mM NaH2PO4, 20 mM KCl, 2 mM MgSO4), 24 μl ONPG (4 mg/ml), 0.4 μl β‐mercaptoethanol) and incubated at 30°C for 50 min in Eppendorf tubes. Reactions were stopped by addition of 56 μl 1 M Na2CO3. The Eppendorf tubes were centrifuged for 1 min at full speed to pellet the cell debris, and 200 μl supernatant was removed and absorbance read at 415 nm using a microplate reader. Miller units were calculated by the equation Miller units = (1,000 × A415)/(50 × 0.04 × ((A595 − 0.055) × 5). Three biological replicates were measured. Error bars are ± 1 SD.
Protein expression and purification
Rosetta2 (λDE3) BL21 E. coli cells were transformed with Rsp5‐containing pCDF‐duet or pMAL plasmids. Culture growth was performed at 37°C in terrific broth medium supplemented with the selective antibiotics. Induction was performed with 1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) at 16°C for 16–20 h. Harvested cells were re‐suspended in washing buffer that contains 50 mM Tris, 150 mM NaCl, 3 mM β‐mercaptoethanol supplemented with lysozyme and AEBSF (4‐(2‐aminoethyl)benzenesulphonyl fluoride hydrochloride) protease inhibitor. Complete lysis and DNA shearing were achieved by sonication following centrifugation to isolate the soluble fraction. Proteins were affinity purified on a column of amylose beads (New England Biolabs) and washed with 1 l of buffer. Elution was performed with washing buffer supplemented with 20 mM of maltose. MBP‐tag removal was performed with tobacco etch virus (TEV) protease.
His6‐Ubc4 and His6‐ubiquitin were purified using the same protocol with nickel resin instead of amylose. Size‐exclusion chromatography was used for further purification of the affinity‐purified proteins. Expression and purification of Rpn10 was previously described in Keren‐Kaplan & Prag (2012).
Size‐exclusion chromatography
Size‐exclusion chromatography was performed on a Superdex 200 16/60 calibrated column with an AKTAprime plus (GE healthcare) FPLC system. Quantities of injected proteins ranged between 4 and 60 mg, depending on the experiment and flow speed was set to 1–1.5 ml/min.
Cross‐linking experiments
To eliminate non‐specific cross‐linking, a mild cross‐linker disuccinimidyl suberate (DSS) was used. Reaction was carried out by incubating 10 μM of protein with a buffer containing 25 mM HEPES, 150 mM NaCl and 0.5 mM DSS. Quenching was performed at increasing time points by boiling samples with SDS‐loading buffer. Quenched samples were resolved on SDS–PAGE followed by Coomassie blue staining for detection.
Mass spectrometry
Mass spectroscopy analyses were performed at the Sanford Burnham Prebys Medical Discovery Institute Proteomics Facility. Briefly, gel bands containing ubiquitylated MBP‐Rsp5232–809 were excised and subjected to de‐staining, reduction and alkylation. Gel bands were digested with trypsin, and peptides were extracted, concentrated and desalted. Desalted peptides were vacuum‐dried and re‐dissolved in LC/MS loading buffer (2% acetonitrile in 0.1% formic acid in water). Tryptic peptides were analysed by 1D LC‐MS/MS analysis utilizing advance low‐flow electrospray ionization source and a Magic C18 AQ reversed‐phase column (Michrom Bioresources Inc.) with a 2‐h LC gradient coupled to the OrbiTrap XL mass spectrometer (Thermo Fisher Scientific). Search results were viewed, sorted, filtered and analysed using Sorcerer Enterprise v.4.0.4 release (Sage‐N Research Inc.) and Peptide/Protein prophet v.4.4.0 (ISB).
Analytical ultracentrifugation
Physical parameters of proteins and buffers were calculated with SEDNTREP based on amino acid sequence and buffer composition. Experiments were performed in‐house using our ProteomLab XL‐I Beckman Coulter centrifuge. Purified proteins were loaded in double sector quartz cells that were mounted in a 4 holes An‐60‐Ti rotor. Data were averaged from five replicates of A280 measurements distanced by 0.003 cm along the cell radius, and all experiments were carried out at 20°C. For sedimentation velocity, 400 μl of SEC purified constructs was loaded at concentrations indicated in Table EV2. Rotor speed acceleration was set up to 35,000 rpm, and full‐length cell A280 coverage was performed every 1 min for a total of 200 scans. Continuous c(s) distribution of species was analysed in Sedfit (Schuck, 2000). For sedimentation equilibrium, SEC purified constructs were loaded at an OD280 of 1 in a volume of 110 μl. After 20 h, measurements were taken every two hours until equilibrium was reached at 8,000 and then at 12,000 rpm. Analysis was performed with a model of single ideally sedimenting solute. Proteins that showed larger average mass than their monomeric size were then analysed in a model of multiple sedimenting species with mass conservation constraints (Vistica et al, 2004). In this model, we substituted molar mass with full integers of the monomeric size to test alternative models of self‐association until reaching the best fits. Plots were prepared by exporting the Sedfit/Sedphat analysed data to GUSSI which was kindly distributed by Dr. Chad Brautigam (http://biophysics.swmed.edu/MBR/software.html).
Microscale thermophoresis (MST) analyses
Self‐binding experiments of Ub‐HECT or UbI44E‐HECT were carried out with Monolith NT.115 (Nano Temper Technologies; Wienken et al, 2010). Briefly, samples consisting of a 10 μl fixed concentration of NT647‐labelled Rsp5 were mixed with 10 μl of titrated of unlabelled identical protein (16 samples of the indicated titrations). After a short incubation, the samples were loaded into standard glass capillaries before data collection. Experiments were carried out in 50 mM Tris (pH 7.6), 150 mM NaCl, 10 mM MgCl2 and 0.05% Tween 20. Data collection was performed with 90% of the LED and 20% of MST power.
Fluorescein labelling of ubiquitin
Ubiquitin labelling was achieved by substitution of K48 to Cys. Following purification, 50–100 μM of purified UbK48C was incubated with a fourfold excess of fluorescein‐5‐maleimide in buffer containing 150 mM NaCl, 20 mM Tris–HCl (pH 7.5), and 1 mM tris(2‐carboxyethyl)phosphine for 2 h at room temperature and quenched by adding a 10‐fold excess β‐mercaptoethanol for 5 min. The labelled protein was purified using a PD miniTrap G25 gravity column pre‐equilibrated in 150 mM NaCl, 20 mM Tris (pH 7.5) and 1 mM tris(2‐carboxyethyl)phosphine following the protocol of the manufacturer. The labelled ubiquitin was visualized on SDS–PAGE gel and scanned in a Typhoon laser at 488 nm. Final concentrations were measured spectrophotometrically at 280 and 495 nm and stored at −80°C until use.
Ubiquitylation in vitro and in cells
Rpn10 or Rvs167 ubiquitylation was set up by incubating 10–60 μg/ml of Rpn10/Rvs167 with 10 μg/ml of UBA1, 20 μg/ml of Ubc4, 10 μg/ml of fluorescein‐labelled UbK48C and 40–100 μg/ml of various His6‐MBP fused Rsp5 constructs supplemented with 13 mM ATP, 10 mM MgCl and 1 mM DTT. Reaction samples were quenched at increasing time intervals by boiling the sample in Laemmli gel‐loading buffer and resolved by SDS–PAGE. Ub conjugates were visualized in a Typhoon laser scanner at 488 nm. The final concentrations were measured spectrophotometrically at 280 and 495 nm and stored at −80°C until use. In the Nedd4 functional assay in vitro, proteins were resolved by SDS–PAGE, transferred to a nitrocellulose membrane and blotted against primary Rabbit α‐Rpn10 and secondary IR labelled mouse α‐rabbit. For ubiquitylation in cells, HeLa cells stably expressing shRNA targeting the Nedd4 3′UTR were transfected (24–48 h) with FLAG‐tagged human FGFR1 and human Nedd4 (WT, or mutants), in the presence DMEM+10% FBS. Cells were then lysed in lysis buffer, as described. Briefly, to detect receptor ubiquitylation, 1 mg of cleared cell lysates was treated with 1% SDS and boiled for 5 min to dissociate protein complexes. The boiled lysates were then diluted 11 times with lysis buffer before IP of the receptor with anti‐FLAG antibodies and blotting with anti‐ubiquitin (anti‐Ub, Santa Cruz #P4D1) antibodies, or blotting for FLAG (Sigma). Control lysates were blotted for human Nedd4 (Abcam #ab27979) and actin (MP Biomedical clone C4 691001).
Cell culture and transfections
Chinese hamster ovary (CHO) and human embryonic kidney (HEK293) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM glutamine, 10% foetal calf serum and antibiotics, incubated at 37°C in 5% CO2 as previously described (Dvir et al, 2014b). HeLa cells stably knocked‐down (via 3′UTR) for Nedd4 were grown in DMEM+10% FBS and transfected as described above. For electrophysiology, CHO cells were seeded on poly‐L‐lysine‐coated glass coverslips in a 24‐multiwell plate and were transfected with the tandem KCNE1‐KCNQ1 MK24 (1 μg) in the absence or presence of WT or Mut Nedd 4 (0.5 μg) using X‐tremeGENE 9 transfection reagent (Roche). pIRES‐CD8 (0.3 μg) was co‐transfected as a surface marker. For TIRF imaging, CHO cells were seeded on poly‐L‐lysine‐coated 1.5‐mm glass coverslips in a 12‐well multiwell plate and transfected with 0.5 μg of KCNQ1‐YFP and 0.5 μg KCNE1 in the absence or presence of 0.5 μg WT or mutant Nedd4. For Western blotting, CHO cells were seeded on 60‐mm dishes and transfected with 5 μg WT or Mut Nedd4 using the using 15 μl X‐tremeGENE 9 transfection reagent (Roche).
TIRF microscopy
Fluorescent images of CHO cells were taken at room temperature by TIRF microscopy as previously described in Dvir et al (2014b). CHO cells were imaged 48 h after transfection at room temperature (20–22°C). Images were taken with a Zeiss inverted microscope (Axio Observer Z1) equipped with an alpha Plan‐Fluar 100× oil immersion objective (NA 1.45). Fluorescent cells were excited with the 514‐nm line of an air‐cooled argon‐ion‐laser (LASOS 771) with light intensities of 5% for YFP. Emission images were acquired via a 575‐nm filter set (Zeiss, filter sets 53) at exposure time of 100 ms and gain*400 with an EM CCD camera (QuantEM:512SC) controlled by an AxioVision software version 4.8.2 that was also used with the Fiji software for quantitation.
Electrophysiology and data analysis
CHO cells were recorded in the whole‐cell configuration of the patch‐clamp technique as previously described (Dvir et al, 2014b). Cells were held at −90 mV. To establish the current‐voltage relations, voltage was stepped for 3 s from −60 mV to +60 mV in 10 mV increments followed by repolarization to −60 mV for 1.5 s. Data analysis was performed using the Clampfit program (pClamp10, Axon Instruments), Microsoft Excel and Prism 5.0 (GraphPad). Conductance (G) was calculated as G = I/(V − Vrev). G was then normalized to the maximal conductance. Activation curves were fitted to a single Boltzmann distribution according to G/Gmax = 1/{1 + exp[(V50 − V)/s]}, where V50 is the voltage at which the current is half‐activated and s is the slope factor. All data were expressed as mean ± SEM. Statistically significant differences were assessed by one‐way ANOVA. Significant interactions were followed by Bonferroni's multiple comparison test post hoc comparisons.
In silico models
Conservation of Rsp5 surface residues was calculated in the ConSurf webserver (Armon et al, 2001) and presented in Pymol. Generation of the Ub‐HECT trimeric model was performed by superimposing the Rsp5 HECT:Ub complex onto the E6AP trimeric structure (3OLM and 1C4Z, respectively). Energy minimization was done in Refmac5 (Murshudov et al, 1997). Fragment‐based structural alignment of the Rsp5, Smurf2 and Nedd4 α1‐helices [3OLM (Kim et al, 2011), 1ZVD (Ogunjimi et al, 2005) and 2XBF (Maspero et al, 2011), respectively] was prepared in Pymol. Sequence alignment was constructed by ClustalW.
Author contributions
IA and GP conceived and designed the hypothesis; IA constructed most clones, performed all biochemical and biophysical experiments and analyses and wrote the paper; WST performed the electrophysiology and TIRF experiments; KM and DAW performed the proteomic analysis; OL‐K and IP provided technical assistance. TK‐K purified proteins for in vitro assays. KJS‐L provided technical and intellectual support for the yeast two‐hybrid experiments. AP performed FGFR1 ubiquitylation experiments; MK and DR provided intellectual support. RW and BM produced labelled ubiquitin and provided technical assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File
Acknowledgements
We thank Simona Polo for the kind gift of the human Nedd4 plasmid. Robert Kass (Columbia University) and Bernard Attali (Tel Aviv University) are acknowledged, respectively, for the kind gift of the tandem construct KCNE1‐KCNQ1 MK24 and the help in performing the TIRF and electrophysiology experiments. We thank Zvulun Elazar and Alik Demishtein (Weizmann Institute of Science) for kindly providing us with Nedd4 antibodies. We thank Peter Schuck (NIBIB‐NIH) for help and constructive comments on AUC. We thank Moran Jerabek‐Willemsen from NanoTemper Technologies for its guidance with the MST experiment. This research was supported by grants from the Israeli Science Foundation (grants numbers 1695/08 and 464/11), from the EC FP7 Marie Curie International Reintegration Grant (PIRG03‐GA‐ 2008‐231079), from the Israeli Ministry of Health (5108), and from the Marianna and Jorge Saia Fund for HIV and Parkinson Diseases to GP. The Constantiner Institute for Molecular Genetics and the Anat Krauskopf Travel Fund supported IA. Parts of this work were funded by grants from the US National Institutes of Health to DAW (GM105802‐01A1 and P30 grants CA030199‐32 and GM085764‐03), and the Canadian Institute of Health Research to (MOP‐130422) and Canadian Foundation for Innovation (CRC, Tier I) to DR, and grants from the Israel Science Foundation and the Israel Cancer Research Fund to MK.
The EMBO Journal (2017) 36: 425–440
See also: S Hill & G Kleiger (February 2017)
References
- Abriel H, Staub O (2005) Ubiquitylation of ion channels. Physiology (Bethesda) 20: 398–407 [DOI] [PubMed] [Google Scholar]
- Armon A, Graur D, Ben‐Tal N (2001) ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J Mol Biol 307: 447–463 [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G (1996) KvLQT1 and IsK (minK) proteins associate to form the IKS cardiac potassium current. Nature 384: 78–80 [DOI] [PubMed] [Google Scholar]
- Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, Krogan NJ (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boase NA, Kumar S (2015) NEDD4: the founding member of a family of ubiquitin‐protein ligases. Gene 557: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongiorno D, Schuetz F, Poronnik P, Adams DJ (2014) Regulation of voltage‐gated ion channels in excitable cells by the ubiquitin ligases Nedd4 and Nedd4‐2. Channels 5: 79–88 [DOI] [PubMed] [Google Scholar]
- Chen C, Matesic LE (2007) The Nedd4‐like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev 26: 587–604 [DOI] [PubMed] [Google Scholar]
- Dunn R, Hicke L (2001) Domains of the Rsp5 ubiquitin‐protein ligase required for receptor‐mediated and fluid‐phase endocytosis. Mol Biol Cell 12: 421–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn R, Klos DA, Adler AS, Hicke L (2004) The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo. J Cell Biol 165: 134–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir M, Peretz A, Haitin Y, Attali B (2014a) Recent molecular insights from mutated IKS channels in cardiac arrhythmia. Curr Opin Pharmacol 15: 74–82 [DOI] [PubMed] [Google Scholar]
- Dvir M, Strulovich R, Sachyani D, Ben‐Tal Cohen I, Haitin Y, Dessauer C, Pongs O, Kass R, Hirsch JA, Attali B (2014b) Long QT mutations at the interface between KCNQ1 helix C and KCNE1 disrupt I(KS) regulation by PKA and PIP2 . J Cell Sci 127: 3943–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French ME, Kretzmann BR, Hicke L (2009) Regulation of the RSP5 ubiquitin ligase by an intrinsic ubiquitin‐binding site. J Biol Chem 284: 12071–12079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, Karin M (2006) Activation of the E3 ubiquitin ligase Itch through a phosphorylation‐induced conformational change. Proc Natl Acad Sci USA 103: 1717–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Liu Z, Wang Y, Cheng H, Yang Q, Guo A, Ren J, Xue Y (2012) UUCD: a family‐based database of ubiquitin and ubiquitin‐like conjugation. Nucleic Acids Res 41: 445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Harvey KF, Kumar S (1999) Nedd4‐like proteins: an emerging family of ubiquitin‐protein ligases implicated in diverse cellular functions. Trends Cell Biol 9: 166–169 [DOI] [PubMed] [Google Scholar]
- Hein C, Springael JY, Volland C, Haguenauer‐Tsapis R, Andre B (1995) NPI1, an essential yeast gene involved in induced degradation of Gap1 and Fur4 permeases, encodes the Rsp5 ubiquitin‐protein ligase. Mol Microbiol 18: 77–87 [DOI] [PubMed] [Google Scholar]
- Hoeller D, Crosetto N, Blagoev B, Raiborg C, Tikkanen R, Wagner S, Kowanetz K, Breitling R, Mann M, Stenmark H, Dikic I (2006) Regulation of ubiquitin‐binding proteins by monoubiquitination. Nat Cell Biol 8: 163–169 [DOI] [PubMed] [Google Scholar]
- Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP (1999) Structure of an E6AP‐UbcH7 complex: insights into ubiquitination by the E2‐E3 enzyme cascade. Science 286: 1321–1326 [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G (2006) Ubiquitin‐binding domains. Biochem J 399: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isasa M, Katz EJ, Kim W, Yugo V, González S, Kirkpatrick DS, Thomson TM, Finley D, Gygi SP, Crosas B (2010) Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome. Mol Cell 38: 733–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen T, Membrez M, Nicolas CS, Pitard B, Staub O, Olesen SP, Baró I, Abriel H (2007) The KCNQ1 potassium channel is down‐regulated by ubiquitylating enzymes of the Nedd4/Nedd4‐like family. Cardiovasc Res 74: 64–74 [DOI] [PubMed] [Google Scholar]
- Kamadurai HB, Qiu Y, Deng A, Harrison JS, Macdonald C, Actis M, Rodrigues P, Miller DJ, Souphron J, Lewis SM, Kurinov I, Fujii N, Hammel M, Piper R, Kuhlman B, Schulman BA (2013) Mechanism of ubiquitin ligation and lysine prioritization by a HECT E3. Elife 2: e00828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O (2001) A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15: 204–214 [DOI] [PubMed] [Google Scholar]
- Kee Y, Lyon N, Huibregtse JM (2005) The Rsp5 ubiquitin ligase is coupled to and antagonized by the Ubp2 deubiquitinating enzyme. EMBO J 24: 2414–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, Huibregtse JM (2007) Regulation of catalytic activities of HECT ubiquitin ligases. Biochem Biophys Res Commun 354: 329–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren‐Kaplan T, Attali I, Motamedchaboki K, Davis BA, Tanner N, Reshef Y, Laudon E, Kolot M, Levin‐Kravets O, Kleifeld O, Glickman M, Horazdovsky BF, Wolf DA, Prag G (2012) Synthetic biology approach to reconstituting the ubiquitylation cascade in bacteria. EMBO J 31: 378–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren‐Kaplan T, Prag G (2012) Purification and crystallization of mono‐ubiquitylated ubiquitin receptor Rpn10. Acta Crystallogr Sect F Struct Biol Cryst Commun 68: 1120–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HC, Steffen AM, Oldham ML, Chen J, Huibregtse JM (2011) Structure and function of a HECT domain ubiquitin‐binding site. EMBO Rep 12: 334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E (1999) Epitope tagging of yeast genes using a PCR‐based strategy: more tags and improved practical routines. Yeast 15: 963–972 [DOI] [PubMed] [Google Scholar]
- Lee JRE, Oestreich AJ, Payne JA, Gunawan MS, Norgan AP, Katzmann DJ (2009) The HECT domain of the ubiquitin ligase Rsp5 contributes to substrate recognition. J Biol Chem 284: 32126–32137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léon S, Erpapazoglou Z, Haguenauer‐Tsapis R (2008) Ear1p and Ssh4p are new adaptors of the ubiquitin ligase Rsp5p for cargo ubiquitylation and sorting at multivesicular bodies. Mol Biol Cell 19: 2379–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Xie P, Zhao F, Shu J, Li L, Zhan Y, Zhang L (2015a) F‐box protein Fbxo3 targets Smurf1 ubiquitin ligase for ubiquitination and degradation. Biochem Biophys Res Commun 458: 941–945 [DOI] [PubMed] [Google Scholar]
- Li M, Rong Y, Chuang YS, Peng D, Emr SD (2015b) Ubiquitin‐dependent lysosomal membrane protein sorting and degradation. Mol Cell 57: 467–478 [DOI] [PubMed] [Google Scholar]
- Lin A, Hou Q, Jarzylo L, Amato S, Gilbert J, Shang F, Man HY (2011) Nedd4‐mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J Neurochem 119: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wan L, Liu P, Inuzuka H, Liu J, Wang Z, Wei W, Liu J, Wan L, Liu P, Inuzuka H, Liu J, Wang Z, Wei W (2014) SCFβ‐TRCP ‐mediated degradation of NEDD4 inhibits tumorigenesis through modulating the PTEN/Akt signaling pathway. Oncotarget 5: 1026–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, Lin YY, Qian J, Tao SC, Zhu J, Pickart C, Zhu H (2008) Functional dissection of a HECT ubiquitin E3 ligase. Mol Cell Proteomics 7: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maspero E, Mari S, Valentini E, Musacchio A, Fish A, Pasqualato S, Polo S (2011) Structure of the HECT: ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep 12: 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maspero E, Valentini E, Mari S, Cecatiello V, Soffientini P, Pasqualato S, Polo S (2013) Structure of a ubiquitin‐loaded HECT ligase reveals the molecular basis for catalytic priming. Nat Struct Mol Biol 20: 696–701 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Ogunjimi AA, Briant DJ, Pece‐Barbara N, Le Roy C, Di Guglielmo GM, Kavsak P, Rasmussen RK, Seet BT, Sicheri F, Wrana JL (2005) Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell 19: 297–308 [DOI] [PubMed] [Google Scholar]
- Pandya RK, Partridge JR, Love KR, Schwartz TU, Ploegh HL (2010) A structural element within the HUWE1 HECT domain modulates self‐ubiquitination and substrate ubiquitination activities. J Biol Chem 285: 5664–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud A, Alberts P, Hayes M, Guettler S, Clarke I, Sicheri F, Dirks P, Ciruna B, Rotin D (2011) Nedd4‐1 binds and ubiquitylates activated FGFR1 to control its endocytosis and function. EMBO J 30: 3259–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud A, Alberts P, Mari S, Tong J, Murchie R, Maspero E, Safi F, Moran MF, Polo S, Rotin D (2014) Tyrosine phosphorylation of NEDD4 activates its ubiquitin ligase activity. Sci Signal 7: ra95 [DOI] [PubMed] [Google Scholar]
- Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP (2002) A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature 416: 451–455 [DOI] [PubMed] [Google Scholar]
- Prag G, Misra S, Jones EA, Ghirlando R, Davies BA, Horazdovsky BF, Hurley JH (2003) Mechanism of ubiquitin recognition by the CUE domain of Vps9p. Cell 113: 609–620 [DOI] [PubMed] [Google Scholar]
- Rodrigues EM, Scudder SL, Goo MS, Patrick GN (2016) Aβ‐Induced synaptic alterations require the E3 ubiquitin ligase Nedd4‐1. J Neurosci 36: 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi VP, Klein JM, Edwards DJ, Haas AL (2014) The active form of E6‐associated protein (E6AP)/UBE3A ubiquitin ligase is an oligomer. J Biol Chem 289: 1033–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384: 80–83 [DOI] [PubMed] [Google Scholar]
- Schuck P (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, De Craene JO, André B (1999) The yeast Npi1/Rsp5 ubiquitin ligase lacking its N‐terminal C2 domain is competent for ubiquitination but not for subsequent endocytosis of the gap1 permease. Biochem Biophys Res Commun 257: 561–566 [DOI] [PubMed] [Google Scholar]
- Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D (1996) WW domains of Nedd4 bind to the proline‐rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J 15: 2371–2380 [PMC free article] [PubMed] [Google Scholar]
- Staub O, Rotin D (2006) Role of ubiquitylation in cellular membrane transport. Physiol Rev 86: 669–707 [DOI] [PubMed] [Google Scholar]
- Steyer JA, Almers W (2001) A real‐time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol 2: 268–275 [DOI] [PubMed] [Google Scholar]
- Tardiff DF, Jui NT, Khurana V, Tambe MA, Thompson ML, Chung CY, Kamadurai HB, Kim HT, Lancaster AK, Caldwell KA, Caldwell GA, Rochet JC, Buchwald SL, Lindquist S (2013) Yeast Reveal a ‘Druggable’ Rsp5/Nedd4 network that ameliorates α‐synuclein toxicity in neurons. Science 342: 979–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL (2011) Ubiquitin ligase Nedd4 promotes alpha‐synuclein degradation by the endosomal‐lysosomal pathway. Proc Natl Acad Sci USA 108: 17004–17009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, Carr SA (2012) Refined preparation and use of anti‐diglycine remnant (K‐ε‐GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics 12: 825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, Schuck P (2004) Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem 326: 234–256 [DOI] [PubMed] [Google Scholar]
- Wang W, Xia J, Kass RS (1998) MinK‐KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J Biol Chem 273: 34069–34074 [DOI] [PubMed] [Google Scholar]
- Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W (2010) Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain‐mediated auto‐inhibition. J Biol Chem 285: 12279–12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S (2010) Protein‐binding assays in biological liquids using microscale thermophoresis. Nat Commun 1: 100 [DOI] [PubMed] [Google Scholar]
- Wiesner S, Ogunjimi AA, Wang HR, Rotin D, Sicheri F, Wrana JL, Forman‐Kay JD (2007) Autoinhibition of the HECT‐type ubiquitin ligase Smurf2 through its C2 domain. Cell 130: 651–662 [DOI] [PubMed] [Google Scholar]
- Woelk T, Oldrini B, Maspero E, Confalonieri S, Cavallaro E, Di Fiore PP, Polo S (2006) Molecular mechanisms of coupled monoubiquitination. Nat Cell Biol 8: 1246–1254 [DOI] [PubMed] [Google Scholar]
- Wu L, Peng J, Kong H, Yang P, He F, Deng X, Gan N, Yin F (2015) The role of ubiquitin/Nedd4‐2 in the pathogenesis of mesial temporal lobe epilepsy. Physiol Behav 143: 104–112 [DOI] [PubMed] [Google Scholar]
- Yi JJ, Berrios J, Newbern JM, Snider WD, Philpot BD, Hahn KM, Zylka MJ (2015) An autism‐linked mutation disables phosphorylation control of UBE3A. Cell 162: 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F, Xu J, Harris RC (2009) Nedd4 mediates ErbB4 JM‐a/CYT‐1 ICD ubiquitination and degradation in MDCK II cells. FASEB J 23: 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu KP, Sartori MA, Kamadurai HB, Ordureau A, Jiang C, Mercredi PY, Murchie R, Hu J, Persaud A, Mukherjee M, Li N, Doye A, Walker JR, Sheng Y, Hao Z, Li Y, Brown KR, Lemichez E, Chen J et al (2016) System‐wide modulation of HECT E3 ligases with selective ubiquitin variant probes. Mol Cell 62: 121–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH (1999) A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400: 687–693 [DOI] [PubMed] [Google Scholar]
- Zou X, Levy‐Cohen G, Blank M (2015) Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim Biophys Acta 1856: 91–106 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Review Process File